

Abstract
Endoglin is an accessory receptor for TGFß and can associate with Alk5 or Alk2. Although prior studies indicated that endoglin and Alk5 were not directly involved in epithelial-mesenchymal transformation (EMT) in the heart, the expression pattern of endoglin prompted a re-examination. We here show that loss of endoglin expression mediated by either antisense DNA or siRNA results in a direct perturbation of EMT and reduced expression of EMT markers including slug, runx2, RhoA, and latrophilin-2. An examination of BrdU incorporation shows that, while endoglin regulates proliferation at an early stage, reduced endothelial cell proliferation does not account for the loss of mesenchyme. As Alk5 interacts with endoglin, we utilized siRNA and a specific inhibitor, HTS466284 (HTS), to perturb this receptor as well. Alk5 inhibition produced similar effects to inhibition of endoglin. There was a reduction in mesenchymal cell formation and loss of EMT marker expression similar to that seen with endoglin. Alk5 kinase inhibition produced a similar loss of EMT marker expression but showed a contrasting upregulation of the proliferation and remodeling markers, Cyclin B2 and ß-catenin. Alk5 and endoglin both mediate endothelial cell proliferation in younger explants but, by stage 16, loss of endoglin no longer alters proliferation rates. These data show that both Alk5 and endoglin are directly involved in the process of EMT, that they interact with both TGFß-regulated activation and invasion pathways and that the roles of these receptors change during cardiac development.
Keywords: EMT, TRßRI, TGFbeta 3, Runx-2
Introduction
EMT is the process by which epithelial cells disaggregate and change shape while invading and migrating into three-dimensional environments to form a specific organ or structure or break down an epithelial barrier. EMT occurs during metastatic cancer and throughout development (Thompson and Newgreen, 2004). In the developing heart, the atrioventricular (AV) canal and the outflow tract (OT) region undergo an EMT to form the valves of the heart (Eisenberg and Markwald, 1995; Runyan et al., 1992). In these areas, the cardiac jelly (extracellular matrix) separates the myocardium from the endothelium and molecular cues from the myocardium signal the endothelium to activate (Krug et al., 1985; Markwald et al., 1977; Runyan and Markwald, 1983). During the activation step, endothelial cells organized in a single cell layer lose cell-cell contacts, undergo hypertrophy and polarization, and increase the expression of ECM molecules (Bolender et al., 1980; Crossin and Hoffman, 1991; Krug et al., 1985). In the chick at stage 17, activated endothelial cells begin to invade the cardiac jelly becoming mesenchymal cells. Studies suggest that only a subset of activated endothelial cells become mesenchymal cells (Krug et al., 1985; Runyan et al., 1990; Wunsch et al., 1994). Among the many molecules shown to regulate the EMT process, there is a common element among them involving the Transforming growth factor β (TGF-β) family (Potts and Runyan, 1989; Potts et al., 1991, Nakajima et al. 1993, Barnett et al., 1994; Brown et al., 1996; Brown et al., 1999).
The primary members of the TGF-β family are the ligands TGFβ1, 2 and 3 (Azhar et al., 2003). Studies in chick embryos show that they have distinct roles in mediating EMT and that there is some specificity of receptors in mediating isoform-specific activities (Boyer et al., 1999; Boyer and Runyan, 2001). Transforming growth factor receptors include a series of serine-threonine kinases collectively identified as Type I receptors or activin-like kinases (Alk) and a type II receptor (TGFβRII). Type I and Type II receptors form a complex that can signal through Smads and other signal transduction molecules (Wrana, 1998). Larger receptors include TGFβRIII (β-glycan) and the membrane glycoprotein, endoglin (CD105), but these have traditionally been presented as accessory proteins interacting with the Type I/Type II complex (Cheifetz et al., 1992; Lopez-Casillas et al., 1991). Endoglin binds TGFβ1, TGFß3, activin-A, bone morphogenetic protein (BMP)-7 and BMP-2 in the presence of the respective ligand binding receptors (Barbara et al., 1999). Endoglin is present in the AV canal of the chick at the time of EMT but the functional significance of this observation could not be demonstrated at the time (Vincent et al., 1998). Sorenson et al. (2003) showed that endoglin null mice have hypocellular cardiac cushions in the AV canal. However, based on collagen gel assays, they concluded that the role of endoglin in EMT was likely indirect.
In humans and mice, endoglin expression is present as two isoforms that differ in the amino acid composition of their cytoplasmic tails, L-endoglin and S-endoglin (Bellon et al., 1993; Gougos and Letarte, 1990; Perez-Gomez et al., 2005a; Perez-Gomez et al., 2005b). Human L-endoglin abrogates the inhibitory action of TGF-β in the proliferation of promonocytic cells while S-endoglin partially inhibits proliferation (Lastres et al., 1996). In humans, mutations of endoglin are associated with hereditary hemorrhagic telangiectasia type 1 (HHT1), a disease characterized by blood vessel malformation (Berg et al., 1996; Guttmacher et al., 1995; McAllister et al., 1995; McAllister et al., 1994). In mice, Endolgin-null mice (Eng -/-) showed arterio-venous malformation (HHT phenotype) and heart defects associated with valve formation and heart septation (Arthur et al., 2000; Bourdeau et al., 1999; Sorensen et al., 2003).
Most studies of endoglin are targeted towards its role as a marker of endothelial cell proliferation in angiogenesis. In this process, endoglin modulates proliferation by interacting with the TBRI receptors, activin-receptor like kinase 1 (Alk1) and Alk5 (Franzen et al., 1993; Wrana et al., 1994). Activation of Alk1 by TGF-β induces endothelial cell proliferation while TGF-β activation of Alk5 inhibits migration and proliferation of endothelial cells. Studies performed with embryonic endothelial cells from heterozygous endoglin mice (End +/-) showed that endoglin balances endothelial cell proliferation by promoting Alk1 and inhibiting the Alk5 pathway when it binds to TGFβ3 in association with TBRII (Blanco et al., 2005; Francisco J. Blanco, 2005; Lebrin et al., 2004). However, Alk5-deficient endothelial cells are not only defective in their TGFβ/Alk5 responses but in their TGFβ/Alk1 responses (Goumans et al., 2003). In these studies, Alk5 proved to be necessary for the recruitment of Alk1 into the TGF-β receptor complex. Alk5 kinase activity was crucial for the activation of Alk1 during endothelial cell proliferation during angiogenesis. Studies by Lai et al. (2000) showed a requirement for Alk2 in EMT and suggested that Alk5 was not involved in this process although it was present in the AV canal. In light of our findings concerning endoglin function, we chose to re-examine Alk5 as well.
In the present study, we re-investigate the roles of endoglin and Alk5 in the process of EMT in the AV cushion. Our results show that blocking endoglin or Alk5 inhibits EMT in cultured AV explants. As endoglin regulates endothelial proliferation by interaction with Alk5 and Alk1, we explored whether the loss of mesenchymal cells, was due to a decrease of cell proliferation. We determined that endoglin does mediate endothelial proliferation in the AV cushion but a role in EMT regulation can be distinguished. Loss of Alk5 expression by treatment with siRNA decreases cell proliferation and mesenchymal cell numbers while inhibition of Alk5 kinase activity decreases mesenchymal cell numbers at all stages and has differential effects on endothelial cell proliferation at different developmental stages. These data suggest that the role of Alk5 changes during development. Loss of EMT markers argues that both endoglin and Alk5 mediate the process of EMT and suggest that they interact together in the AV canal.
Material and Methods
In situ hybridization
Chick embryos (stages 14 to 18) were collected and fixed overnight at 4° C in 4 % paraformaldehyde. Embryos were processed for in situ hybridization as in the Harland Xenopus protocol (Harland, 1991). Antisense RNA was transcribed with T3 RNA polymerase and the sense probe with the T7 RNA polymerase from the same sub clone used for siRNa cocktail (see siRNA method).
Collagen gel culture
Collagen gel explants were prepared as previously described (Runyan and Markwald, 1983, Romano and Runyan, 1999). Briefly, rat tail collagen in acidic, diluted Medium 199 (M199, pH 4.0, 0.1-% concentration) was polymerized by addition of 10X M199 and 10X NaHCO3 (2.2 g/100ml) in 16mm multiwell dishes (Nunc). After polymerization each gel was rinsed once with 300-500 μl M199 and then incubated with complete medium (M199, 1.0% chick serum, ITS supplement (Gibco) and Antibiotic-Antimicotic (Gibco) to condition the gel until used. AV canal segments were collected from staged chick embryos into Tyrode′s solution (Sigma) and transferred into 5 ml polystyrene round-bottom tubes for each treatment. Excess medium was decanted from the gel surface and heart segments were placed on the gel surface by pipette. After attachment overnight, cultures were treated with added medium. A video showing gel preparation and AV canal dissection is available on request from the authors.
Cell counts
To determine the effects of the treatments in EMT, we counted all mesenchymal cells produced by each AV explant (cells observed below the gel surface using Hoffman Differential Contrast microscopy). Mesenchymal cell numbers were averaged per treatment for 10-30 explants. The control mean was set as 100% and the mean and standard error for each treatment was compared to the control value determined for that experiment. A similar comparison was performed to evaluate cellular activation (cells separated from each other in the endothelium but not invaded into the gel matrix). To determine the effects of the treatments in cell proliferation, cells on the surface of the gels incorporating BrdU in the nucleus were counted and compared to all DAPI-stained nuclei seen on the gel surface. Cells within the myocardial explant and within the gel matrix were ignored. Again, the mean for controls cultures was set to 100% and comparisons within each experiment were compared to that value.
siRNA treatment of AV canal explants
siRNA was made by two methods. For endoglin, Silencer siRNA construction kits from Ambion (#1625 and 1620) were used to make both a cocktail of siRNAs and a targeted sequence. For Alk5, kit # 1620 (targeted sequence) was used. To design ALK5 siRNA targets, we used the Ambion website (http://www.ambion.com/techlib/misc/siRNA_finder.html) to select specific sequences. For an endoglin target sequence, we used the RNAi target sequence prediction of the BBSRC ChickEST Database (http://www.chick.umist.ac.uk/). To the selected target sequence, we added a T7 promoter sequence and used the same Ambion siRNA construction kit.
The siRNA cocktail for endoglin was made from a fragment of 488 bp of the chicken endoglin complete sequence AY702002 corresponding to base pairs 1033 to 152 (extracellular domain). RNA was extracted from stage 15 AV and cDNA was synthesized after DNase treatment. A PCR reaction replicated the endoglin fragment. This piece was subcloned into pBluescript II SK (+/-) vector using Xho I and EcoR I. Top10 bacteria were transformed with this construct and sequencing was performed to confirm the endoglin sequence. siRNA cocktail was transfected into AV explants with oligofectamine from Invitrogen. The siRNA (10 nM) and the oligofectamine (6 μl) were diluted separately in 100 μl of M199 1X medium. They were mixed together and incubated for 15 min at room temperature to allow siRNA/oligofectamine complexes to form.
For treatment, AV explants in M199 medium were collected in 100 μl of medium and added to the mixture of siRNA and oligofectamine. Explants in 300 μl of the mixture were incubated at 37° C, 5% CO2 for 45 min to 1hr. Explants were then placed on conditioned collagen gels and incubated under the same conditions overnight. The next day, a boost was given to the explants with 10 nM of siRNA solution and incubated at 37° C, 5% CO2 for 45 min to 1hr and the siRNA solution was replaced with complete media. For transfection of siRNA derived from 21 bp RNA (target sequence), we used 4 μl of the siPORT NeoFX (Ambion) or oligofectamine and the rest of the treatment was identical. Twentyfour hours after initial transfection and incubation, explants cultures were fixed for 1/2 hr. with 4% paraformaldehyde at room temperature and washed with PBS. Fixed cultures were used for cell counts. Similar explants used for Real Time PCR were homogenized in 1 ml of Trizol and stored at - 80°C or processed immediately for RNA isolation.
The siRNA target sequence for endoglin was 5’-AAGGTCTGGCGCTTCCGCTTC-3’; for Alk5, 5’-AAGGAACTACTTTGAAGGATT-3’ and for control siRNA, a scrambled 21 oligonucleotide template containing the same number of the different bases of the endoglin siRNA target that did not blast to any gene in the chicken genome, 5’-AGACTGTCGCGTGCTCTGTCC-3’.
BrdU Staining and DNA fragmentation
After siRNA treatment and boost, AV explants on collagen gels were incubated with 100 μM of BrdU (BD Biosciences Pharmingen) in complete medium for 3 - 4 hrs. at 37° C, 5% CO2. Then, washed with PBS four times for 5 min. and fixed with 4% paraformaldehyde for 1/2 hr at room temperature. Explants were rinsed three times with PBS and washed 4X five min at room temperature. They were permeabilized with 0.1% Triton X-100/PBS for 15 min and treated with 25-30 ul of DNAse (0.1 U/ul) at 37° C for 1/2 hr. Explants were washed 5X for 5 min. at room temperature and blocked with 1% BSA/0.1% TritonX-100/PBS for 1 hr at room temperature or overnight at 4°C. Explants were incubated with 2.5 ng/μl of anti-BRDU antibody for 1hr at room temperature or overnight at 4° C. After antibody treatment, they were washed with 0.1% TritonX-100 in PBS five times 10 min and incubated for 1-2 hrs with 1:200 dilution of anti-mouse secondary antibody MCR546 (2 mg/ml) at room temperature. Explants were washed with 0.1% TritonX-100 in PBS five times 10 min. They were rinsed with PBS three times and then incubated with 300 μl of Dapi (0.1 μg/ml) for 20 min. Samples were rinsed with PBS three times and then mounted on slides with Polymount. BrdU labeled cells were counted and photographed using a Deltavision Deconvolution microscope.
Apoptosis was measured using a DNA Ladder Assay PCR Kit designed for the detection of nucleosomal ladders in apoptotic cells (Maxim Biotech, Inc., San Francisco, cat. # APO-DNA1). Fifteen stage 16 AV explants were treated with endoglin antisense, 13 with reverse sense and 13 untreated and incubated at 37° C, 5% CO2 for 45 min in 300 ul of M199 medium containing the respective treatments. A positive control sample of 13 AV explants was incubated at 42° C in M199 medium for 45 min. After this incubation period, explants were transferred to collagen gels and incubated for 8 hours at 37° C, 5% CO2. DNA from each of the samples was extracted with the BDtract Genomic DNA Isolation Kit (Maxim Biotech, Inc., cat # SA-4002). Analysis was carried out according to the protocol instructions of APO-DNA1 kit. Equal amounts of genomic DNA were used for the ligation of the adaptors and the PCR reaction. The positive control of the kit was used to compare the levels of apotosis in the experimental samples. 10 ul from each PCR reaction corresponding to each of the respective treatments and the kit’s positive control were loaded into each well of a 2% agarose.
Quantitative Real Time PCR
AV explants were homogenized with Trizol (Invitrogen) and processed for RNA isolation according to the manufacturer′s protocol. RNA was DNased (Turbo DNA-free Ambion Kit #1907) and cDNA was synthesized using an iScript cDNA synthesis kit (# 170-8891, BioRad). cDNA was measured with a Mini-Fluorometer (Turner Biosystems) after staining with OliGreen ssDNA Quantitation (Molecular Probes) and equal aliquots of cDNA of control and experimental samples were added to triplicate reaction mixtures. Real time PCR reactions were carried out using primers in table 1, the super mix Platinum SYBR Green qPCR Super Mix UDG (invitrogen) and the Rotor Gene 3000 System from Corbett Research. Analysis of the data was carried out using the Rotor Gene 6 software provided by the manufacturer.
Table 1.
Real Time PCR primers
| Primer | Forward sequence | Reverse Sequence |
|---|---|---|
| Endoglin | 5’-CAACAACCAAGGGCTGGGG-3’ | 5’-TGGAGATGGGACGGGTATGC-3’ |
| ALK5 | 5’-GCGGCCGCATTACAATGTTAC-3’ | 5’-TGGCCTGTCTCGAGGAATCA-3’ |
| Runx2 | 5’-AACCCAAACTTGCCCAACCAGA-3’ | 5’-GCCTCCAAACGGACTCATCCAT-3’ |
| Slug | 5’-GCAGACCCACTCGGATGTGAA-3’ | 5’-CGCAGCAGCCAGATTCCTCA-3’ |
| Latrophilin 2 | 5’-CTGAGGGAACCGACAGCTAC-3’ | 5’-CGGAGAGTCTCTGAGGTTGG-3’ |
| RhoA | 5’-GTTGGCTTTGTGGGATAC-3’ | 5’-CAGAAATGCTTCACTTCCG-3’ |
| VE- cadherin | 5’-TTCACAGCTCTTCACCCAGGCA-3’ | 5’-CCCCAAGCAGCAATTTTGAGGA-3’ |
| β-catenin | 5’- TCCCCCATTGAGAATATCCA -3’ | 5’- GTTGCAACACCCTCATTCCT -3’ |
| Cyclin B2 | 5’-AGGGGTGGAGAATGCCGTGA-3’ | 5’-TGCCAGGTCCTTTCGTAGCCTT-3’ |
| Fibulin 2 | 5’-CAGCCAGGAGTGTGCCAATGTT-3’ | 5’-CGGAAAGTGCAAAGAATGCCCT-3’ |
| Id1 | 5’-CGCTGCTGTATGACATGAAGGG-3’ | 5’CAGGTCCCAGATGTAGTCGATCAC-3’ |
Inhibition of Alk5 kinase activity
AV explants from stage 15 and 16 embryos were dissected in Tyrode′s, rinsed in M1991X medium and placed on preconditioned collagen gels. The explants were incubated at 37° C, 5% CO2 for 8hrs to allow them to adhere to the collagen. 100 nM of HTS466284 (HTS; gift of Dr. Daruka Mahadevan) in 300 μl of complete medium was added to the experimental explants at eight hours (solid inhibitor was dissolved in DMSO and was serially diluted in M199 to reach the final concentration). The timing of HTS exposure allowed for normal endothelial outgrowth from the explants but was added at a time when there was little or no mesenchymal cell invasion into the gel. Explants received a final DMSO concentration of 0.5-2 nl/ml. Controls were treated with an equal volume of DMSO in complete medium. Explants were incubated overnight. After 16-18 hr, the inhibitor was replaced by with complete medium containing BrdU in the cultures needed for cell counting and complete medium in the cultures needed for RNA extraction. After 3 to 4 hrs of incubation, the cultures were processed for BrdU staining and/or RNA extraction respectively (as above).
Results
Developmental expression of Endoglin in the AV canal
In humans and mice endoglin expression occurs during the time when the heart valves are formed and it is expressed in the endothelium as well as in the mesenchyme of the AV canal (Bourdeau et al., 2000; Qu et al., 1998). Studies of endoglin expression in the chick by immunohistochemistry showed that endoglin protein levels in the AV canal progressively decreased in the myocardium between developmental stages 14 to 16 and then rose at stage 17 in the endothelium and cushions (Vincent et al., 1998). We explored by in situ and real-time PCR whether expression levels of endoglin mRNA changed in concert with protein levels during development in the AV canal. Our in situ data show that endoglin mRNA expression is detected in the endothelium at stage 15 (earliest stage examined), decreases at stage 16 and then increases at stage 17 when mesenchymal cells first start invading the cushions in the AV canal (Figs. 1A, B and C). Endoglin expression in the heart is present in the endothelium of the outflow tract region and the pro-epicardium (Fig. 1E) where EMT occurs. The pro-epicardium gives rise to the coronary vessels of the heart (Mikawa and Fischman, 1992; Olivey et al., 2004; Poelmann et al., 1993). In the chick, we also see endoglin expression in the developing blood vessels of the brain and of the rest of the body (Fig. 1F). These data agree with the pattern of expression of endoglin in the mouse where it is expressed in the cushions and major and minor developing blood vessels (Bourdeau et al., 1999). Endoglin mRNA was also localized in the mesenchymal cells of the AV canal at stage 17 and 18 (Fig.1G) as well as in the mesenchymal cells of the OFT region. We confirmed the apparent decrease of endoglin expression at stage 16 by Real Time-PCR using RNA isolated from staged AV explants. These results show that endoglin mRNA expression levels decrease between stages 14 to 16 and increase at stage 17 (Fig. 2A). The expression of endoglin in the AV canal (endothelial and mesenchymal cells), OT and proepicardium is consistent with a role for endoglin in the process of EMT.
Figure 1.

Endoglin expression by in situ hybridization in the heart at the time of EMT. (A). Endoglin is expressed in the endothelium (arrow) of the AV canal in a stage 15 embryo. (B). A stage 16 embryo shows a decrease in endoglin expression in the AV canal compared to stage 15 and 17 embryos. (C). Endoglin expression increases in the AV canal at stage 17. (D). Stage 15 embryo reacted with an endoglin sense probe shows no expression of endoglin. (E). Stage 17 embryo shows expression in the endothelium of the outflow tract (OT) region and in the proepicardium (PE) where EMT occurs. (F). Embryo at stage 17 shows endoglin expression in blood vessels including the posterior cardinal veins (pCV), intersegmantal vessels (ISv) between the somites and the microvessels (mv) in the head. (G). Section of the AV canal at stage 18 shows endoglin expression in the mesenchyme and the endothelium of the Av canal.
Figure 2.

Endoglin expression changes during development but is required for EMT. (A). Levels of endoglin mRNA expression in AV canal segments measured by Real Time PCR. Dissected AV canal segments were collected for mRNA analysis. Endoglin expression levels decreased at stage 16 and increased at stage 17 when mesenchymal cells begin to invade the cushions. Data represent duplicate experiments with three repetitive reactions per stage. Data was normalized to total cDNA in each reaction. Changes are graphed relative to stage 14 and relative values are displayed above each bar. (B). Cells from antisense endoglin treated explants (60 pooled explants, stages 15 -17 at time of collection) showed a loss of mesenchymal and activated cells (40% reduction) after 24 hrs compared to control oligonucleotides. (C) Staged explants were specifically examined for activation and invasion after siRNA endoglin treatment. Loss of endoglin produced the greatest loss of mesenchyme in stage 15 explants but a loss was observed in explants collected from each of the other stages. Activation of endothelial cells on the surface was unaffected in stage 16 explants and significant in both the younger and older explants. siRNA effects on mesenchymal and activated cells are compared to effects of control siRNA. Activated cells are defined as cells without attached neighboring cells remaining on the surface of the gels. A minimum of 15 explants were counted for each siRNA experiment. Error bars indicate the SEM. Significant p values, smaller than 0.05, are indicated by an asterisk (*) in each panel.
Blocking endoglin expression reduces the number of mesenchymal cells in AV explant cultures
As endoglin expression is present in the AV canal during EMT, we examined whether blocking endoglin expression would affect either the activation process before EMT or the number of mesenchymal cells produced in vitro. We used both antisense DNA and siRNA endoglin to block endoglin expression in AV explants (stages 13-17) and measure changes in the number of activated endothelia or invaded mesenchymal cells within the collagen gel matrix. AV explants were transfected with either antisense DNA or siRNA for 45 min at 37° C, 5% CO2. The explants were then cultured on collagen gels overnight and fixed after 18 - 24 or 48 hours of incubation with antisense DNA and for 24 hrs with siRNA. Results with antisense DNA were qualitatively and quantitatively similar to siRNA in showing a loss of mesenchyme at 18-24 hr (Figs. 2B, 2C and 3) but by 48 hr mesenchymal cell numbers were indistinguishable from controls (data not shown).
Figure 3.

Blocking endoglin expression by either Antisense DNA or siRNA decreases the number of mesenchymal cells. Images of collagen explant cultures were taken with a plane of focus just beneath the endothelial layer to show mesenchymal cells within the gel. A. Control, culture treated with reverse sense, endoglin oligodeoxynucleotides shows a typical distribution of mesenchymal cells after 24 hours of treatment. B. Endoglin antisense DNA decreased the number of mesenchymal cells in cultured AV explants after 24 hrs of treatment. C. Control siRNA treatment with normal mesenchymal cell invasion in stage 16 cultured AV explants. D. siRNA endoglin decreased the number of mesenchymal cells in cultured AV explants. The endothelial monolayer produces the out of focus polygonal pattern and the dark mass in each picture is the myocardial portion of the explant. Arrows mark selected mesenchymal cells in each micrograph.
Cultures treated with siRNA were examined to see whether changes in activation (endothelial cell separation) were coincident with changes in mesenchymal cell formation. Endoglin siRNA decreased the number of mesenchymal cells significantly compared to controls by 90% at stage 15, 40% at stage 16, and 50% at stage 17 (Figs. 2C and 3). A loss of activation was observed at stages 15 and 17 but not at stage 16 (Fig. 2C). At stages 15 and 16 there was a relatively greater loss of mesenchymal cell number than a reduction in activation.
Loss of mesenchyme in the cultures can occur by a direct inhibition of EMT, cell death in endothelial or mesenchymal cells or a reduced rate of proliferation. To explore programmed cell death, we performed a DNA fragmentation assay between endoglin antisense DNA-treated explants and explants treated with the control oligonucleotide. There was little DNA fragmentation and no difference between treated and control cultures (data not shown). We conclude that cell death is not a viable explanation for the loss of mesenchymal cells in these cultures. While changes in cell proliferation are explored below, the decrease in number of mesenchymal cells by both antisense and siRNA argues that endoglin has a functional role in the formation of the mesenchymal cell population.
Endoglin regulates genes known to function in the process of EMT
To begin to understand how endoglin functions in the EMT process during heart valve development, we performed initial gene microarrays to identify potential endoglin targets during EMT in the AV canal. AV explants from chicken embryos of stages 15, 16, and 17 were treated with either antisense endoglin or reverse sense sequence. The microarray data identified endoglin numerous ESTs as well as several well-known genes involved in the process of EMT including β-catenin and RhoA. ß-catenin is considered to be a crosslink between TGFβ2 and Wnt-signaling pathways in the induction of the AV canal EMT in the mouse (Liebner et al., 2004) as well as a gene regulated by Wnt-9a during mesenchymal cell proliferation in the AV canal (Person et al., 2005). RhoA is involved in the EMT process, regulated by TGFβ2 and TGFβ3 and it regulates β-catenin expression in the chick AV canal (Tavares et al., 2006). Our previous work identified slug and VE-cadherin as targets of TGFß2 (Romano and Runyan, 2000). Runx2, a TGFß-related transcription factor, is expressed in the AV canal (Gitler et al., 2003) and blocking its expression decreases the number of mesenchymal cells (unpublished data). Latrophilin-2 is a G protein-coupled receptor involved in EMT in the AV canal (Doyle et al., 2006). Fibulin-2 is an extracellular matrix maker that is expressed by mesenchymal cells in the AV cushions during EMT in the AV canal of the mouse (Takeshi Tsuda, 2001).
Real Time PCR was used to determine whether loss of endoglin expression in AV explants at stage 16 perturbs expression of these markers. To examine whether changes in marker expression were specific to AV canal endothelium engaged in EMT, changes in selected markers were also evaluated in ventricular explants. Real time PCR measurement showed that inhibition of endoglin expression by siRNA in the AV canal down regulates expression of Runx2, slug, latrophilin2, Rho A, VE-cadherin, β-catenin, fibulin2 as well as cyclin B2 and Id1, two markers of cell proliferation (Fig.4A). Ventricular explants produced endothelia but not mesenchyme after 24 hr in culture and showed an increase in Runx2 expression after loss of endoglin (Supplemental Figures 1 and 2). As formation of a RISC complex and specific degradation of mRNA by siRNA is not detectable in less than 10 hr (unpublished data), we examined loss of marker mRNA expression after treatment with antisense DNA targeting endoglin mRNA at earlier time points (Runyan et al., 2000). Real time PCR measurement of selected markers at 4 hr and 8 hr after treatment showed significant loss of marker expression at 4 hr and a greater loss of marker expression at 8 hr (Fig. 4B). While the change in marker expression at these intervals does not determine whether endoglin directly or indirectly regulates marker expression, it suggests that changes in marker expression are the result of a loss of signal transduction requiring endoglin.
Figure 4.

Inhibition of Endoglin expression decreases expression of EMT markers but alters proliferation only in younger explants. (A). AV canal explants were collected for measurement of mRNA levels after 24 hr of treatment with endoglin or control siRNA. Endoglin siRNA effectively inhibited expression of endoglin and concomitantly reduced the TGFß receptor Alk5, EMT-related markers Runx2, Slug, Latrophilin-2, RhoA, and Fibulin 2. The endothelial and mesenchymal cell proliferation markers, ß-catenin, Id1 and cyclin B2, and an endothelial cell adhesion marker, VE-cadherin, are also reduced after loss of endoglin. The mean expression level for each marker in the control samples was set at 1.0. (B). To explore loss of endoglin at earlier intervals, cultures (30 explants/treatment) were incubated with antisense DNA or control DNA and examined at 4 and 8 hr. Endoglin expression was reduced 90-95% at both time points. These treatments blocked the expression of markers similarly to siRNA and the effect was stronger at 8 hr than at 4 hr. Triplicate reactions were performed for each gene and compared to control samples that were normalized to 1.0. Error bars indicate the standard deviation. (C). Endoglin regulates endothelial cell proliferation in younger explants but not in older explants. To assess changes during development, heart explants were taken from various stages of development. We noted a transition in responses between stage 15 (∼52 hr) and stage 16 (∼55hr) embryos. Endothelial cell proliferation is reduced in explants derived from stage 15 when endoglin expression is inhibited. Older explants, closer to the onset of EMT in the AV canal (stage 16), show no change in proliferation upon siRNA inhibition of endoglin. N= 8 for each SEM bar. * indicates p<0.01, n.s.: not significant.
Endoglin-mediated regulation of proliferation is stage specific
Several studies showed that endoglin modulates the process of endothelial cell proliferation and migration during angiogenesis. Lebrin and coworkers found that Endoglin balances signaling pathways between TGF-β/Alk1 and TGF-β/Alk5 (Lebrin et al., 2004). Endothelial cells that lack endoglin do not proliferate because TGFβ/Alk1 signaling is reduced and TGFβ/Alk5 signaling is increased creating an imbalance in cell proliferation. Our results (above) show that blocking endoglin expression decreases cyclin B2 expression in the AV canal and confirm a role for endoglin in endothelial cell proliferation. Therefore, we explored whether the decrease in mesenchymal cell numbers was due to a decrease in cell proliferation. Endoglin expression in AV explants derived from stage 15 and 16 embryos was inhibited with siRNA and explants were labeled with BRDU to measure the rate of endothelial cell proliferation. Our results show that inhibition of endoglin expression significantly reduced the number of BrdU-labeled endothelial cells in stage 15 explants (Fig. 4C). However, older, stage 16 explants had no significant change in endothelial cell labeling. This stage specificity is concomitant with a change in protein levels (Vincent et al., 1998) and a change in mRNA expression levels in the endothelium and mesenchyme (Fig 1A, B, C, G and 2A). Inhibition of endoglin expression at both stages produced a decrease in the number of mesenchymal cells (Fig. 2B). These data suggest that loss of mesenchymal cell numbers at the earlier stage could be due to a changed rate of proliferation but that reduced mesenchymal cell formation in the older stage is not attributable to altered proliferation. These data indicate that at stage 16 endoglin has distinct roles in endothelial cell proliferation and EMT in the AV canal.
Alk5 kinase inhibitor blocks mesenchymal cell formation in vitro
Endoglin is known to interact with two TGFß Type I receptors, Alk1 and Alk5 (Blanco et al., 2005; Guerrero-Esteo et al., 2002). It is recognized that TGFβ1 and TGFß3 inhibit endothelial cell proliferation through the Alk5 pathway (Goumans et al., 2002). The interaction of endoglin with the Alk5 and Alk1 pathways balances endothelial cell proliferation by potentiating the Alk1 pathway and inhibiting the Alk5 pathway (Lebrin et al., 2004). We used 100 nM HTS, a specific Alk5 kinase inhibitor (Singh et al., 2003), to block Alk5 kinase activity in AV explants stages 15 and 16 and measure proliferation rate by incorporation of BrdU. Our results show that inhibiting the kinase activity of Alk5 with this specific inhibitor does not significantly change endothelial cell proliferation in explants at either stage (Fig.5A). However, the number of mesenchymal cells decreased significantly in both stages (Fig. 5B and 6A and 6B). This is consistent with a role for Alk5 in mediating TGFß stimulated EMT. These results dissociate the number of mesenchymal cells within the gels from cell proliferation.
Figure 5.

Inhibition of Alk5 kinase activity does not affect significantly the rate of endothelial cell proliferation in AV canal explants but it does block EMT. (A). Alk5 kinase activity inhibited with 100 nM HTS does not significantly alter endothelial cell proliferation in explants derived from either stage 15 or 16. Stage 15 N= 10 and stage 16 N= 15. (B) Inhibition of Alk5 kinase activity by HTS at both stages 15 and16 reduces the number of mesenchymal cells in the collagen gel matrix. N=13 for each bar. * indicates p<0.01.
Figure 6.

Alk5 regulates the process of EMT in the AV canal. Both inhibition of Alk5 kinase activity with HTS or loss of Alk5 by siRNA blocks the invasion of mesenchymal cells. Pictures are focused beneath the endothelial cell layer to show the presence of mesenchymal cells in the gels. (A). Control stage 16 AV explant treated with DMSO demonstrates normal level of mesenchymal cells in the collagen matrix. (B). AV explant treated with the Alk5 kinase inhibitor has almost no mesenchymal cell invasion. (C). Stage 16 explant treated with control siRNA has normal invasion of mesenchymal cells. (D). AV explant treated with Alk5 siRNA blocked the invasion of mesenchymal cells into the collagen matrix. Mesenchymal cells are marked by arrowheads.
Loss of Alk5 expression decreases proliferation rate and mesenchymal cell number
As inhibition of kinase activity by HTS may not prevent important intermolecular associations by Alk5 and the inhibitor may also inhibit Alk4 kinase, we explored whether loss of Alk5 expression would similarly affect endothelial cell proliferation and EMT in the AV canal. siRNA was used to inhibit Alk5 expression in stage 15 and 16 AV explants and cell proliferation was measured by incorporation of BrdU. Results showed that loss of Alk5 significantly decreased the rate of endothelial cell proliferation (Fig. 7A) and the number of mesenchymal cells in both stages (Fig. 7B). However, the number of activated endothelial cells decreased at stage 15 but not at stage 16 (Figs. 6C, 6D and 7B) as seen with endoglin siRNA in both stages. These results confirm a requirement for Alk5 in EMT but suggest that regulation of cell proliferation and cell activation in the AV canal by Alk5 is more complex.
Figure 7.
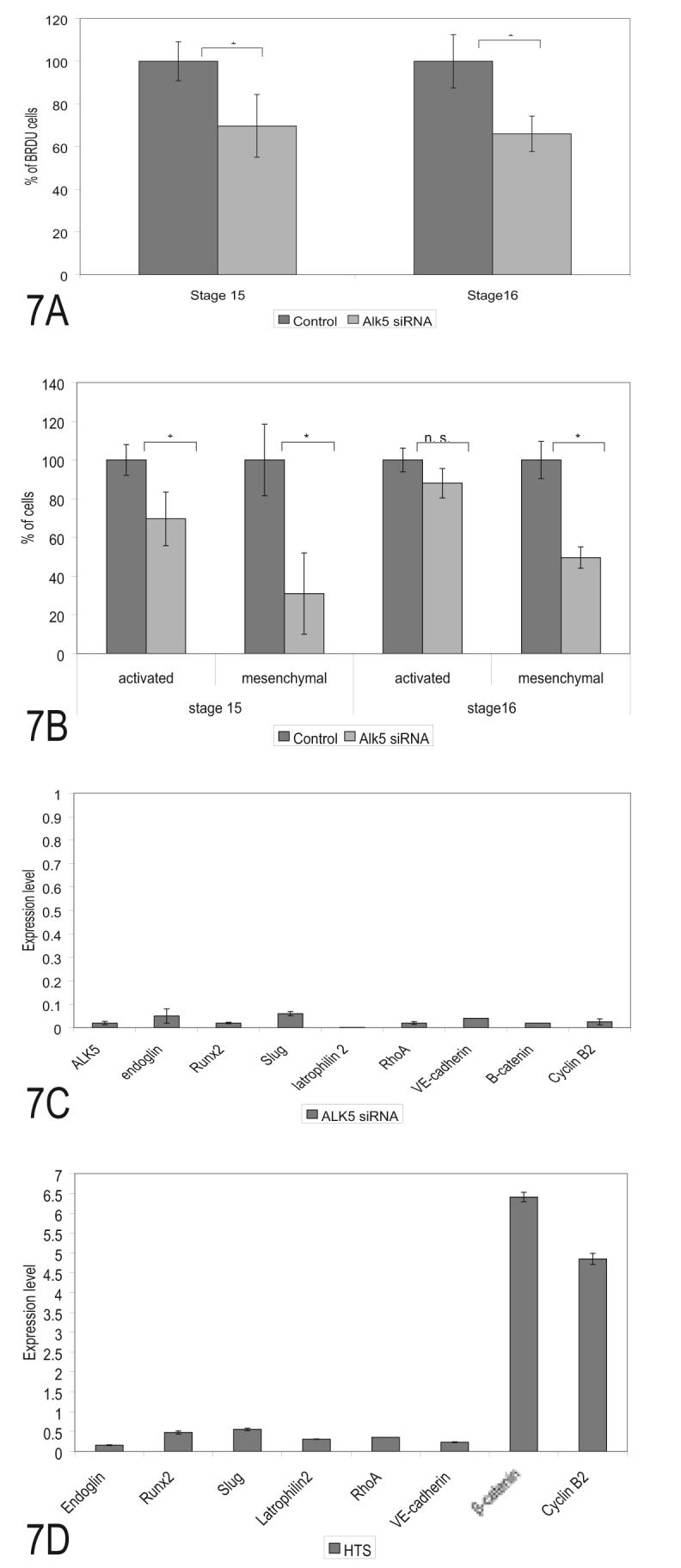
Loss of Alk5 expression affects endothelial cell proliferation, invasion and marker expression. (A) Alk5 siRNA treatment significantly decreased endothelial BRDU incorporation in stage and stage and 16 explants compared to the controls. (B). Alk5 siRNA significantly decreased the number of activated cells in stage 15 explants but not stage 16 explants while decreasing the number of mesenchymal cells in both stages. The loss of mesenchymal cells was greater than the change in cell proliferation at each stage. (C). siRNA inhibition of Alk5 in stage16 AV canals decreased mRNA expression of EMT markers including endoglin, Runx2, slug, latrophilin-2, and RhoA. Cell adhesion marker VE-cadherin and the proliferation markers β-catenin and Cyclin B2, were also reduced by loss of ALK5. (D). In contrast to loss of Alk5 expression, inhibition of Alk5 kinase activity by HTS (100 nM added 8 hrs after incubation) produced an inhibition of EMT markers and an elevation of cell proliferation markers. Kinase activity by this TGFß receptor is part of the regulation of EMT and appears to be keeping cell proliferation in check. Ten explants were counted for each lane in A and B and *= p<.05. Thirty explants were collected for each treatment in C and D and error bars indicate the SEM of triplicate real time PCR measurements.
Blocking Alk5 expression and inhibiting its kinase activity down-regulated the expression of EMT markers in the AV canal
We examined marker regulation to compare with the endoglin data and to explore differences between loss of Alk5 and kinase inhibition. Real time-PCR was performed on stage 16 AV explants after siRNA (Fig. 7C) or HTS (Fig. 7D) treatment overnight. The timing of HTS addition was chosen to allow outgrowth of endothelia onto the collagen gel surface prior to EMT and to approximate the time that functional perturbation produced by siRNA would be expected to start. Loss of EMT marker expression after each treatment was similar to that seen after inhibition of endoglin expression and is consistent with the model of endoglin-Alk5 interaction seen in endothelia. Loss of Alk5 by siRNA decreased cyclin B2 expression, consistent with BrdU incorporation (Fig. 7A). However, inhibition of Alk5 kinase activity with HTS increased the expression of cyclin B2 and ß-catenin (Fig. 7D). This contrasting regulation of two endothelial cell proliferation markers by loss of Alk5 or inhibition of Alk5 kinase suggests that Alk5 kinase activity is required to keep cell proliferation in check or that HTS interacts with another kinase in the regulation of these to markers.
Singh et al (2003) showed that HTS had an IC50 of 60 nM for Alk5. To demonstrate that HTS kinase inhibitor at 100 nM was an appropriate intervention, a dose response experiment was undertaken. Using two EMT markers (Runx2 and Slug) and a proliferation marker (Id1) under the same treatment protocol, we found small differences in dose-response between each marker. Runx2 showed a maximum inhibition of expression at 50nM while Slug was maximally inhibited at 200nM and Id1 was most strongly affected at 400 nM (Fig. 8). Differences in inhibition were relatively small in the range 50-200 nM. At 400 nM HTS, inhibition of Runx2 was no longer observed but inhibition of slug and Id1 remained. The 400 nM dose was also observed to completely block endothelial outgrowth from stage 16 explants when HTS was added to collagen gels prior to the addition of explants (data not shown). Together, these results suggest that ALK5 kinase is necessary for the regulation of EMT and proliferation genes and that HTS is not a completely specific inhibitor of Alk5 kinase activity.
Figure 8.
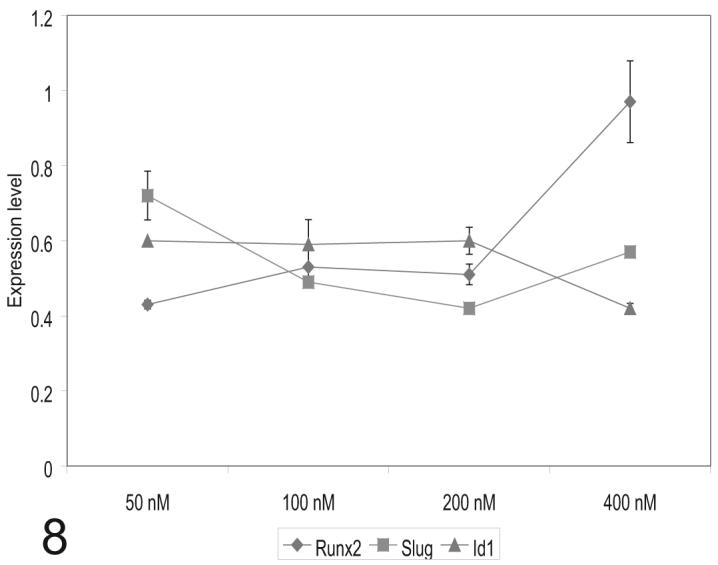
Alk5 kinase inhibitor, HTS, effects vary by dose, tissue developmental stage and time of treatment. Stage 16 explants were treated with varying does of HTS after 8 hr in culture. This interval was selected to disrupt the process of EMT at a time similar to the perturbation provided by siRNA treatment. Under these conditions, the maximal inhibition of Runx2 was at 50 nM, for Slug at 200nM and 400nM for Id1. These data suggest that 100 nM HTS is an appropriate dose for comparison to siRNA treatment for endoglin and Alk5.
Discussion
In the present study, we show evidence for the first time that endoglin and Alk5, members of the TGFβ receptor family, directly regulate the process of EMT in the AV canal. Loss of endoglin or Alk5 expression or inhibition of Alk5 kinase activity, each decrease the number of mesenchymal cells and concomitantly down-regulate expression of EMT markers in AV explants. We further show that changes in endoglin expression in the AV canal during development coincide with the acquisition of a role for endoglin in mediating EMT.
While changes in cell proliferation with the loss of endoglin could account for the decrease of mesenchymal cells during EMT, we found clear evidence in older explants that the decrease in mesenchymal cell number is independent of changes in endothelial cell proliferation. DNA fragmentation assays also showed that the loss of mesenchyme after inhibition of endoglin expression was not due to programmed cell death. Thus, endoglin appears to play a direct signal transduction role in the regulation of EMT. We further show that changes in endoglin expression in the AV canal during development coincide with this role for endoglin in mediating EMT. Together, these results suggest an interaction between Alk5 and endoglin in mediating EMT. Differences in cyclin B2 and ß-catenin expression between loss of endoglin and Alk5 kinase activity suggest that the interaction is not exclusive.
These data vary significantly from published work on both endoglin and Alk5. The endoglin null mouse has cardiac cushions that are acellular or hypocellular consistent with a role in EMT but, in contrast to the data presented here, Sorensen and co-workers (2003) found that AV explant cultures from these mice were indistinguishable from controls at 48 hr. We observed significant differences in chick cultures at 24 hours but saw a recovery by 48 hrs. As our interventions may be temporally limited, recovery in the chick cultures could correspond to renewal of mRNA and protein expression, but this does not explain the observation in murine cultures. We speculate that a similar recovery may have also occurred in the mouse cultures by the induction of a redundant mechanism. As there is no apparent recovery in vivo, differences between extracellular matrix substrates in vivo and in vitro may be significant. The striking loss of mesenchymal cells in the cushions of the Eng-/- mouse argues that endoglin may also function as a mediator of EMT in mammals in vivo. Expression of endoglin in the AV canal has been observed in humans. Studies by the Letarte group showed that endoglin expression is up-regulated during heart septation and valve formation in humans in cushion tissue mesenchyme at greater levels than the other TGF-β receptors (Qu et al., 1998).
Mutations in endoglin are associated with hereditary hemorrhagic telangiectasia (HHT1) (McAllister et al., 1994: 1995). It is possible that the inhibition of EMT seen here with loss of endoglin has a relationship to the etiology of the arterio-venous malformations associated with HHT1. As noted by Braverman et al (1990) the formation of the lesions is associated with a loss of peri-endothelial cells. It is not clear that peri-endothelial cells are formed by EMT from the endothelium but there is evidence to suggest that endothelial cells “transdifferentiate” into vascular wall cells in a process similar to the EMT seen in the heart (De Ruiter et al., 1997). Angiogenic mechanisms include also an invasion and migration of endothelial-derived cells similar to the EMT described here. Reports that angiogenesis is reduced in endoglin heterozygote mice and that endoglin is associated with cell migration of endothelial cells may be relevant to both EMT and the etiology of the arterio-venous malformations associated with HHT1 (Conley et al., 2004; Jerkic et al., 2006).
Studies by Barnett and colleagues similarly suggested that Alk5 does not regulate EMT in the AV cushion of the chick heart. In the experiments of Lai et al. (2000), an antibody against the extracellular domain of Alk5 (with a demonstrated blocking activity in regulation of a PAI-1 reporter in mink lung epithelial cells) failed to inhibit EMT in stage 13 to 18 AV explants. As both inhibition of kinase activity and reduction in expression of the molecule inhibit EMT in this study, the failure of the antibody to inhibit EMT suggests that the extracellular epitope recognized by the antibody is not a critical part of the EMT signal. Additional studies by Desgrosellier et al. (2000) using adenoviral constructs containing constitutively active Alk5 (caAlk5) receptor were injected into the heart lumen of stage 10 - 12 chick embryos. AV explants from these embryos were cultured in collagen gels and overexpression of caAlk5 had no effect on mesenchymal cell numbers. The failure of overexpression to induce added EMT suggests a requirement for a specific interaction. The caAlk5 construct was generated by replacing threonine 204 of the GS domain in Alk5 with an aspartic acid, which yields a product that has an increased kinase activity (Labbe et al., 2000; Ward et al., 2002; Wieser et al., 1995). However, it was shown by others that a constitutively active human Alk5 with the same substitution (T204D) does not interact with endoglin when co-transfected in COS-7 cells (Guerrero-Esteo et al., 2002). One explanation for the inability of caAlk5 to enhance EMT in the explants, despite an active kinase domain, is its inability to interact with endoglin. Though kinase inhibitor experiments suggest that Alk5 kinase activity is important, the common regulation of EMT markers by the loss of either Alk5 or endoglin is consistent with a requirement for an interaction between these molecules.
There is a complexity of TGFß signaling and receptor activity in the AV canal that remains to be elucidated. We previously showed that two distinct TGFß signal transduction pathways are involved in the induction of EMT. Inhibition of TGFß2 blocks endothelial cell separation and expression of the zinc finger transcription factor, slug (Boyer et al., 1999; Romano and Runyan, 2000). Inhibition of TGFßRIII receptor in AV canal cultures similarly blocks cell separation and EMT (Brown et al., 1999, Boyer et al., 1999; 2001). Inhibition of TGFß3, TGFßRII or Alk2 all result in cultures where endothelial cells are activated and separated but are unable to invade the collagen gel matrix (Potts et al., 1991; Brown et al., 1996, Boyer et al., 1999; 2001; Lai et al., 2000; unpublished data). At its simplest level, these molecules can be divided into two distinct and sequential pathways using TGFß2 for separation/activation and TGFß3 for invasion. However, the present study appears to blur this distinction.
In this study, loss of either endoglin or Alk5 yields images of collagen gel cultures with rounded rather than fibroblastic endothelial cells and the younger cultures tested with siRNA towards Alk5 showed a loss of activation as well as cell invasion. Though endoglin binds TGFß3 with greater affinity than TGFß2 (Cheifetz et al., 1992), these data suggest a relationship with the TGFß2-mediated activation process as well. The regulation of slug was previously shown to be TGFß2-specific (Romano and Runyan, 1999) and loss of either endoglin or Alk5 produced a loss of expression in the present study. Measurements of activation and invasion showed an effect by loss of endoglin on both steps of EMT. A recent examination by microarray also supports this suggestion (unpublished data). We found that genes and ESTs altered by loss of endoglin were equally likely to be found in subsets of markers specifically regulated by blocking antibodies to TGFß2 or TGFß3. The basis for an interaction between endoglin, Alk5 and the TGFß2/TGFßRIII pathway is poorly understood but it was shown in chondrocytes that endoglin and TGFßRIII (betaglycan) could form a heteromeric complex independently or involving TGFßRII (Parker et al., 2003). If such a complex is formed in the developing heart as part of cell separation and activation, it does not require TGFßRII, as separation, but not invasion, proceeds normally when TGFßRII is inhibited (Boyer et al., 2001). The regulation of cell invasion markers such as Runx2, Latrophilin-2 by endoglin and Alk5 is consistent with an interaction with the TGFßRII as a regulator of invasion (Brown et al. 1996) but we note that Jiao et al. (2006) recently demonstrated normal EMT in a mouse null for TGFßRII. The recovery of cell invasion in mouse endoglin null explants and the normal invasion seen in TGFßRII expants suggests that such an association is not critical for invasion in vitro. It would be interesting to determine whether the loss of both TGFßRII and endoglin in the mouse would produce a failure of EMT without the recovery seen in the endoglin null explants.
One explanation for the role of Alk5 in the EMT process is based on the observation that Alk5 kinase activity is required for TGFß/Alk1 signaling for cell proliferation (Goumans et al., 2003). Loss of Alk5 could inhibit Alk2-mediated EMT in a similar manner. Observed differences in morphology argue against this explanation but they do not disprove it. When we explored a dose response to HTS using selected markers of proliferation and EMT, we found a much different pattern of regulation of Runx2, suggesting that the kinase inhibitor may be interacting with other kinases as well despite the reported specificity of the molecule (Singh et al., 2003). While the inhibitor activity is consistent with loss of Alk5 by siRNA for EMT markers, the differences in the dose response assays between Id1 and Cyclin B2 and the contrast between the HTS and siRNA treatments suggests that the role of Alk5 and other kinases in regulation of proliferation is more complicated.
The data are most consistent with a role for endoglin at both stages of EMT. First, as a modifier of TGFß2 signal transduction, in concert with Alk5 and TGFRIII, at the cell separation stage. Markers such as slug and VE-cadherin in addition to morphology argue this point. Their sensitivity to loss of endoglin and Alk5 as well as TGFß2 supports an interaction. TGFß3 is required for cell invasion and involves both TGFßRII and Alk2 in the chick (Potts et al., 1991, Brown et al., 1996, Boyer et al., 1999; Lai et al., 2000; Boyer and Runyan, 2001). RhoA is a marker of cell separation and invasion regulated by both TGFß2 and TGFß3 (Tavares et al., 2006). It is sensitive to both endoglin and Alk5 function in the present study. Runx2 and latrophilin-2 are markers primarily for cell invasion (Gitler et al., 2001; Doyle et al., 2006) and they are also sensitive to the perturbation of both endoglin and Alk5. Though changes in marker expression during invasion could be consequences of altered TGFß2 signaling, a large majority of responses to TGFß isoforms in this system are isoform-specific. In light of the potential for endoglin to interact with TGFß3 (Cheifetz et al., 1992) and Alk2 (Mo et al., 2002) and for Alk5 to interact with Alk2 (Blanco et al., 2005), the regulation of invasion markers seen here is likely to be direct. Ongoing efforts to characterize downstream markers of the separation and invasion steps will be useful in resolving potential components of receptor signaling complexes.
Supplementary Material
Legend for Supplemental Figure 1:
Endoglin siRNA effects on stage 16 AV and ventricle explants on collagen gels. (A). A representative AV explant treated with control siRNA shows normal mesenchymal cell invasion into the collagen matrix (focus is within gel matrix). (B). Endoglin siRNA inhibits mesenchymal cell invasion into the collagen matrix. (C). Micrograph of explant focused within the collagen gel matrix. This ventricle explant, treated with control siRNA, shows no invasion of mesenchymal cells . (D). A micrograph of a ventricle explant treated with endoglin siRNA shows no invasion of mesenchymal cells into collagen matrix (focus is within the gel matrix). (E). A ventricle explant treated with control siRNA shows outgrowth of endothelium (surface view of explant in C). (F). Surface view of explant in D shows little endothelium on gel surface. This ventricle explant treated with endoglin siRNA shows no outgrowth of endothelium. Arrows mark selective mesenchymal cells.
Acknowledgements
The authors thank Dr. Joey Barnett (Vanderbilt) and Drs. Antin, Klewer, Camenisch and Heimark (Arizona) for useful discussions during the conduct of this work. We thank Crystal Willingham for technical assistance. This work was supported by HL63926 from NHLBI and 0650076Z from the American Heart Association, Pacific Mountain Affiliate. Dr. Mercado-Pimentel was supported by NIEHS training grant ES007091. Microscopy facilities were supported in part by NIEHS grant ES06694
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arthur HM, Ure J, Smith AJ, Renforth G, Wilson DI, Torsney E, Charlton R, Parums DV, Jowett T, Marchuk DA, Burn J, Diamond AG. Endoglin, an ancillary TGFbeta receptor, is required for extraembryonic angiogenesis and plays a key role in heart development. Dev Biol. 2000;217:42–53. doi: 10.1006/dbio.1999.9534. [DOI] [PubMed] [Google Scholar]
- Azhar M, Schultz Jel J, Grupp I, Dorn GW, 2nd, Meneton P, Molin DG, Gittenberger-de Groot AC, Doetschman T. Transforming growth factor beta in cardiovascular development and function. Cytokine Growth Factor Rev. 2003;14:391–407. doi: 10.1016/s1359-6101(03)00044-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbara NP, Wrana JL, Letarte M. Endoglin is an accessory protein that interacts with the signaling receptor complex of multiple members of the transforming growth factor-beta superfamily. J Biol Chem. 1999;274:584–94. doi: 10.1074/jbc.274.2.584. [DOI] [PubMed] [Google Scholar]
- Bellon T, Corbi A, Lastres P, Cales C, Cebrian M, Vera S, Cheifetz S, Massague J, Letarte M, Bernabeu C. Identification and expression of two forms of the human transforming growth factor-beta-binding protein endoglin with distinct cytoplasmic regions. Eur J Immunol. 1993;23:2340–5. doi: 10.1002/eji.1830230943. [DOI] [PubMed] [Google Scholar]
- Berg JN, Guttmacher AE, Marchuk DA, Porteous ME. Clinical heterogeneity in hereditary haemorrhagic telangiectasia: are pulmonary arteriovenous malformations more common in families linked to endoglin. J Med Genet. 1996;33:256–7. doi: 10.1136/jmg.33.3.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco FJ, Santibanez JF, Guerrero-Esteo M, Langa C, Vary CP, Bernabeu C. Interaction and functional interplay between endoglin and ALK-1, two components of the endothelial transforming growth factor-beta receptor complex. J Cell Physiol. 2005 doi: 10.1002/jcp.20311. [DOI] [PubMed] [Google Scholar]
- Bolender DL, Seliger WG, Markwald RR. A histochemical analysis of polyanoinic compounds found in the extracellular matrix encountered by migrating cephalic neural crest cells. Anat Rec. 1980;196:401–12. doi: 10.1002/ar.1091960405. [DOI] [PubMed] [Google Scholar]
- Bourdeau A, Dumont DJ, Letarte M. A murine model of hereditary hemorrhagic telangiectasia. J Clin Invest. 1999;104:1343–51. doi: 10.1172/JCI8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourdeau A, Faughnan ME, Letarte M. Endoglin-deficient mice, a unique model to study hereditary hemorrhagic telangiectasia. Trends Cardiovasc Med. 2000;10:279–85. doi: 10.1016/s1050-1738(01)00062-7. [DOI] [PubMed] [Google Scholar]
- Boyer AS, Ayerinskas II, Vincent EB, McKinney LA, Weeks DL, Runyan RB. TGFbeta2 and TGFbeta3 have separate and sequential activities during epithelial-mesenchymal cell transformation in the embryonic heart. Dev Biol. 1999;208:530–45. doi: 10.1006/dbio.1999.9211. [DOI] [PubMed] [Google Scholar]
- Boyer AS, Runyan RB. TGFbeta Type III and TGFbeta Type II receptors have distinct activities during epithelial-mesenchymal cell transformation in the embryonic heart. Dev Dyn. 2001;221:454–9. doi: 10.1002/dvdy.1154. [DOI] [PubMed] [Google Scholar]
- Braverman IM, Keh A, Jacobson BS. Ultrastrucuture and three-dimensional organization of the telangiectases of hereditary hemorrhagic telangiectasia. J. Invest. Dermatol. 1990;95:422–427. doi: 10.1111/1523-1747.ep12555569. [DOI] [PubMed] [Google Scholar]
- Cheifetz S, Bellon T, Cales C, Vera S, Bernabeu C, Massague J, Letarte M. Endoglin is a component of the transforming growth factor-beta receptor system in human endothelial cells. J Biol Chem. 1992;267:19027–30. [PubMed] [Google Scholar]
- Conley BA, Koleva R, Smith JD, Kacer D, Zhang DW, Bernabeu C, Vary CPH. Endoglin controls cell migration and composition of focal adhesions - Function of the cytosolic domain. J. Biol Chem. 2004;279:27440–27449. doi: 10.1074/jbc.M312561200. [DOI] [PubMed] [Google Scholar]
- Crossin KL, Hoffman S. Expression of adhesion molecules during the formation and differentiation of the avian endocardial cushion tissue. Dev Biol. 1991;145:277–86. doi: 10.1016/0012-1606(91)90126-n. [DOI] [PubMed] [Google Scholar]
- De Ruiter MC, Poelmann RE, Van Munsteren JC, Mironov V, Markwald RR, Gittenberger-de-Groot AC. Embryonic endothelial cells transdifferentiate into mesenchymal cells expressing smooth muscle actins in vivo and in vitro. Circ Res. 1997;80:444–451. doi: 10.1161/01.res.80.4.444. [DOI] [PubMed] [Google Scholar]
- Doyle SE, Scholz MJ, Greer KA, Hubbard AD, Darnell DK, Antin PA, Klewer SE, Runyan RR. Latrophilin-2 is a novel component of the epithelial-mesenchymal transition within the atrioventricular canal of the embryonic chicken heart. Dev Dyn. 2006;235:3213–3221. doi: 10.1002/dvdy.20973. [DOI] [PubMed] [Google Scholar]
- Eisenberg LM, Markwald RR. Molecular regulation of atrioventricular valvuloseptal morphogenesis. Circ Res. 1995;77:1–6. doi: 10.1161/01.res.77.1.1. [DOI] [PubMed] [Google Scholar]
- Franzen P, ten Dijke P, Ichijo H, Yamashita H, Schulz P, Heldin CH, Miyazono K. Cloning of a TGF beta type I receptor that forms a heteromeric complex with the TGF beta type II receptor. Cell. 1993;75:681–92. doi: 10.1016/0092-8674(93)90489-d. [DOI] [PubMed] [Google Scholar]
- Gitler AD, Lu MM, Jiang YQ, Epstein JA, Gruber PJ. Molecular markers of cardiac endocardial cushion development. Dev Dyn. 2003;228:643–50. doi: 10.1002/dvdy.10418. [DOI] [PubMed] [Google Scholar]
- Gougos A, Letarte M. Primary structure of endoglin, an RGD-containing glycoprotein of human endothelial cells. J Biol Chem. 1990;265:8361–4. [PubMed] [Google Scholar]
- Goumans MJ, Valdimarsdottir G, Itoh S, Lebrin F, Larsson J, Mummery C, Karlsson S, ten Dijke P. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol Cell. 2003;12:817–28. doi: 10.1016/s1097-2765(03)00386-1. [DOI] [PubMed] [Google Scholar]
- Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. Embo J. 2002;21:1743–53. doi: 10.1093/emboj/21.7.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero-Esteo M, Sanchez-Elsner T, Letamendia A, Bernabeu C. Extracellular and cytoplasmic domains of endoglin interact with the transforming growth factor-beta receptors I and II. J Biol Chem. 2002;277:29197–209. doi: 10.1074/jbc.M111991200. [DOI] [PubMed] [Google Scholar]
- Guttmacher AE, Marchuk DA, White RI., Jr. Hereditary hemorrhagic telangiectasia. N Engl J Med. 1995;333:918–24. doi: 10.1056/NEJM199510053331407. [DOI] [PubMed] [Google Scholar]
- Harland RM. In situ hybridization: an improved whole-mount method for Xenopus embryos. Methods Cell Biol. 1991;36:685–95. doi: 10.1016/s0091-679x(08)60307-6. [DOI] [PubMed] [Google Scholar]
- Jerkic M, Rodriguez-Barbero A, Prieto M, Toporsian M, Penicacho M, Rivas-Elena JV, Obreo J, Wang A, Perez-Barriocanal F, Arevalo M, Letarte M, Lopez-Novoa JM. Reduced angiogenic responses in adult endoglin heterozygous mice. Cardiovasc. Res. 2006;69:845–854. doi: 10.1016/j.cardiores.2005.11.020. [DOI] [PubMed] [Google Scholar]
- Jiao K, Langworthy M, Batts L, Brown CB, Moses HL, Baldwin HS. Tgfß signaling is required for atrioventricular cushion mesenchyme remodeling during in vivo cardiac development. Development. 2006;133:4585–93. doi: 10.1242/dev.02597. [DOI] [PubMed] [Google Scholar]
- Krug EL, Runyan RB, Markwald RR. Protein extracts from early embryonic hearts initiate cardiac endothelial cytodifferentiation. Dev Biol. 1985;112:414–26. doi: 10.1016/0012-1606(85)90414-2. [DOI] [PubMed] [Google Scholar]
- Labbe E, Letamendia A, Attisano L. Association of Smads with lymphoid enhancer binding factor 1/T cell-specific factor mediates cooperative signaling by the transforming growth factor-beta and wnt pathways. Proc Natl Acad Sci U S A. 2000;97:8358–63. doi: 10.1073/pnas.150152697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lastres P, Letamendia A, Zhang H, Rius C, Almendro N, Raab U, Lopez LA, Langa C, Fabra A, Letarte M, Bernabeu C. Endoglin modulates cellular responses to TGF-beta 1. J Cell Biol. 1996;133:1109–21. doi: 10.1083/jcb.133.5.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrin F, Goumans MJ, Jonker L, Carvalho RL, Valdimarsdottir G, Thorikay M, Mummery C, Arthur HM, ten Dijke P. Endoglin promotes endothelial cell proliferation and TGF-beta/ALK1 signal transduction. Embo J. 2004;23:4018–28. doi: 10.1038/sj.emboj.7600386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebner S, Cattelino A, Gallini R, Rudini N, Iurlaro M, Piccolo S, Dejana E. {beta}-Catenin is required for endothelial-mesenchymal transformation during heart cushion development in the mouse. J. Cell Biol. 2004;166:359–367. doi: 10.1083/jcb.200403050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Casillas F, Cheifetz S, Doody J, Andres JL, Lane WS, Massague J. Structure and expression of the membrane proteoglycan betaglycan, a component of the TGF-beta receptor system. Cell. 1991;67:785–95. doi: 10.1016/0092-8674(91)90073-8. [DOI] [PubMed] [Google Scholar]
- Markwald RR, Fitzharris TP, Manasek FJ. Structural development of endocardial cushions. Am J Anat. 1977;148:85–119. doi: 10.1002/aja.1001480108. [DOI] [PubMed] [Google Scholar]
- McAllister KA, Baldwin MA, Thukkani AK, Gallione CJ, Berg JN, Porteous ME, Guttmacher AE, Marchuk DA. Six novel mutations in the endoglin gene in hereditary hemorrhagic telangiectasia type 1 suggest a dominant-negative effect of receptor function. Hum Mol Genet. 1995;4:1983–5. doi: 10.1093/hmg/4.10.1983. [DOI] [PubMed] [Google Scholar]
- McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, Helmbold EA, Markel DS, McKinnon WC, Murrell J, et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet. 1994;8:345–51. doi: 10.1038/ng1294-345. [DOI] [PubMed] [Google Scholar]
- Mikawa T, Fischman DA. Retroviral analysis of cardiac morphogenesis: discontinuous formation of coronary vessels. Proc Natl Acad Sci U S A. 1992;89:9504–8. doi: 10.1073/pnas.89.20.9504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivey HE, Compton LA, Barnett JV. Coronary vessel development: the epicardium delivers. Trends Cardiovasc Med. 2004;14:247–51. doi: 10.1016/j.tcm.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Perez-Gomez E, Eleno N, Lopez-Novoa JM, Ramirez JR, Velasco B, Letarte M, Bernabeu C, Quintanilla M. Characterization of murine S-endoglin isoform and its effects on tumor development. Oncogene. 2005a;24:4450–61. doi: 10.1038/sj.onc.1208644. [DOI] [PubMed] [Google Scholar]
- Person AD, Garriock RJ, Krieg PA, Runyan RB, Klewer SE. Frzb modulates Wnt-9a-mediated beta-catenin signaling during avian atrioventricular cardiac cushion development. Dev Biol. 2005;278:35–48. doi: 10.1016/j.ydbio.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Poelmann RE, Gittenberger-de Groot AC, Mentink MM, Bokenkamp R, Hogers B. Development of the cardiac coronary vascular endothelium, studied with antiendothelial antibodies, in chicken-quail chimeras. Circ Res. 1993;73:559–68. doi: 10.1161/01.res.73.3.559. [DOI] [PubMed] [Google Scholar]
- Qu R, Silver MM, Letarte M. Distribution of endoglin in early human development reveals high levels on endocardial cushion tissue mesenchyme during valve formation. Cell Tissue Res. 1998;292:333–43. doi: 10.1007/s004410051064. [DOI] [PubMed] [Google Scholar]
- Romano LA, Runyan RB. Slug is a mediator of epithelial-mesenchymal cell transformation in the developing chicken heart. Dev Biol. 1999;212:243–54. doi: 10.1006/dbio.1999.9339. [DOI] [PubMed] [Google Scholar]
- Romano LA, Runyan RB. Slug is an essential target of TGFbeta2 signaling in the developing chicken heart. Dev Biol. 2000;223:91–102. doi: 10.1006/dbio.2000.9750. [DOI] [PubMed] [Google Scholar]
- Runyan RB, Markwald RR. Invasion of mesenchyme into three-dimensional collagen gels: a regional and temporal analysis of interaction in embryonic heart tissue. Dev Biol. 1983;95:108–14. doi: 10.1016/0012-1606(83)90010-6. [DOI] [PubMed] [Google Scholar]
- Runyan RB, Potts JD, Sharma RV, Loeber CP, Chiang JJ, Bhalla RC. Signal transduction of a tissue interaction during embryonic heart development. Cell Regul. 1990;1:301–13. doi: 10.1091/mbc.1.3.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runyan RB, Potts JD, Weeks DL. TGF-beta 3-mediated tissue interaction during embryonic heart development. Mol Reprod Dev. 1992;32:152–9. doi: 10.1002/mrd.1080320211. [DOI] [PubMed] [Google Scholar]
- Singh J, Chuaqui CE, Boriack-Sjodin PA, Lee WC, Pontz T, Corbley MJ, Cheung HK, Arduini RM, Mead JN, Newman MN, Papadatos JL, Bowes S, Josiah S, Ling LE. Successful shape-based virtual screening: the discovery of a potent inhibitor of the type I TGFbeta receptor kinase (TbetaRI) Bioorg Med Chem Lett. 2003;13:4355–9. doi: 10.1016/j.bmcl.2003.09.028. [DOI] [PubMed] [Google Scholar]
- Sorensen LK, Brooke BS, Li DY, Urness LD. Loss of distinct arterial and venous boundaries in mice lacking endoglin, a vascular-specific TGFbeta coreceptor. Dev Biol. 2003;261:235–50. doi: 10.1016/s0012-1606(03)00158-1. [DOI] [PubMed] [Google Scholar]
- Takeshi Tsuda HW, Rupert Timpl, Mon-Li Chu Fibulin-2 expression marks transformed mesenchymal cells in developing cardiac valves, aortic arch vessels, and coronary vessels. Developmental Dynamics. 2001;222:89–100. doi: 10.1002/dvdy.1172. [DOI] [PubMed] [Google Scholar]
- Tavares AL, Mercado-Pimentel ME, Runyan RB, Kitten GT. TGFbeta-mediated RhoA expression is necessary for epithelial-mesenchymal transition in the embryonic chick heart. Dev Dyn. 2006;235:1589–1598. doi: 10.1002/dvdy.20771. [DOI] [PubMed] [Google Scholar]
- Vincent EB, Runyan RB, Weeks DL. Production of the transforming growth factor-beta binding protein endoglin is regulated during chick heart development. Dev Dyn. 1998;213:237–47. doi: 10.1002/(SICI)1097-0177(199811)213:3<237::AID-AJA1>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Ward SM, Desgrosellier JS, Zhuang X, Barnett JV, Galper JB. Transforming growth factor beta (TGFbeta) signaling via differential activation of activin receptor-like kinases 2 and 5 during cardiac development. Role in regulating parasympathetic responsiveness. J Biol Chem. 2002;277:50183–9. doi: 10.1074/jbc.M209668200. [DOI] [PubMed] [Google Scholar]
- Wieser R, Wrana JL, Massague J. GS domain mutations that constitutively activate T beta R-I, the downstream signaling component in the TGF-beta receptor complex. Embo J. 1995;14:2199–208. doi: 10.1002/j.1460-2075.1995.tb07214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrana JL. TGF-beta receptors and signalling mechanisms. Miner Electrolyte Metab. 1998;24:120–30. doi: 10.1159/000057359. [DOI] [PubMed] [Google Scholar]
- Wrana JL, Attisano L, Wieser R, Ventura F, Massague J. Mechanism of activation of the TGF-beta receptor. Nature. 1994;370:341–7. doi: 10.1038/370341a0. [DOI] [PubMed] [Google Scholar]
- Wunsch AM, Little CD, Markwald RR. Cardiac endothelial heterogeneity defines valvular development as demonstrated by the diverse expression of JB3, an antigen of the endocardial cushion tissue. Dev Biol. 1994;165:585–601. doi: 10.1006/dbio.1994.1278. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Legend for Supplemental Figure 1:
Endoglin siRNA effects on stage 16 AV and ventricle explants on collagen gels. (A). A representative AV explant treated with control siRNA shows normal mesenchymal cell invasion into the collagen matrix (focus is within gel matrix). (B). Endoglin siRNA inhibits mesenchymal cell invasion into the collagen matrix. (C). Micrograph of explant focused within the collagen gel matrix. This ventricle explant, treated with control siRNA, shows no invasion of mesenchymal cells . (D). A micrograph of a ventricle explant treated with endoglin siRNA shows no invasion of mesenchymal cells into collagen matrix (focus is within the gel matrix). (E). A ventricle explant treated with control siRNA shows outgrowth of endothelium (surface view of explant in C). (F). Surface view of explant in D shows little endothelium on gel surface. This ventricle explant treated with endoglin siRNA shows no outgrowth of endothelium. Arrows mark selective mesenchymal cells.