

Abstract
Nitric oxide (NO) is an important second messenger molecule for blood pressure homeostasis, as a neurotransmitter, and in the immune defense system. Excessive NO can lead to neurodegeneration and connective tissue damage. Three different isozymes of the enzyme nitric oxide synthase regulate NO production in endothelial (eNOS), neuronal (nNOS), and macrophage (iNOS) cells. Whereas creating a lower level of NO in some cells could be beneficial, it also could be detrimental to the protective effects that NO has on other cells. Therefore, it is essential that therapeutic NOS inhibitors be made that are subtype selective. Previously, we reported a series of nitroarginine-containing dipeptide amides as potent and selective nNOS inhibitors. Here we synthesize peptidomimetic hydroxyethylene isosteres of these dipeptide amides for potential increased bioavailability. None of the compounds is as potent or selective as the dipeptide amides, but they exhibit good inhibition and selectivity. When the terminal amino group was converted to a hydroxyl group, potency and selectivity greatly diminished, supporting the importance of the terminal amino group for binding.
1. Introduction
Nitric oxide (NO), an important biomolecule with a wide array of functions, is a cell-signaling agent that is involved in the cardiovascular, gastrointestinal, genitourinary, respiratory, and nervous systems.1 NO is known to be involved in important processes, such as neuronal transmission, cytoprotection, and platelet aggregation. The regulation of NO biosynthesis is the responsibility of the heme-containing metalloenzyme nitric oxide synthase (NOS) (EC 1.14.13.39).2 NOS exists in three distinct isoforms: the constitutively expressed endothelial isoform (eNOS) controls blood pressure by the regulation of smooth muscle relaxation and is involved in the inhibition of platelet and white blood cell adhesion and to suppress the replication of smooth muscle cells.3 Pharmacological inhibition of eNOS in animal models was shown to cause vasoconstriction, hypertension, and enhanced platelet activation.4 “Knockout” mice are more prone to atherogenesis and developing aneurysms.5 These inhibition experiments strongly support the importance of NO production from the endothelial isoform. The isoform originally identified in neuronal cells (nNOS), also constitutive, produces NO that is known to be involved in neurotransmission and is important for brain development and learning,6 modification of pain perception,7 and long-term potentiation.8 The inducible form of the enzyme (iNOS) is expressed in macrophages (white blood cells) as an immune response.9 The NO produced from iNOS acts as a cytotoxic agent against bacterial endotoxins, pro-inflammatory cytokines, protozoa, fungi, and viruses.10,11
Because of its wide range of function, nitric oxide has gained much interest in the field of medicinal chemistry. The overproduction of NO has been implicated in pathophysiological changes in virtually every organ system linking it to a large variety of disease states. Excess generation of NO from nNOS has been linked to the ischemia and neurodegeneration resulting from stroke,12 migraine headache,13 Parkinson’s disease,14 Alzheimer’s disease,15 amyotrophic lateral sclerosis,16 and Huntington’s disease.17 Enhanced NO derived from iNOS has been related to arthritis,18 colitis,19 septic shock,20 inflammatory bowel disease,21 and asthma.22,23 Since overproduction has been linked to the variety of disease states discussed above, it would be beneficial to attenuate the generation of NO directly related to a specific condition. Whereas creating a lower level of NO in some cells could be beneficial, it also could be detrimental to the protective effects that NO has on other cells. Therefore, it is essential that therapeutic NOS inhibitors be made that are subtype selective. Selectivity is especially needed over eNOS because of its importance in the fundamental physiology of blood pressure homeostasis.
Crystal structure studies have shown that the active sites of eNOS and iNOS are nearly identical.24,25 However, the height above the heme cofactor differs among the isoforms creating a difference in active site size that decreases in the order nNOS > iNOS > eNOS.26 Along with the difference in size, there appear to be subtle, albeit relatively minor, structural differences among the substrate binding sites of the three isozymes.27,28,29,30 These slight disparities present avenues that may be exploited to successfully develop isoform-specific NOS inhibitors with broad therapeutic potential. Many amino acids, as well as nonamino acid analogues, are known to be selective nNOS inhibitors.31
Prior to the publication of the NOS crystal structures, we synthesized a library of 152 dipeptide amides containing L- and D-NG-nitroarginine (NNA) and a variety of amino acids.32 Several analogues were relatively potent and selective nNOS inhibitors; L-NNA-L-Dbu-NH2 (1) had a Ki of 130 nM with 1538-fold selectivity for nNOS over eNOS and 192-fold selectivity over iNOS. To simplify the structure, it was found that excision of the carboxamide group did not have a major impact on the potency or selectivity (2, n = 1-3); the propanediamine analogue (2, n = 2), which corresponds to 1, had a Ki of 460 nM with nNOS/eNOS and nNOS/iNOS selectivities of 463 and 256, respectively.33
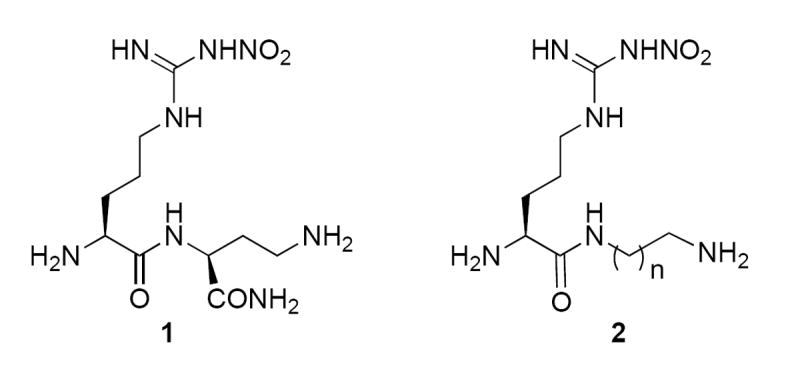
Although dipeptides, such as L-NNA-L-Dbu-NH2, produce exceptional biological activity, they are not desirable as drug candidates. The peptide bond can be hydrolyzed, so it can easily be metabolized in the gut prior to delivery to the desired target enzyme. Because of this, peptide-based compounds tend to exhibit poor bioavailability. A peptidomimetic approach can be taken to potentially increase bioavailability. Here we explore the utilization of a hydroxyethylene isostere of 2 as a bioisosteric replacement for the amide bond.34
2. Results and Discussion
2.1 Design Rationale
The reason the hydroxyethylene isostere is appealing is three-fold. First, the peptide bond is replaced by an alcohol functional group, which is not hydrolyzable, thereby preventing proteolysis. On the basis of X-ray crystallography, the peptide bond nitrogen appears to be involved in a hydrogen bond interaction with the propionate chain of the heme porphyrin through a bridged water molecule.28 The alcohol moiety of the hydroxyethylene isostere also should be able to undergo this interaction. Furthermore, the hydroxyl group might displace the structural water molecule and lead to a direct interaction with the heme propionate group, possibly resulting in higher potency. The loss of the peptide carbonyl should not have a detrimental effect on potency or selectivity because the 2 series with the carbonyl group reduced (i.e., 3) is more potent and selective than the corresponding 2 series.35 Furthermore, an X-ray crystal structure of
 3 (n = 1) bound to nNOS exhibited a similar interaction between the reduced amide nitrogen and the enzyme as was observed with the peptide bond nitrogen of the dipeptide inhibitors.28 In fact, the reduced amide nitrogen may be close enough to interact directly with the propionate chain of the heme porphyrin. These two conclusions produced the hypothesis that the hydroxyethylene isosteres could potentially produce more potent and selective inhibitors than the corresponding amides.
3 (n = 1) bound to nNOS exhibited a similar interaction between the reduced amide nitrogen and the enzyme as was observed with the peptide bond nitrogen of the dipeptide inhibitors.28 In fact, the reduced amide nitrogen may be close enough to interact directly with the propionate chain of the heme porphyrin. These two conclusions produced the hypothesis that the hydroxyethylene isosteres could potentially produce more potent and selective inhibitors than the corresponding amides.
Four different chiral hydroxyethylene isosteric analogues of 3, in addition to their corresponding alcohols in case simple hydrogen bonding interactions were important, were synthesized (4-11). Their syntheses and biological evaluation as inhibitors of NOS isozymes is reported here.

2.2 Chemistry
The synthetic plan was to prepare intermediates containing the terminal hydroxyl group, then convert them into amino groups. The synthetic route designed to obtain the terminal 3-hydroxypropyl analogues (4,5) is shown in Scheme 1.Boc-Orn(Z)-OH (12) was used as the starting material. An Arndt-Eistert homologation was employed to obtain the Weinreb amide (13), followed by reduction to the aldehyde (14) using lithium aluminum hydride.34,36,37 The crucial step in the synthesis was the subsequent Grignard addition to this aldehyde. The addition of allylmagnesium bromide successfully produced the secondary alcohol adducts (5a,b).38 The addition was conducted without any stereocontrol, but the two diastereomers were separated by column chromatography. To determine the absolute stereochemistry of each alcohol, a number of methods were considered. Since the chiral center of the ornithine compound was fixed in the L-configuration, cyclization would allow the two chiral centers to be compared using 1D-NOESY experiments (see Supplementary Material). Sodium hydride-induced condensation of the alcohol with the carbonyl of the Boc-protecting group produced the corresponding perhydro-1,3-oxazin-2-ones (19a,b), which allowed stereochemical identification of each diastereomer (Scheme 2). The anti-diastereomer has the two chiral protons in parallel axial positions; the relevant peaks are multiplets at 3.4 and 4.2 ppm (Figure 1). Irradiation of the peak at 3.4 ppm produced a strong NOE at 4.2 ppm, and irradiation at 4.2 ppm produced a strong NOE at 3.4 ppm. These data are consistent with two 1,3-diaxial protons, as in the anti-diastereomer. The syn-diastereomer has one proton axial (3.4 ppm) and one equatorial (4.3 ppm), which should not exhibit a NOE. When the peak at 3.4 ppm was irradiated, the peak at 4.3 ppm was not affected by the α-proton. Similarly, when the peak at 4.3 ppm was irradiated, no NOE was observed at 3.4 ppm. These results indicate that the compound that produced these spectra is the syn-diastereomer.
Scheme 1.

Scheme 2.

Figure 1.

Two diastereomers isolated from the NaH cyclization of 15a,b
The synthetic route was completed for each isomer separately to obtain both diastereomers of the final product. Employing a hydroboration-oxidation reaction with 9-BBN produced the terminal alcohols (16a,b).39 Deprotection of the Cbz-protecting group by hydrogenation in the presence of 10% palladium on carbon produced the free amines (17a,b), and addition of N-nitromethylthioguanidine afforded the desired analogues (18a,b).40 Removal of the Boc-protecting group using trifluoroacetic acid completed the synthesis of 4 and 5.
The synthetic route designed to obtain the terminal hydroxyethyl analogues (6 and 7) is given in Scheme 3. This method only differs from the scheme to develop the first two compounds at one of the steps. Originally, vinylmagnesium bromide was substituted to provide the compounds one carbon shorter, but this addition was not successful. As an alternative, ozonolysis of the allyl Grignard addition products (15a,b) followed by reduction with sodium borohydride produced the desired alcohols (20a,b).41 Deprotection and nitroguanidination as discussed above completed the synthesis of 6 and 7.39
Scheme 3.

The original plan was to convert the terminal hydroxyl group of 4-7 to a protected amine by employing a Mitsunobu reaction with N-Boc ethyl oxamate. Unfortunately, the secondary alcohol attacked the Mitsunobu intermediate to form a cyclic ether; therefore, a protection scheme was needed to prevent this nucleophilic attack. Scheme 4 shows the synthesis of the terminal aminopropyl analogues 8 and 9. Starting from Grignard adducts 15a,b, the tri-protected adducts (23a,b) were afforded.42 The terminal alcohols (24a,b) were obtained using the same hydroboration-oxidation method presented in Scheme 1.38 Using a previously established method,43 the terminal alcohol was converted to Boc-protected amines 26a,b. Removal of the Cbz-protecting group and the three benzyl groups necessitated harsher hydrogenation conditions than normally needed for benzyl deprotection. Palladium hydroxide on carbon at 60 °C successfully afforded the unprotected ornithine analogues (27a,b). Nitroguanidination followed by removal of the Boc-protecting groups yielded the first set of terminal amine compounds (8,9).39
Scheme 4.

The synthetic route for the final set of terminal amine compounds (10,11) is presented in Scheme 5. Starting from Grignard adducts 15a,b, ozonolysis and reduction produced the terminal alcohols (20a,b).40 The secondary alcohol did not need protection because a four-membered ring cyclization was not favored, so Mitsunobu conditions, followed by LiOH hydrolysis, successfully afforded Boc-protected amines 30a,b.42 Deprotection of the Cbz group, followed by nitroguanidination and Boc deprotection, yielded the final two compounds (10,11).39
Scheme 5.

2.3 Enzyme Inhibition Studies
The eight synthetic targets were tested against all three isoforms of NOS to determine potency and selectivity. The isoforms used are recombinant enzymes, which were overexpressed in E. coli from different sources: murine macrophage iNOS, rat brain nNOS, and bovine eNOS. The biological activities for the terminal alcohol compounds (4-7) are given in Table 1.
Table 1.
Inhibition of NOS isozymes by 4-7
| IC50 (μM) | Selectivity | ||||
|---|---|---|---|---|---|
| Compound | nNOS | iNOS | eNOS | eNOS/nNOS | iNOS/nNOS |
| 4 | 255 | 524 | 2582 | 10.1 | 2.1 |
| 5 | 490 | 266 | 879 | 1.8 | 0.54 |
| 6 | 106 | 331 | 1094 | 10.2 | 3.1 |
| 7 | 168 | 1293 | 1662 | 9.9 | 7.7 |
The potency results are reported as IC50 values, which are the inhibitor concentrations that cause 50% loss of enzyme activity. The selectivities that the inhibitors have for nNOS over the other two isoforms are also presented. This series of compounds showed poor potency against nNOS, with 3 providing the best inhibition, IC50 = 106 μM. Some selectivity toward nNOS was observed, but not significant enough to explore further. It was proposed that the terminal hydroxyl compounds might produce good potency because of a potential hydrogen bond interaction with the structural water molecule. These results provide strong evidence that the enzyme has an affinity for a positively charged amino side chain, not just a hydrogen-bond donor group. Previous inhibitor docking experiments suggest that a potential long-range electrostatic interaction is lost when incorporating a hydroxyl group at the side chain position.44
The amino side chain-containing inhibitors (8-11) produced better results (Table 2). All four of these compounds are much more potent inhibitors of nNOS. Compounds 9 and 10 exhibited sub-micromolar Ki values, 830 and 290 nM, respectively. Compound 10 is comparable in potency to L-NNA-L-Dbu-NH231 and the reduced amide peptidomimetic compounds.34 The chain length of 10 corresponds to the optimal chain length in the reduced amide series (3, n = 1), suggesting a similar binding geometry for the two series of compounds. The aminoalkyl hydroxyethylene compounds exhibited good selectivity over eNOS, but only moderate selectivity over iNOS, although the selectivity of the hydroxyethylene isosteres toward nNOS is significantly lower than that of the previous inhibitors in this class. The potency indicates that this isostere successfully binds to nNOS, but we had thought that the hydroxyl group would displace a structural water molecule and bind directly to the heme cofactor, resulting in higher potency. This increased inhibition was not observed, suggesting that the molecules either are not binding as anticipated or the proposed binding does not lead to greater potency. Recently, we found that the N-hydroxyl analogue of 3 (n = 1; OH on the secondary NH group), which computer modeling predicted also could displace the structural water molecule and bind directly to the heme side chain was shown by X-ray crystallography to do that, yet the Ki value was not changed from the parent compound.45 However, other important interactions were moderated, thereby compensating for the direct interaction with the enzyme. Apparently, a similar result occurs with the hydroxyethylene isosteric molecules.
Table 2.
Inhibition of NOS isozymes by 8-11
| IC50 (μM) | Selectivity | ||||
|---|---|---|---|---|---|
| Compound | nNOS | iNOS | eNOS | eNOS/nNOS | iNOS/nNOS |
| 8 | 23.5 (2.8)a | 361 | 1395 | 59 | 15 |
| 9 | 6.8 (0.83)a | 313 | 1529 | 225 | 33 |
| 10 | 2.4 (0.29)a | 257 | 1141 | 475 | 107 |
| 11 | 9.4 (1.1)a | 167 | 1342 | 143 | 18 |
Data in parentheses represent Ki values
The two diastereomers of each compound are expected to bind differently to the NOS isozymes. A difference was observed, but the results were not consistent across the two sets of aminoalkyl compounds. Between the two aminoethyl side chain inhibitors, the syn-diastereomer (10) proved to be more potent, whereas the anti-diastereomer (9) of the aminopropyl compounds was the more potent isomer. In contrast, 9 showed higher potency than 11. Since the position of the terminal amino group appears to be so crucial for enzyme binding, the resultant position of the terminal amino group ultimately will determine inhibitor potency. With the terminal amino group in the favored position, the hydroxyl groups of the syn-aminoethyl diastereomer (10) and the anti-aminopropyl diastereomer (9) are in preferred positions, whereas the hydroxyl groups of the anti-aminoethyl diastereomer (11) and the syn-aminopropyl diastereomer (8) are in less optimal positions.
3. Summary
A series of chiral hydroxyethylene isosteres of Nω -nitro-L-arginine amides were synthesized and shown to exhibit potent and selective nNOS inhibition, but not as potent or selective as the parent compounds. However, hydroxyethylene isosteres have been shown in the literature to be more bioavailable than the corresponding parent peptide analogues.46
4. Experimental Section
6-(Benzyloxycarbonylamino)-(3S)-(tert-butyloxycarbonylamino)-(N-methoxy-N-methyl)hexamide (13)
To a flame-dried 250 mL flask charged with argon, was added N,O-dimethyl hydroxylamine hydrochloride (1.60 g, 16.4 mmol) in dry THF (20 mL). Triethylamine (3.04 mL, 26.8 mmol) was added dropwise at room temperature, and the mixture was allowed to stir overnight. The resulting white precipitated salt was filtered and washed with dry THF. To a solution of Boc-L-Orn(Z)-OH (12, 4.0 g, 10.9 mmol) in ethylene glycol dimethyl ether (DME) (10 mL), cooled to -20 °C and under an argon atmosphere, was added with vigorous stirring 4-methylmorpholine (1.27 mL, 11.4 mmol) followed by isobutyl chloroformate (1.56 mL, 12.0 mmol). The resulting white precipitate was removed by filtration and washed with DME. The filtrate was added to diazomethane in ether, generated using an Aldrich Diazald Kit (~16 mmol). The solution was allowed to stir for 1-2 h at 0 °C and concentrated in vacuo. The resulting residue was dissolved in dry THF (~50 mL). To this solution was added the filtrate resulting from the previous reaction followed by silver benzoate (0.51 g, 2.20 mmol) in triethylamine (5.8 mL), and the reaction was allowed to stir overnight. The above reaction was performed in duplicate, and the solutions were combined and worked up as one reaction. The resulting dark brown solution was concentrated in vacuo, and the residue was dissolved in ethyl acetate. The insoluble material was removed by filtration through a pad of Celite. The filtrate was washed with saturated sodium bicarbonate (3 × 100 mL), water (100 mL), 1 M potassium bisulfite (2 × 50 mL), brine, dried over anhydrous magnesium sulfate, and concentrated in vacuo. The resulting clear, yellow oil was purified by flash chromatography, (ethyl acetate-hexane 2:1) to afford a clear, colorless oil, (6.15 g, 67%). 1H NMR (400 MHz, CDCl3) δ 7.36 (m, 5H), 5.38 (m, 1H), 5.09 (s, 2H), 4.99 (brs, 1H), 3.90 (m, 1H), 3.68 (s, 3H), 3.20 (m, 2H), 3.17 (s, 3H), 2.70 (m, 1H), 2.59 (m, 1H) 1.28-1.66 (m, 4H), 1.40 (s, 9H). MS (ESI) (m/z): 424.2 (M+1).
6-(Benzyloxycarbonylamino)-(3S)-(tert-butyloxycarbonylamino)hexanal (14)
A 250 mL three-necked round bottom flask was equipped with a thermometer, flame dried, and charged with argon and a 1 M solution of lithium aluminum hydride in THF (29 mL, 29 mmol). To this solution was added a solution of Weinreb amide 13 (6.15 g, 14.5 mmol) in THF (30 mL) dropwise by cannula while keeping the temperature below -60 °C. After addition was complete the Dry Ice bath was removed, and the solution was allowed to stir for 30 min or until all starting material was consumed. The Dry Ice bath was replaced, and the reaction was cautiously quenched with saturated potassium bisulfate (10 mL). The Dry Ice bath was removed once again, and the solution was allowed to warm to room temperature for 2 h, resulting in a clear, yellow solution with a white precipitate. The precipitate was removed by filtration through a pad of Celite, and the filtrate was concentrated in vacuo. The resulting residue was dissolved in ethyl acetate and washed with cold 1 N HCl (3 × 50 mL), saturated sodium bicarbonate (2 × 50 mL), brine, dried over anhydrous magnesium sulfate, and concentrated in vacuo. The resulting yellow oil was purified by flash chromatography (hexane-ethyl acetate 3:1) to afford a clear, colorless oil that crystallized under vacuum (2.05 g, 38%). 1H NMR (400 Mhz, CDCl3) δ 9.61 (s, 1H), 7.26 (m, 5H), 5.41 (m, 1H), 5.05 (m, 1H), 5.00 (s, 2H), 3.93 (m, 1H), 3.08 (m, 2H), 2.43 (m, 2H), 1.15-1.55 (m, 4H), 1.34 (s, 9H). MS (ESI) (m/z): 365.5 (M+1).
N-[(4S)-(tert-Butyloxycarbonylamino)-(6S and 6R)-hydroxynon-8-enyl-(N-(benzyloxycarbonyl)]amine (15a,b)
A three-necked 250 mL round bottom flask was equipped with a thermometer, flame dried, and charged with argon. To a stirred 1 M solution of zinc chloride in ether (16 mL, 16 mmol), was added THF (40 mL). The solution was cooled to 0 °C, and a 1 M solution of allylmagnesium bromide was added (24 mL, 24 mmol). After being stirred for 30 min, the suspension was cooled to − 30 °C, and a solution of aldehyde 14 in THF (30 mL) was added slowly, keeping the internal temperature below -20 °C. The reaction was stirred and allowed to warm to room temperature overnight. The mixture was then cooled to -30 °C, cautiously quenched with 5% citric acid (100 mL), and diluted with ethyl acetate (200 mL). The resulting organic layer was washed with water (50 mL), saturated sodium bicarbonate (50 mL), dried over anhydrous magnesium sulfate, and concentrated in vacuo. The resulting cloudy oil was purified by flash chromatography (hexane-ethyl acetate 4:1) to afford two separate diastereomers as clear, colorless oils (15a, S,S-isomer [1.0 g, 44%] and 15b, S,R-isomer [0.72 g, 32%]).
15a: 1H NMR (400 MHz, CDCl3) δ 7.30 (m, 5H), 5.79 (m, 1H), 5.25 (m, 1H), 5.05 (s, 2H), 5.05 (m, 2H), 4.81 (m, 1H), 3.96 (m, 1H), 3.62 (m, 1H), 3.11 (m, 2H), 2.18 (m, 2H) 1.22-1.56 (m, 4H), 1.40 (s, 9H). MS (ES) (m/z): M+H+ calcd for C22H34N2O5, 406.5 found 407.5.
15b: 1H NMR (400 MHz, CDCl3) δ 7.31 (m, 5H), 5.76 (m, 1H), 5.35 (m, 1H), 5.08 (m, 2H), 5.03 (s, 2H), 4.84 (m, 1H), 3.66 (m, 2H), 3.13 (m, 2H), 2.19 (m, 2H), 1.25-1.55 (m, 4H), 1.39 (s, 9H). MS (ESI) (m/z): 407.3 (M+1).
The following procedures were completed using both isomers, but one example procedure is given for each.
N-[(4S)-(tert-Butyloxycarbonylamino)-(6R,9)-dihydroxynonyl-(N-(benzyloxycarbonyl)]amine (16b)
A sample of allyl alcohol 15b (0.12 g, 0.29 mmol) was added to a 50 mL round bottom flask, dried overnight using a vacuum pump, and charged with argon. To this sample was added a 0.5 M solution of 9-BBN in THF (5.8 mL, 2.9 mmol) at room temperature, and the reaction was allowed to stir overnight. The mixture was then cooled to 0 °C, and 2 M potassium hydroxide (5 mL, 10 mmol) was added, followed by 30% hydrogen peroxide (5 mL). After being stirred for 5 min, the solution was diluted with ethyl acetate (25 mL), washed with water (3 × 50 mL) and brine, dried over anhydrous magnesium sulfate, and concentrated in vacuo. The cloudy oil was purified by flash chromatography (methylene chloride-methanol 50:1) to afford a clear, colorless oil (35 mg, 29%). This procedure was repeated for the other isomer.
R-isomer: 1H NMR (400 MHz, CDCl3) δ 7.31 (m, 5H), 5.19 (m, 1H), 5.10 (m, 1H), 5.04 (s, 2H), 4.78 (m, 1H), 3.62 (m, 2H), 3.34 (m, 1H), 3.18 (m, 2H), 1.65 (m, 2H), 1.26-1.60 (m, 8H), 1.40 (s, 9H). MS (ESI) (m/z): 425.0 (M+1).
S-isomer: 1H NMR (400 MHz, CDCl3) δ 7.35 (m, 5H), 6.52 (bs, 1H), 5.08 (s, 2H), 4.99 (m, 1H), 3.84 (m, 2H), 3.71 (m, 2H), 3.20 (m, 2H), 1.64 (m, 2H), 1.34-1.58 (m, 8H), 1.41 (s, 9H). MS (ESI) (m/z): 425.0 (M+1).
N-[(4S)-(tert-Butyloxycarbonylamino)-(6R,9)-dihydroxynonyl]amine (17b)
To a 25 mL round bottom flask was added Cbz-protected alcohol 16b (0.035 g, 0.082 mmol), which was dissolved in methanol (10 mL). To this solution was added 10% Pd/C (10 mg), and the suspension was exposed to hydrogen gas overnight. The suspension was then filtered through a pad of Celite and concentrated in vacuo to afford a yellow oil. The crude yellow oil was used in the next reaction without purification. This procedure was repeated for the other isomer.
N-[(4S)-(tert-Butyloxycarbonylamino)-(6R,9)-(dihydroxynonyl)]nitroguanidine (18b)
To a solution of crude amine 17b (0.23 g, 0.082 mmol) in absolute ethanol (5 mL), was added N-nitromethylthioguanidine47 (0.012 g, 0.091 mmol). The solution was heated at 40 °C for 5 min and allowed to stir overnight at room temperature. The reaction mixture was concentrated in vacuo and purified by flash chromatography (ethyl acetate-hexane 7:1) to afford a clear, colorless oil (0.025 g, 81%). This procedure was repeated for the other isomer.
R-isomer: 1H NMR (400 MHz, CD3OD) δ 3.72 (m, 1H), 3.56 (t, J = 8.0 Hz, 2H), 3.30 (m, 1H), 3.24 (m, 2H), 1.84 (m, 2H), 1.32-1.70 (m, 8H), 1.44 (s, 9H). MS (ESI) (m/z): 378.3 (M+1).
S-isomer: 1H NMR (400 MHz, CD3OD) δ 3.61 (m, 1H), 3.56 (t, J = 8.0 Hz, 2H), 3.31 (m, 1H), 3.23 (m, 2H), 1.34-1.70 (m, 10H), 1.42 (s, 9H). MS (ESI) (m/z): 378.3 (M+1).
N-[(4S)-Amino-(6S,9)-dihydroxynonyl]nitroguanidine (4) and N-(4S-Amino-(6R,9)-dihydroxynonyl)nitroguanidine (5)
Compound 18a (0.012 g, 0.032 mmol) or 18b (0.025 g, 0.066 mmol) was dissolved in 50% TFA in methylene chloride (2 mL) and allowed to stir for 3 h at room temperature. The mixture was concentrated in vacuo, and the resulting yellow oil was dissolved in ethanol. Concentrated HCl was added dropwise until the solution became cloudy, and the solution was concentrated once again. The resulting yellow oil was purified by HPLC to afford a white solid that became sticky when exposed to air (0.010 g, 99% for 4; 0.02 g, 99% for 5).
4: 1H NMR (500 MHz, D2O) δ 3.69 (m, 1H), 3.46 (m, 2H), 3.32 (m, 1H), 3.20 (m, 2H), 1.30-1.79 (m, 10H). 13C NMR (100 MHz, D2O) δ 159.0, 70.1, 61.6, 51.0, 40.7, 38.2, 33.6, 29.7, 27.4. HRMS (ES) (m/z): M+H+ calcd for C10H24N5O4, 278.1828 found 278.1818. . (syn-isomer).
5: 1H NMR (500 MHz, D2O) δ 3.72 (m, 1H), 3.46 (m, 2H), 3.35 (m, 1H), 3.19 (m, 2H), 1.32-1.72 (m, 10H). 13C NMR (100 MHz, D2O) δ 159.0, 67.5, 61.6, 51.9, 51.5, 40.6, 37.6, 33.2, 29.3, 27.5. HRMS (ES) (m/z): M+H+ calcd for C10H24N5O4, 278.1828 found 278.1838 . (anti-isomer).
(6R)-Allyl-(4S)-3-(benzyloxycarbonylamino)propyl-(perhydro-1,3-oxazin-2-one) (19b)
To a solution of allyl alcohol 15b (0.065 g, 0.16 mmol) in dry DMF (2 mL) was added NaH (60% in mineral oil, 10 mg, 0.23 mmol). The reaction was allowed to stir overnight, cooled to 0 °C, slowly quenched with saturated ammonium chloride (10 mL), and diluted with ethyl acetate (20 mL). The organic layer was washed with water (3-5 times) and brine, dried over magnesium sulfate, and concentrated in vacuo. The resulting yellow oil was purified by flash chromatography (hexane:ethyl acetate 2:1) to afford a clear, colorless oil. (0.024 g, 45%). This procedure was repeated for the other isomer.
R-isomer: 1H NMR (500 MHz, CDCl3) δ 7.36 (m, 5H), 6.14 (brs, 1H), 5.81 (m, 1H), 5.14 (m, 2H), 5.10 (s, 2H), 5.03 (m, 1H), 4.26 (m, 1H), 3.46 (m, 1H), 3.22 (m, 2H), 2.48 (m, 1H), 2.37 (m, 1H), 1.95 (m, 1H), 1.27-1.72 (m, 6H).
S-isomer: 1H NMR (500 MHz, CDCl3) δ 7.35 (m, 5H), 6.85 (brs, 1H), 5.80 (m, 1H), 5.13 (m, 2H), 5.10 (s, 2H), 4.35 (m, 1H), 3.47 (m, 1H), 3.21 (m, 2H), 2.50 (m, 1H), 2.36 (m, 1H), 1.82 (m, 1H), 1.22-1.76 (m, 6H).
N-[(4S)-(tert-Butyloxycarbonylamino)-(6R,8)-dihydroxyoctyl-(N-(benzyloxycarbonyl)]amine_(20b)
To a 100 mL three-necked round bottom flask was added a solution of allyl alcohol 15b (0.30 g, 0.74 mmol) in methanol (10 mL). Ozone was bubbled through the solution at a constant rate for 1-2 h or until all starting material was consumed. The reaction was then worked up by the addition of sodium borohydride (0.28 g, 7.4 mmol). After being stirred for several hours, the mixture was concentrated in vacuo, and the resulting residue was dissolved in ethyl acetate. The solution was then washed with saturated ammonium chloride, brine, dried over anhydrous magnesium sulfate, and concentrated in vacuo to afford a cloudy oil. The oil was purified by flash chromatography (methylene chloride-methanol 50:1) to afford a clear, colorless oil (0.15 g, 49%). This procedure was repeated for the other isomer.
R-isomer: 1H NMR (400 MHz, CDCl3) δ 7.35 (m, 5H), 5.23 (brs, 1H), 5.07 (s, 2H), 4.92 (brs, 1H), 4.30 (m, 1H), 4.19 (m, 2H), 3.24 (m, 4H), 2.14 (m, 2H), 1.96 (m, 1H), 1.25-1.76 (m, 6H), 1.46 (s, 9H). MS (ESI) (m/z): 433.3 (M+Na+).
S-isomer: 1H NMR (400 MHz, CDCl3) δ 7.35 (m, 5H), 5.28 (s, 2H), 4.98 (m, 1H), 4.64 (m, 1H), 4.56 (m, 1H), 3.78 (m, 2H), 3.31 (m, 1H), 3.17 (m, 2H), 1.19-1.78 (m, 8H), 1.43 (s, 9H). MS (ESI) (m/z): 433.3 (M+Na+).
N-[(4S)-(tert-Butyloxycarbonylamino)-(6R,8)-dihydroxyoctyl]amine (21b)
To solution of 20b (0.050 g, 0.12 mmol) in methanol (10 mL) was added Pd/C (0.050 g). The suspension was exposed to hydrogen gas and allowed to stir overnight. The suspension was then filtered through a pad of Celite and concentrated in vacuo to afford a yellow oil. The crude yellow oil was used in the next reaction without purification. This procedure was repeated for the other isomer.
N-[(4S)-(tert-Butyloxycarbonylamino)-(6R,8)-(dihydroxyoctyl)]nitroguanidine (22b)
To a solution of crude alcohol 21b in absolute ethanol (5 mL) was added N-nitromethylthioguanidine (16 mg, 0.12 mmol). The solution was heated at 40 °C for 5 min and allowed to stir overnight at room temperature. The reaction mixture was concentrated in vacuo and purified by flash chromatography (ethyl acetate-hexane 7:1) to afford a clear, colorless oil (20 mg, 46%). This procedure was repeated for the other isomer.
R-isomer: 1H NMR (400 MHz, CD3OD) δ 3.80 (m, 1H), 3.68 (m, 2H), 3.26 (m, 3H), 1.64 (m, 2H), 1.34-1.58 (m, 6H), 1.44 (s, 9H). MS (ESI) (m/z): 364.5 (M+1).
S-isomer: 1H NMR (400 MHz, CD3OD) δ 3.78 (m, 1H), 3.67 (m, 2H), 3.29 (m, 2H), 3.23 (m, 1H) 1.63 (m, 2H), 1.30-1.55 (m, 6H), 1.43 (s, 9H). MS (ESI) (m/z): 364.5 (M+1).
N-[(4S)-Amino-(6S,8)-dihydroxyoctyl]nitroguanidine (6) and N-(4S-Amino-(6R,8)-dihydroxyoctyl)nitroguanidine (7)
Compound 22a (0.016 g, 0.044 mmol) or 22b (0.021 g, 0.058 mmol) was dissolved in 50% TFA in methylene chloride (2 mL) and was allowed to stir for 3 h at room temperature. The mixture was concentrated in vacuo, and the resulting yellow oil was dissolved in ethanol. Concentrated HCl was added dropwise until the solution became cloudy, and the solution was concentrated once again. The resulting yellow oil was purified by HPLC to afford a white solid that became sticky when exposed to air (0.015 g, 99% for 6; 0.017 g, 99% for 7).
6: 1H NMR (500 MHz, D2O) δ 3.82 (q, J = 7.9 Hz, 1H), 3.55 (t, J = 7.1 Hz, 2H), 3.36 (m, 1H), 3.17 (m, 2H), 1.53-1.70 (m, 8H). 13C NMR (100 MHz, D2O) δ 159.0, 64.7, 58.3, 49.0, 40.6, 38.8, 37.9, 29.4, 23.5. HRMS (ESI) (m/z): M+H+ calcd for C9H22N5O4, 264.1672 found 264.1663. (syn-isomer).
7: 1H NMR (500 MHz, D2O) δ 3.81 (m, 1H), 3.55 (t, J = 6.7 Hz, 2H), 3.32 (m, 1H), 3.16 (m, 2H), 1.45-1.78 (m, 8H). 13C NMR (100 MHz, D2O) δ 159.0, 67.3, 58.1, 50.9, 40.7, 39.3, 38.4, 29.7, 23.4. HRMS (ESI) (m/z): M+H+ calcd for C9H22N5O4, 264.1672 found 264.1682. (anti-isomer).
N-[(4S)-[N-Benzyl-N-(tert-Butyloxycarbonyl]amino-(6R)-benzyloxynon-8-enyl-[(N-benzyl-N-(benzyloxycarbonyl)]amine (23b)
To a 100 mL round bottom flask, flame dried and charged with argon, was added allyl alcohol 15b (0.63 g, 1.55 mmol). The oil was dissolved in anhydrous DMF (10 mL) and treated with 60% sodium hydride in mineral oil (0.62 g, 15.5 mmol). The suspension was cooled to 0 °C followed by addition of tetrabutylammonium iodide (1.91 g, 5.17 mmol) and benzyl bromide (0.61 mL, 5.17 mmol). The mixture was allowed to warm to room temperature overnight, cooled to 0 °C, and cautiously quenched with saturated ammonium chloride. The solution was extracted with methylene chloride (3 × 50 mL), and the combined organic layers were concentrated in vacuo to afford a mixture of a clear oil containing a white solid. The mixture was dissolved in methylene chloride and was purified by flash chromatography (hexane-ethyl acetate 6:1) to afford a clear, colorless oil (0.46 g, 44%). This procedure was repeated for the other isomer
R-isomer: 1H NMR (400 MHz, CDCl3) δ 7.05-7.45 (m, 20H), 5.84 (m, 1H), 5.18 (s, 2H), 5.08 (m, 2H), 4.08-4.50 (m, 7H), 2.94-3.42 (m, 3H), 2.32 (m, 2H), 1.06-1.89 (m, 6H), 1.42 (s, 9H). MS (ESI) (m/z): 677.4 (M+1)..
S-isomer: 1H NMR (400 MHz, CDCl3) δ 7.05-7.45 (m, 20H), 5.60 (m, 1H), 5.12 (s, 2H), 4.98 (m, 2H), 3.80-4.65 (m, 7H), 2.90-3.35 (m, 3H), 2.11 (m, 2H), 1.18-1.55 (m, 6H), 1.43 (s, 9H). MS (ESI) (m/z): 677.4 (M+1).
N-[(4S)-[N-Benzyl-N-(tert-Butyloxycarbonyl]amino-(6R)-benzyloxy-9-hydroxynonyl-{N-benzyl-N-(benzyloxycarbonyl)}]amine (24b)
To a 100 mL round bottom flask, flame dried and charged with argon, was added tribenzyl compound 23b (0.46 g, 0.68 mmol). The oil was then dissolved in a 0.5 M solution of 9-BBN in THF (13.6 mL, 6.8 mmol) and was allowed to stir at room temperature overnight. The flask was cooled to 0 °C, and 2.0 M aqueous KOH (10 mL, 20 mmol) was added followed by 30% hydrogen peroxide (10 mL). After being stirred for 5 min, the solution was diluted with ethyl acetate (50 mL). The resulting organic layer was washed with water (5 × 50 mL) and brine and concentrated in vacuo to afford a white, cloudy oil. The crude product was purified by flash chromatography (methylene chloride-methanol 40:1). Some impurities remained, so the crude material was purified by flash chromatography a second time (hexane-ethyl acetate 3:1) to afford a soap-like white oil (0.39 g, 82%). This procedure was repeated for the other isomer.
R-isomer: 1H NMR (400 MHz, CDCl3) δ 6.88-7.50 (m, 20H), 5.15 (s, 2H), 4.39 (s, 2H), 4.39 (m, 1H), 4.21 (s, 2H), 4.15 (s, 2H), 3.56 (m, 2H), 2.92-3.33 (m, 3H), 1.86 (m, 2H), 1.05-1.72 (m, 8H), 1.44 (s, 9H). MS (ESI) (m/z): 717.9 (M+Na+).
S-isomer: 1H NMR (400 MHz, CDCl3) δ 6.95-7.58 (m, 20H), 5.14 (s, 2H), 3.80-4.66 (m, 7H), 3.53 (m, 2H), 2.80-3.33 (m, 3H), 1.10-1.85 (m, 10H), 1.44 (s, 9H). MS (ESI) (m/z): 717.9 (M+Na+).
N-[(4S)-[N-Benzyl-N-(tert-Butyloxycarbonyl]amino-(6R)-benzyloxy-9-{N-tert-butyloxycarbonyl)-N-[2-ethoxy-(oxoacetyl)amino}nonyl-{N-benzyl-N-(benzyloxycarbonyl)}]amine (25b)
A two-necked 100 mL round bottom flask was equipped with a water-jacketed condenser, flame dried, and placed under an argon atmosphere. Triphenylphosphine (0.16 g, 0.62 mmol) and N-Boc-ethyl oxamate (0.14 g, 0.62 mmol) were added followed by a solution of tribenzyl alcohol 24b (0.39 g, 0.56 mmol) in dry THF (10 mL). The solution was cooled to 0 °C, and diisopropyl azodicarboxylate (0.12 mL, 0.62 mmol) was added dropwise. The reaction was then heated to 60 °C and was allowed to stir overnight. The resulting light yellow liquid was concentrated in vacuo and purified by flash chromatography (hexane-ethyl acetate 7:1) to afford a clear, colorless oil (0.24 g, 48%). This procedure was repeated for the other isomer.
R-isomer: 1H NMR (400 MHz, CDCl3) δ 7.04-7.49 (m, 20H), 5.16 (s, 2H), 3.97-4.50 (m, 9H), 3.65 (m, 2H), 2.93-3.34 (m, 3H), 1.82 (m, 2H), 1.22-1.72 (m, 9H), 1.51 (s, 9H), 1.48 (s, 9H). MS (ESI) (m/z): 917.3 (M+Na+).
S-isomer: 1H NMR (400 MHz, CDCl3) δ 6.96-7.72 (m, 20H), 5.12 (s, 2H), 4.95 (m, 2H) 3.78-4.71 (m, 9H), 2.88-3.36 (m, 3H), 0.64-1.82 (m, 11H), 1.24 (s, 9H), 1.23 (s, 9H). MS (ESI) (m/z): 917.3 (M+Na+).
N-[(4S)-[N-Benzyl-N-(tert-Butyloxycarbonyl]amino-(6R)-benzyloxy-9-(N-tert-butyloxycarbonylamino)nonyl-{N-benzyl-N-(benzyloxycarbonyl)}]amine (26b)
To a solution of oxamate 25b (0.50g, 0.56 mmol) in THF (4 mL) a solution of lithium hydroxide (0.14 g, 6.0 mmol) in water (3 mL) was added at room temperature. After 3 h the reaction mixture was diluted with water and extracted with methylene chloride. The organic layer was dried over anhydrous sodium sulfate and concentrated in vacuo. The resulting cloudy oil was purified by flash chromatography (hexane-ethyl acetate 1:1) to afford a clear, colorless oil (0.40 g, 90%). This procedure was repeated for the other isomer.
R-isomer: 1H NMR (400 MHz, CDCl3) δ 7.05-7.50 (m, 20H), 5.18 (m, 2H), 4.94 (m, 1H), 3.94-4.64 (m, 9H), 2.92-3.34 (m, 5H), 1.92 (m, 2H), 1.12-1.72 (m, 8H), 1.44 (s, 18H). MS (ESI) (m/z): 794.6 (M+1).
S-isomer: 1H NMR (400 MHz, CDCl3) δ 6.90-7.62 (m, 20H), 5.15 (m, 2H), 5.04 (m, 1H), 4.10-4.78 (m, 9H), 2.68-3.41 (m, 5H), 1.08-1.66 (m, 10H), 1.42 (s, 18H). MS (ESI) (m/z): 794.6 (M+1).
(4S)-[N-(tert-Butyloxycarbonylamino)-(6R)-hydroxy-9-(N-tert-butyloxycarbonylamino)nonylamine (27b)
To solution of 26b (1.01 g, 1.27 mmol) in ethanol (50 mL), was added Pd(OH)2/C (0.25 g). The suspension was exposed to hydrogen gas, heated to 60 °C, and allowed to stir overnight. The suspension was then filtered through a pad of Celite and concentrated in vacuo to afford a yellow oil. The crude yellow oil was used in the next reaction without purification. This procedure was repeated for the other isomer.
N-{(4S)-[(tert-Butyloxycarbonylamino]-(6R)-hydroxy-9-(N-tert-butyloxycarbonylamino)nonyl}nitroguanidine (28b)
To a solution of crude amine 27b in absolute ethanol (20 mL) was added N-nitromethylthioguanidine (0.17 g, 1.27 mmol) and triethylamine (1 mL). The solution was heated at 40 °C for 5 min and was allowed to stir overnight at room temperature. The reaction mixture was concentrated in vacuo and purified by flash chromatography (ethyl acetate-hexane 7:1) to afford a clear, colorless oil (0.20 g, 33%). This procedure was repeated for the other isomer.
R-isomer: 1H NMR (400 MHz, CDCl3) δ 8.59 (brs, 1H), 7.67 (m, 2H), 4.94 (m, 2H), 3.65 (m, 2H), 3.39 (m, 1H), 3.23 (m, 1H), 3.05 (m, 2H), 1.32-1.70 (m, 10H), 1.38 (s, 9H), 1.36 (s, 9H). MS (ESI) (m/z): 477.4 (M+1).
S-isomer: 1H NMR (400 MHz, CDCl3) δ 8.60 (brs, 1H), 7.57 (m, 2H), 5.02 (brs, 1H), 4.79 (brs, 1H) 3.63 (m, 2H), 3.38 (m, 1H), 3.24 (m, 1H), 3.07 (m, 2H), 1.26-1.74 (m, 10H), 1.39 (s, 18H). MS (ESI) (m/z): 477.4 (M+1).
N-[(4S,9)-Diamino-(6S)-hydroxynonyl]nitroguanidine (8) and N-[(4S,9)-Diamino-(6R)-hydroxynonyl]nitroguanidine (9)
Compound 28a (0.035 g, 0.073 mmol) or 28b (0.050 g, 0.10 mmol) was dissolved in 50% TFA in methylene chloride, and was allowed to stir for 3 h at room temperature. The mixture was concentrated in vacuo, and the resulting yellow oil was dissolved in ethanol. Concentrated HCl was added dropwise until the solution became cloudy, and the solution was concentrated once again. The resulting yellow oil was purified by HPLC to afford a white solid that became sticky when exposed to air (0.026 g, 99% for 8; 0.036 g, 99% for 9).
8: 1H NMR (500 MHz, D2O) δ 3.72 (m, 1H), 3.35 (m, 1H), 3.17 (m, 2H), 2.86 (m, 2H), 1.34-1.71 (m, 10H). 13C NMR (100 MHz, D2O) δ 159.0, 67.0, 51.9, 51.5, 40.6, 39.4, 37.7, 33.6, 29.3, 23.2. HRMS (ES) (m/z): M+H+ calcd for C10H24N6O3, 277.1988 found 277.1997. (syn-isomer).
9: 1H NMR (500 MHz, D2O) δ 3.68 (m, 1H), 3.31 (m, 1H), 3.16 (m, 2H), 2.85 (m, 2H), 1.32-1.76 (m, 10H). 13C NMR (100 MHz, D2O) δ 159.0, 69.5, 50.8, 40.7, 39.4, 38.2, 34.0, 29.6, 23.0. HRMS (ES) (m/z): M+H+ calcd for C10H24N6O3, 277.1988 found 277.2000. (anti-isomer).
N-[(4S)-(tert-Butyloxycarbonylamino)-(6R)-hydroxy-8-{N-tert-butyloxycarbonyl)-N-[2-ethoxy-(oxoacetyl)amino}octyl-(N-(benzyloxycarbonyl)]amine (29b)
A two-necked 100 mL round bottom flask was equipped with a water-jacketed condenser, flame dried, and placed under an argon atmosphere. Triphenylphosphine (0.070 g, 0.26 mmol) and N-Boc-ethyl oxamate (0.059 g, 0.26 mmol) were added followed by a solution of the Cbz-protected ethyl alcohol 20b (0.10 g, 0.24 mmol) in dry THF (5 mL). The solution was cooled to 0 °C, and diisopropyl azodicarboxylate (0.053 mL, 0.26 mmol) was added dropwise. The reaction was then heated to 60 °C and was allowed to stir overnight. The resulting light yellow liquid was concentrated in vacuo and purified by flash chromatography (hexane-ethyl acetate 5:1) to afford a clear colorless oil (0.050 g, 31%). This procedure was repeated for the other isomer.
R-isomer: 1H NMR (400 MHz, CDCl3) δ 7.37 (m, 5H), 5.10 (s, 2H), 4.93 (m, 1H), 4.62 (m, 1H), 4.32 (q, J = 8.0 Hz, 2H), 3.79 (m, 2H), 3.65 (m, 2H), 3.21 (m, 3H), 1.72 (m, 2H), 1.39-1.60 (m, 6H), 1.51 (s, 9H), 1.42 (s, 9H), 1.35 (t, J = 7.9 Hz, 3H). MS (ESI) (m/z): 632.5(M+Na+).
S-isomer: 1H NMR (400 MHz, CDCl3) δ 7.34 (m, 5H), 5.07 (s, 2H), 4.84 (m, 1H), 4.53 (m, 1H), 4.33 (q, J = 8.0 Hz, 2H), 3.79 (m, 3H), 3.60 (m, 2H), 3.19 (m, 2H), 1.66 (m, 2H), 1.38-1.58 (m, 6H), 1.51 (s, 9H), 1.43 (s, 9H), 1.35 (t, J = 8.0 Hz, 3H). MS (ESI) (m/z): 632.5 (M+Na+).
N-[(4S)-(tert-Butyloxycarbonylamino)-(6R)-hydroxy-8-{N-(tert-butyloxycarbonyl)amino}octyl-(N-(benzyloxycarbonyl)]amine (30b)
To a solution of oxamate 29b (0.05 g, 0.08 mmol) in THF (4 mL), a solution of lithium hydroxide (0.14 g, 6 mmol) in water (3 mL) was added at room temperature. After 3 h the reaction mixture was diluted with water and extracted with methylene chloride. The organic layer was dried over anhydrous sodium sulfate and concentrated in vacuo. The resulting cloudy oil was purified by flash chromatography (hexane-ethyl acetate 1:1) to afford a clear, colorless oil (0.04 g, 98%). This procedure was repeated for the other isomer
R-isomer: 1H NMR (400 MHz, CDCl3) δ 7.35 (m, 5H), 5.08 (s, 2H), 4.95 (m, 2H), 4.68 (m, 1H), 3.78 (m, 1H), 3.63 (m, 2H), 3.43 (m, 1H), 3.19 (m, 2H), 3.10 (m, 1H), 1.16-1.73 (m, 8H), 1.43 (s, 18H). MS (ESI) (m/z): 510.3 (M+1)..
S-isomer: 1H NMR (400 MHz, CDCl3) δ 7.34 (m, 5H), 5.07 (s, 2H), 4.92 (m, 2H), 4.70 (m, 1H), 3.75 (m, 1H), 3.67 (m, 2H), 3.37 (m, 1H), 3.18 (m, 3H), 1.16-1.72 (m, 8H), 1.43 (s, 9H), 1.42 (s, 9H). MS (ESI) (m/z): 510.3 (M+1).
N-[(4S)-(tert-Butyloxycarbonylamino)-(6R)-hydroxy-8-{N-(tert-butyloxycarbonyl)amino}octylamine (31b)
To a solution of 30b (0.050 g, 0.10 mmol) in methanol (10 mL) was added Pd/C (0.050 g). The suspension was exposed to hydrogen gas and allowed to stir overnight. The suspension was then filtered through a pad of Celite and concentrated in vacuo to afford a yellow oil. The crude yellow oil was used in the next reaction without purification. This procedure was repeated for the other isomer.
N-[(4S)-(tert-Butyloxycarbonylamino)-(6R)-hydroxy-8-{N-(tert-butyloxycarbonyl)amino}octyl]nitroguanidine (32b)
To a solution of crude amine 31b in absolute ethanol (5 mL) was added N-nitromethylthioguanidine (13 mg, 0.10 mmol). The solution was heated at 40 °C for 5 min and allowed to stir overnight at room temperature. The reaction mixture was concentrated in vacuo and purified by flash chromatography (ethyl acetate-hexane 7:1) to afford a clear, colorless oil (15 mg, 32%). This procedure was repeated for the other isomer.
R-isomer: 1H NMR (400 MHz, CD3OD) δ 3.65 (m, 1H), 3.11-3.38 (m, 5H), 1.60 (m, 2H), 1.25-1.50 (m, 6H), 1.43 (m, 18H). MS (ES) (m/z): 463.3 (M+1).
S-isomer: 1H NMR (400 MHz, CD3OD) δ 3.66 (m, 2H), 3.24 (m, 2H), 3.14 (m, 2H), 1.63 (m, 2H), 1.40-1.50 (m, 6H), 1.43 (m, 9H), 1.42 (m, 9H). MS (ES) (m/z): 463.3 (M+1).
N-[(4S,8)-Diamino-(6S)-hydroxyoctyl)]nitroguanidine (10) and N-[(4S,8)-Diamino-(6R)-hydroxyoctyl)]nitroguanidine (11)
Compound 32a (0.010 g, 0.022 mmol) or 32b (0.015 g, 0.032 mmol) was dissolved in 50% TFA in methylene chloride (2 mL), and allowed to stir for 3 h at room temperature. The mixture was concentrated in vacuo, and the resulting yellow oil was dissolved in ethanol. Concentrated HCl was added dropwise until the solution became cloudy, and the solution was concentrated once again. The resulting yellow oil was purified by HPLC to afford a white solid that became sticky when exposed to air (0.007 g, 95% for 10; 0.010 g, 93% for 11).
10: 1H NMR (500 MHz, D2O) δ 3.82 (m, 1H), 3.36 (m, 1H), 3.15 (m, 2H), 2.97 (m, 2H), 1.52-1.77 (m, 8H). 13C NMR (100 MHz, D2O) δ 159.0, 65.6, 51.9, 51.5, 40.6, 38.0, 36.9, 34.0, 30.1. HRMS (ES) (m/z): M+H+ calcd for C9H23N6O3, 263.1832 found 263.1837. (syn-isomer).
11: 1H NMR (500 MHz, D2O) δ 3.80 (m, 1H), 3.34 (m, 1H), 3.19 (m, 2H), 2.97 (m, 2H), 1.42-1.82 (m, 8H). 13C NMR (100 MHz, D2O) δ 159.0, 65.6, 51.9, 51.5, 40.5, 38.0, 36.8, 34.0, 29.4. HRMS (ES) (m/z): M+H+ calcd for C9H23N6O3, 263.1832 found 263.1837. (anti-isomer).
Enzyme assay
The NOS isoforms used were recombinant enzymes overexpressed in E. coli. Murine macrophage iNOS,48 rat nNOS,49 and bovine eNOS50 were overexpressed and isolated as reported. Interspecies similarity of each isozyme is quite high (80-94% identical);51 therefore, the source of the NOS used in various studies has been thought not to be important. The formation of nitric oxide was monitored using a hemoglobin capture assay as described previously.52 Briefly, a solution of nNOS or eNOS containing 10 µM L-arginine, 1.6 mM CaCl2, 11.6 µg /mL calmodulin, 100 µM DTT, 100 µM NADPH, 6.5 µM H 4B, 3 mM oxyhemoglobin, and varying concentrations of inhibitor in 100 mM Hepes (pH 7.4) was monitored at 30 °C. For the determination of inhibition of iNOS, no additional Ca2+ or calmodulin were added. The assay was initiated by the addition of enzyme, and the absorption of UV light at 400 nm was recorded over one minute. As NO was evolved and coordinated to the hemoglobin, the absorption at 400 nm increased, producing a value for the enzyme velocity under these conditions.
Enzyme inhibition studies
A value for the initial rate was obtained when no inhibitor was added (v0). The velocity of the enzyme (v) was then determined in the presence of varying concentrations of inhibitor, until a concentration of inhibitor that reduced the enzyme velocity to half its initial value (v/v0 ~ 0.5) was discovered. Concentrations of inhibitor above and below this value were tested and a graph of v/v0 versus inhibitor concentration ([I]) was plotted. Extrapolation of this graph allowed the determination of an IC50 value. The Ki was estimated from the IC50 using the equations below. The Km values used were: 1.3 μM (nNOS), 8.3 μM (iNOS) and 1.7 μM (eNOS).
Supplementary Material
NOESY spectra for determination of absolute diastereomeric structures of 19a,b. HPLC traces with two different eluents to demonstrate purity of 4-11.
Acknowledgments
We are grateful to the National Institutes of Health (GM 49725 to R.B.S. and GM52419 and HL30050 to Prof. Bettie Sue Masters, in whose laboratory P.M. and L.J.R. work) for financial support of this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Alderton WK, Cooper CE, Knowles RG. Biochem J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kerwin JF, Jr, Heller M. J Med Chem. 1995;38:4342–62. [Google Scholar]
- 3.Napoli C, Ignarro LJ. Nitric Oxide. 2001;5:88–97. doi: 10.1006/niox.2001.0337. [DOI] [PubMed] [Google Scholar]
- 4.Cayette AJ, Palacino JJ, Horten K, Cohen RA. Arterioscler Thromb. 1994;14:753–759. doi: 10.1161/01.atv.14.5.753. [DOI] [PubMed] [Google Scholar]
- 5.Kuhlencordt PJ, Gyurko R, Han F, Scherrer-Crosbie M, Aretz TH, Hajjar R, Picard MH, Huang PL. Circulation. 2001;104:448–454. doi: 10.1161/hc2901.091399. [DOI] [PubMed] [Google Scholar]
- 6.Drummond GI. Cyclic Nucleotides in the Nervous System. Raven Press; New York: 1984. pp. 40–125. [Google Scholar]; Garthwaite J. In: Nitric Oxide from L-Arginine: A Bioregulatory System. Moncada S, Higgs EA, editors. Elsevier; Amsterdam: 1990. pp. 115–137. [Google Scholar]
- 7.Merighi A, Aimar P, Lossi L, Pasti L, Carmignoto G. Oxid Stress and Disease. 2000;5:17–53. [Google Scholar]; Forman LJ, Tringo L, Sun M. Brain Res Bull. 1997;44:125–129. doi: 10.1016/s0361-9230(97)00100-7. [DOI] [PubMed] [Google Scholar]
- 8.Lev-Ram V, Wong ST, Storm DR, Tsien RY. Proc Natl Acad Sci USA. 2002;99:8389–8393. doi: 10.1073/pnas.122206399. [DOI] [PMC free article] [PubMed] [Google Scholar]; Grassi S, Pettorossi VE. Neuroscience. 2000;101:157–164. doi: 10.1016/s0306-4522(00)00334-1. [DOI] [PubMed] [Google Scholar]
- 9.MacMicking J, Xie QW, Nathan C. Annu Rev Immunol. 1997;15:323–50. doi: 10.1146/annurev.immunol.15.1.323. [DOI] [PubMed] [Google Scholar]
- 10.Hibbs JB, Jr, Taintor RR, Vavrin Z, Rachlin EM. Biochem Biophys Res Commun. 1988;157:87–94. doi: 10.1016/s0006-291x(88)80015-9. [DOI] [PubMed] [Google Scholar]
- 11.Bogdan C. Nature Immunol. 2001;2:907–916. doi: 10.1038/ni1001-907. [DOI] [PubMed] [Google Scholar]
- 12.Eliasson MJ, Huang Z, Ferrante RJ, Sasamata M, Molliver ME, Snyder SH, Moskowitz MA. J Neuroscience. 1999;19:5910–5918. doi: 10.1523/JNEUROSCI.19-14-05910.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]; Huang Z. Science. 1994;265:1883–1885. doi: 10.1126/science.7522345. [DOI] [PubMed] [Google Scholar]
- 13.Ashina M. Expert Opin Pharmacother. 2002;3:395–399. doi: 10.1517/14656566.3.4.395. [DOI] [PubMed] [Google Scholar]; Sarchielli P, Alberti A, Floridi A, Gallai V. J Neurol Sci. 2002;198:9–15. doi: 10.1016/s0022-510x(02)00035-7. [DOI] [PubMed] [Google Scholar]
- 14.Gatto EM, Riobo NA, Carreras MC, Poderoso JJ. Oxid Stress Dis. 2000;5:291–311. [Google Scholar]
- 15.Takashi T, Omi K, Eizo I. Neurol Res. 2004;26:563–566. [Google Scholar]; Dorheim M-A, Tracey WR, Pollock JS, Grammas P. Biochem Biophys Res Commun. 1994;205:659–665. doi: 10.1006/bbrc.1994.2716. [DOI] [PubMed] [Google Scholar]
- 16.Urushitani M, Shimohama S. Amyotrophic Lateral Sclerosis and Other Motor Neuron Disorders. 2001;2:71–81. doi: 10.1080/146608201316949415. [DOI] [PubMed] [Google Scholar]
- 17.DiGirolamo G, Farina M, Riberio ML, Ogando D, Aisemberg J, de los Santos AR, Marti ML, Franchi AM. Br J Pharmacol. 2003;139:1164–1170. doi: 10.1038/sj.bjp.0705315. [DOI] [PMC free article] [PubMed] [Google Scholar]; Perez-Severiano F, Escalante B, Vergara P, Rios C, Segovia J. Brain Res. 2002;951:36–42. doi: 10.1016/s0006-8993(02)03102-5. [DOI] [PubMed] [Google Scholar]
- 18.van’t Hof RJ, Ralston SH. Immunology. 2001;103:255–261. doi: 10.1046/j.1365-2567.2001.01261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kankuri E, Vaali K, Knowles RG, Lahde M, Korpela R, Vapaatalo H, Moilanen E. J Pharmacol Exp Ther. 2001;298:1128–1132. [PubMed] [Google Scholar]; Hokari R, Kato S, Matsuzaki K, Kuroki M, Iwai A, Kawaguchi A, Nagao S, Miyahara T, Itoh K, Sekizuka E, Nagata H, Ishii H, Miura S. Free Rad Biol Med. 2001;31:153–63. doi: 10.1016/s0891-5849(01)00565-2. [DOI] [PubMed] [Google Scholar]; Cho CH. J Physil Paris. 2001;95:253–256. doi: 10.1016/s0928-4257(01)00034-1. [DOI] [PubMed] [Google Scholar]
- 20.Vallance P, Charles I. Sepsis. 1998;1:93–100. [Google Scholar]
- 21.Kubes P, McCafferty DM. Am J Med. 2000;109:150–158. doi: 10.1016/s0002-9343(00)00480-0. [DOI] [PubMed] [Google Scholar]
- 22.Sanders SP. Am J Respir Cell Mol Biol. 1999;21:147–149. doi: 10.1165/ajrcmb.21.2.f158. [DOI] [PubMed] [Google Scholar]
- 23.Silkoff PE, Robbins RA, Gaston B, Lundberg JO, Townley RG. J Allergy Clin Immunol. 2000;105:438–448. doi: 10.1067/mai.2000.104938. [DOI] [PubMed] [Google Scholar]
- 24.Li H, Raman CS, Glaser CB, Blasko E, Young TA, Parkinson JF, Whitlow M, Poulos TL. J Biol Chem. 1999;274:21276–21284. doi: 10.1074/jbc.274.30.21276. [DOI] [PubMed] [Google Scholar]
- 25.Fischmann TO, Hruza A, Da Niu X, Fossetta JD, Lunn CA, Dolphin E, Prongay AJ, Reichert P, Lundell DJ, Narula SK, Weber PC. Nat Struct Biol. 1999;6:233–242. doi: 10.1038/6675. [DOI] [PubMed] [Google Scholar]
- 26.Gerber NC, Rodriguez-Crespo I, Nishida CR, Ortiz de Montellano PR. J Biol Chem. 1997;272:6285–6290. doi: 10.1074/jbc.272.10.6285. [DOI] [PubMed] [Google Scholar]
- 27.Ji H, Li H, Flinspach M, Poulos TL, Silverman RB. J Med Chem. 2003;46:5700–5711. doi: 10.1021/jm030301u. [DOI] [PubMed] [Google Scholar]
- 28.Flinspach M, Li H, Jamal J, Yang W, Huang H, Hah J-M, Gomez-Vidal JA, Litzinger EA, Silverman RB, Poulos TL. Nature (Struct Mol Biol) 2004;11:54–59. doi: 10.1038/nsmb704. [DOI] [PubMed] [Google Scholar]
- 29.Flinspach M, Li H, Jamal J, Yang W, Huang H, Silverman RB, Poulos TL. Biochemistry. 2004;43:5181–5187. doi: 10.1021/bi0361867. [DOI] [PubMed] [Google Scholar]
- 30.Li H, Flinspach ML, Igarashi J, Jamal J, Yang W, Gómez-Vidal JA, Litzinger EA, Huang H, Erdal EP, Silverman RB, Poulos TP. Biochemistry. 2005;44:15222–15229. doi: 10.1021/bi0513610. [DOI] [PubMed] [Google Scholar]
- 31.Erdal EP, Litzinger EA, Seo J, Zhu Y, Ji H, Silverman RB. Curr Top Med Chem. 2005;5:603–624. doi: 10.2174/1568026054679317. [DOI] [PubMed] [Google Scholar]
- 32.Huang H, Martasek P, Roman LJ, Masters BSS, Silverman RB. J Med Chem. 1999;42:3147–3153. doi: 10.1021/jm990111c. [DOI] [PubMed] [Google Scholar]
- 33.Huang H, Martásek P, Roman LJ, Silverman RB. J Med Chem. 2000;43:2938–2945. doi: 10.1021/jm000127z. [DOI] [PubMed] [Google Scholar]
- 34.Vabeno J, Lejon T, Nielsen CU, Steffansen B, Chen W, Ouyang, Borchardt RT, Luthman K. J Med Chem. 2004;47:1060–1069. doi: 10.1021/jm031022+. [DOI] [PubMed] [Google Scholar]; Roggo S. Curr Top Med Chem. 2002;2:359–370. doi: 10.2174/1568026024607490. [DOI] [PubMed] [Google Scholar]
- 35.Hah J-M, Roman LJ, Martásek P, Silverman RB. J Med Chem. 2001;44:2667–2670. doi: 10.1021/jm0101491. [DOI] [PubMed] [Google Scholar]
- 36.Limal D, Quesnel A, Briand J-P. Tetrahedron Lett. 1998;39:4239–42. [Google Scholar]
- 37.Goel OP, Krolls U, Stier M, Kesten S. Org Synth. 1988;45:67–69. [Google Scholar]
- 38.Thompson WJ, Tucker TJ, Schwering JE, Barnes JL. Tetrahedron Lett. 1990;31:6819–6822. [Google Scholar]
- 39.Plunkett MJ, Ellman JA. J Org Chem. 1995;60:6006–6007. [Google Scholar]
- 40.Ohta A, Aoyagi Y, Kurihara Y, Yuasa K, Shimazaki M. Heterocycles. 1987;26:3181–3191. [Google Scholar]
- 41.Jirousek MR, Gillig JR, Neel DA, Rito CJ, O’Bannon D, Heath WF, McDonald JH, III, Faul MM, Winneroski LL, Melikian-Badalian A, Baevsky M, Ballas LM, Hall SE. Bioorg Med Chem Lett. 1995;5:2093–2096. [Google Scholar]
- 42.Kanai K, Sakamoto I, Ogawa S, Suami T. Bull Chem Soc Jpn. 1987;60:1529–1531. [Google Scholar]
- 43.Beree F, Michelot G, Le Corre M. Tetrahedron Lett. 1998;39:8275–8276. [Google Scholar]
- 44.Mbadugha BNA, Seo J, Ji H, Martásek P, Roman LJ, Shea TM, Li H, Poulos TL, Silverman RB. Bioorg Med Chem. 2006;14:3681–3690. doi: 10.1016/j.bmc.2006.01.044. [DOI] [PubMed] [Google Scholar]
- 45.Seo J, Igarashi J, Li H, Martásek P, Roman LJ, Poulos TL, Silverman RB. J Med Chem. 2007;50:2089–2099. doi: 10.1021/jm061305c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boyd SA, Fung AKL, Baker WR, Mantei RA, Armiger YL, Stein HH, Cohen J, Egan DA, Barlow JL, et al. J Med Chem. 1992;35:1735–46. doi: 10.1021/jm00088a007. [DOI] [PubMed] [Google Scholar]
- 47.Fishbein L, Gallaghan JA. J Am Chem Soc. 1954;76:1877–1879. [Google Scholar]
- 48.Hevel JM, White KA, Marletta M. J Biol Chem. 1991;266(34):22789–22791. [PubMed] [Google Scholar]
- 49.Roman LJ, Sheta EA, Martásek P, Gross SS, Liu Q, Masters BSS. Proc Natl Acad Sci USA. 1995;92(18):8428–8432. doi: 10.1073/pnas.92.18.8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martasek P, Liu Q, Roman LJ, Gross SS, Sessa WC, Masters BSS. Biochem Biophys Res Commun. 1996;219(2):359–365. doi: 10.1006/bbrc.1996.0238. [DOI] [PubMed] [Google Scholar]
- 51.Sessa WC. J Vasc Res. 1994;31:131–143. doi: 10.1159/000159039. [DOI] [PubMed] [Google Scholar]
- 52.Hevel JM, Marletta MA. Methods Enzymol. 1994;133:250–258. doi: 10.1016/s0076-6879(94)33028-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
NOESY spectra for determination of absolute diastereomeric structures of 19a,b. HPLC traces with two different eluents to demonstrate purity of 4-11.