

Abstract
To firmly establish the pathway involved in ligand-induced degradation of the AHR, cell lines derived from mouse rat or human tissues were exposed to inhibitors specific to the proteasome or calpain proteases and exposed to TCDD. The level of endogenous AHR and CYP1A1 protein was then evaluated by quantitative Western blotting. Treatment of cells with the calpain inhibitors: calpeptin, calpain inhibitor III, or PD150606 either individually or in combinations up to 75μM did not reduce TCDD-induced degradation of the AHR, the induction of endogenous CYP1A1 or the nuclear accumulation of the AHR. The activity of the inhibitors was verified with an in vivo calpain assay. In contrast, exposure of cells to the specific proteasome inhibitors: epoxomicin (1-5μM), proteasome inhibitor I (5-10μM) or lactacystin (5-15μM) completely inhibited TCDD-induced degradation of the AHR. Inhibition of AHR degradation with these compounds did not reduce the induction of endogenous CYP1A1. In addition, exposure of the Hepa-1 line to the various proteasome inhibitors caused an accumulation of the AHR in the nucleus in the absence of TCDD exposure. Finally, Western blot analysis of the DNA bound AHR showed that its molecular mass was unchanged in comparison to the unliganded (cytoplasmic) AHR. Thus, these studies conclusively implicate the proteasome and not calpain proteases in the ligand-induced degradation of the mouse, rat and human AHR and suggest that the pharmacological use of proteasome inhibitors may impact the time course and magnitude of gene regulatory events mediated through the AHR.
Keywords: Aryl hydrocarbon receptor, calpain, proteasome, protein degradation, TCDD, ubiquitin
1. INTRODUCTION
The aryl hydrocarbon receptor (AHR) is a ligand activated transcription factor that is a member of the basic-helix-loop-helix/PAS family of proteins (reviewed in 1, 2]. The prototypical ligand for the AHR is the environmental contaminant 2,3,7,8-tetrachlorodibenzo-p-dixion (TCDD) and exposure in numerous model systems leads to carcinogenesis as well as developmental and reproductive toxicities [reviewed in 3, 4]. Since the concentration of an endogenous signaling protein will ultimately determine the responsiveness of the cell to a given signal, there has been interest in understanding how modulation of AHR protein levels impact normal cellular functions as well as the biological responses to TCDD [reviewed in 5, 6]. For example, studies have shown that reductions in the level of AHR protein in culture can impact the magnitude of AHR-mediated induction of CYP1A1 and affect cell growth rates [7, 8]. Conversely, it has been reported that a constitutively active AHR expressed in transgenic mice causes a myriad of effects including reduced life span and stomach tumors [9, 10]. AHR knock out mice (Ah-/-) also provide insight into the importance of endogenous AHR protein in normal developmental processes. Ah-/- mice exhibit a variety of growth defects, including immune system impairment [11, 12], reduced mammary gland development [13], lower incidence of large infrontal bones [14], liver fibrosis [11], hepatic defects [15, 16], and impaired reproductive outcome and fetal viability [11, 13, 17]. Importantly, some of the phenotypes observed in the Ah-/- mice are similar to those reported in TCDD-treated animals, suggesting that the endogenous AHR responds to endogenous signals. Notably, Ah-/- mice exhibit defects in reproduction and ovary development, and these defects are also observed in wild type mice treated with TCDD [17-20]. Collectively, these findings support the hypothesis that a reduction in the level of endogenous AHR protein may reduce responsiveness to endogenous signaling molecules and possibly contribute to the biological effects mediated by TCDD.
Importantly, numerous studies have established that TCDD exposure leads to a rapid and sustained reduction in the level of endogenous AHR protein both in vitro and in vivo [5]. In addition, inhibition of AHR degradation results in increased levels of gene regulation in various culture models [21-23]. Presently, the ubiquitin-proteasome pathway has been implicated in the ligand-induced degradation of the AHR due to the ability of the proteasome inhibitors, MG-132 and lactacystin to inhibit AHR degradation [21, 23], as well as studies showing that AHR degradation is blocked in cells that contain a temperature sensitive mutation in the E1 ubiquitin conjugating enzyme [23]. More recently it has been shown that the ligand-bound AHR may be a target for the ubiquitin-proteasome pathway only after ligand binding, association with ARNT and DNA binding [23, 24]. Interestingly, these studies also indicate that C-terminal truncations of the AHR are targeted for degradation via the proteasome, but sugges that they may be targeted there by a mechanism distinct from the full-length AHR. However, in spite of these compelling results, the ubiquitin ligase enzyme and site of ligand-mediated AHR ubiquitylation remain undefined. Because of this, it has recently been reported that calpain proteases may also contribute to the ligand-mediated degradation of the AHR [25]. Since cellular proteases are now being targeted by novel pharmacologicals in a number of disease states [reviewed 26, 27], studies were initiated to evaluate the impact of numerous calpain and proteasome inhibitors on the ligand-induced degradation of the endogenous AHR in cell culture lines derived from human, mouse and rat tissues.
2.0 MATERIALS AND METHODS
2.1. Antibodies and Reagents
Specific antibodies against the mouse AHR (A-1 and A-1A) are identical to those described previously and have been extensively validated for specificity to all mammalian AHRs [28, 29]. For Western blot analysis goat anti rabbit antibodies conjugated to horseradish peroxidase (GAR-HRP) were utilized (Jackson Immunoresearch). Polyclonal rabbit ß-actin antibodies and were purchased from Sigma-Aldrich. Polyclonal rabbit CYP1A1 antibodies were purchased from Santa Cruz. TCDD was purchased from Cambridge Isotope Laboratories. All protease inhibitors were purchased from Calbiochem and were solubilized in DMSO. When possible protease inhibitors were purchased already solubilized in DMSO under inert gas. t-BOC-L-leucyl-L-methionine amide (t-BOC-leu-met) was purchased from Molecular Probes. All solubilized compounds were stored in a desiccator at -20°C.
2.2 Buffers
PBS is 0.8% NaCl, 0.02% KCl, 0.14% Na2HPO4, 0.02% KH2PO4, pH 7.4. SDS sample buffer is 60mM Tris, pH 6.8, 2% SDS, 15% glycerol, 2mM EDTA, 5mM EGTA 10mM DTT, 0.005% bromphenol blue. 1X Cell lysis buffer is 50mM Tris, pH 6.8, 10% glycerol, 1mM EDTA, 2.5mM EGTA, and 0.5% NP-40. MENG is 25mM MOPS, pH 7.0, 5mM EDTA, 10% glycerol and 0.2% NaN3. TTBS is 50mM Tris, 0.2% Tween 20, 150mM NaCl, pH 7.5. TTBS+ is 50mM Tris, 0.5% Tween 20, 300mM NaCl, pH 7.5. BLOTTO is 5% dry milk in TTBS.
2.3 Cells and growth conditions
Human retinal pigmented epithelial (ARPE-19) cells and rat retinal-pigmented epithelial (RPE-J) cells were purchased from American Type Culture Collection (ATCC). These cells were not derived from tumors and represent essentially normal tissue. All cells were propagated at 37°C in cell culture media specified by ATCC. Murine Hepa-1 cells were a generous gift from James Whitlock (Stanford). All cells were passaged at 1-week intervals and used in experiments during a 2-month period.
2.4 Production of total cell lysates, nuclear lysates and nuclear extracts
All cells were treated by adding the test compound directly to the media. Following treatment, cell monolayers were washed twice with PBS and detached from plates by trypsinization (0.05% trypsin/0.5mM EDTA). For total cell lysates, cell pellets were washed with PBS and sonicated directly in 75-150μl ice-cold cell lysis buffer for 12 seconds. Lysates were immediately heated for 3 min at 100°C and then sonicated an additional 5 seconds. Samples were stored at -20°C until analysis. For nuclear and cytoplasmic lysates, cells were harvested as above, and disrupted by vortexing in cell lysis buffer. Samples were centrifuged at 5000rpm and supernatants removed. Pellets (nuclei) were washed once with ice-cold cell lysis buffer (without NP-40) and then sonicated in cell lysis buffer. Protein concentrations were determined by the coomassie plus assay (Pierce) with BSA as the standard. Samples were denatured by adding an equal concentration of SDS sample buffer and boiling. Nuclear extracts were prepared by disrupting cell pellets in MENG buffer using a dounce homogenizer. Following disruption, samples were centrifuged at 1000 × g and pellets washed once with MENG buffer. Proteins were extracted by incubating the pellets in MENG containing 400mM KCl on ice for 30 minutes and centrifuging at 10,000 × g. Protein concentrations were determined as detailed above and nuclear extracts stored at -80°C until analysis.
2.5 Western blot analysis and quantification of protein
Protein samples were resolved by denaturing electrophoresis on discontinuous polyacrylamide slab gels (SDS-PAGE) and were electrophoretically transferred to nitrocellulose. Immunochemical staining was carried out with varying concentrations of primary antibody (see text and figure legends) in BLOTTO buffer supplemented with DL-histidine (20 mM) for 1-2 hours at 22°C. Blots were washed with 5 changes of TTBS+ for a total of 50 minutes. The blot was then incubated in BLOTTO buffer containing a 1:12,000 dilution of GAR-HRP for 1 hour at 22°C and washed in 5 changes of TTBS+ as above. Prior to detection, the blots were washed in PBS for 5 minutes. Bands were visualized with the enhanced chemiluminescence (ECL) kit as specified by the manufacturer. Multiple exposures of each set of samples were produced. The relative concentration of target protein was determined by computer analysis of the autoradiographs and normalization to the internal standard (actin) as detailed previously [29, 30]. Each experiment was repeated at least three times.
2.6 In vivo calpain assay
Cells were pre-treated with test compounds for the times indicated in the text and then incubated with 10μM t-BOC-leu-met for 30 minutes. Cells were then washed with PBS and wet-mounted on glass slides. Cell fluorescence was evaluated at 405nM and fields photographed with a digital camera to generate the raw data. All fields in a given experiment were photographed using the same exposure times.
2.7 Immunofluorescence staining, microscopy and image analysis
All immunocytochemical procedures (cell plating, fixation, and staining) were carried out as previously described [28-30]. Cells were observed on an Olympus IX70 microscope. On average, 15-20 fields (5-20 cells each) were evaluated on each coverslip and 3-4 fields were photographed with a digital camera at the same exposure time to generate the raw data. Experiments were repeated at least two times. The nuclear fluorescence intensities of cells were determined using MicroSuite image analysis software. Typically, 50-75 individual nuclei were quantified in 3-4 different fields after being photographed for identical times. Statistics were carried out as detailed below.
2.8 Statistics
Target protein bands were normalized to internal standards (actin) and the normalized densitometry units compared by ANOVA and Tukey-Kramer multiple comparison tests using InStat software (GraphPad Software Inc. San Diego, CA). Results are presented as mean ± SEM of 3 individual experiments, unless noted otherwise. A probability value of <0.05 was considered significant.
3.0 RESULTS
3.1 Effect of calpain proteases inhibitors on TCDD-induced degradation of the AHR and induction of endogenous CYP1A1
Following ligand binding, the AHR is rapidly degraded both in vivo and in vitro [7, 21-23, 31, 32]. Although a number of studies have implicated the UPP as the degradation mechanism by using proteasome inhibitors [21, 23, 33-35], it has recently been proposed that calpain proteases may also be involved [25]. To assess the role of calpains in AHR-mediated signaling, Hepa-1 cells were pre-treated with 15μM of the specific calpain inhibitors, calpeptin, calpian inhibitor III, or PD150606 for 1 hr and then exposed to TCDD for an additional 4 hrs. The level of AHR, CYP1A1 and actin protein was then evaluated in total cells lysates by Western blotting. Figure 1A shows that incubation of the cells with the various calpain inhibitors did not decrease the level of TCDD-induced degradation of the AHR or the level of induction of CYP1A1 in the Hepa-1 line. Since 15μM of the various inhibitors did not impact AHR degradation or gene regulation as previously reported [25], studies were repeated using either 25μM or 50μM of each inhibitor. Representative Western blot results are presented in figures 1B-D and the quantified results from three independent studies are presented in Figure 1E.. Consistent with the results shown in Figure 1A, incubation of the cells with up to 50μM inhibitor did not decrease the level of TCDD-induced degradation of the AHR or the level of induction of CYP1A1. In fact, it can be observed that there is a small but significant increase in the level of basal CYP1A1 expression in the absence of TCDD when the level of the inhibitors was increased above 25μM. Importantly, identical results were observed in rat and human retinal-pigmented epithelial cells (RPE) when they were exposed to the various inhibitors at concentrations of up to 50μM.1
Figure 1. Effect of individual calpain inhibitors on TCDD-induced degradation of the AHR and induction of CYP1A1.
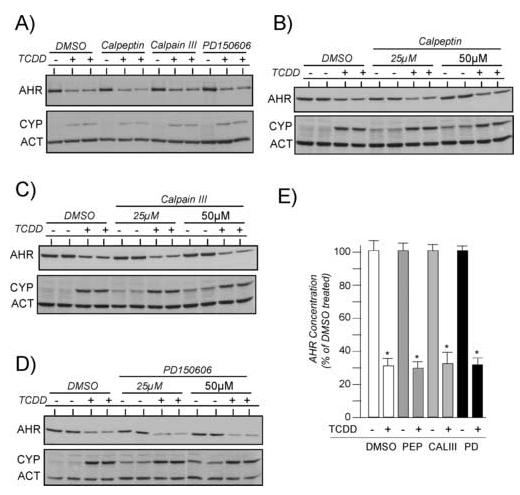
Duplicate plates of Hepa-1 cells were treated with 0.05% DMSO or the indicated calpain inhibitors for 1 hr at 37°C. Cells were then exposed to TCDD (2nM) for an additional 4 hrs and total cell lysates prepared. Equal amounts of total cell lysates were resolved by SDS-PAGE, blotted and stained with A-1A anti-AHR IgG (1.0μg/ml), anti-ß-actin IgG (1:1000) or anti-CYP1A1 IgG (1:1000). Reactivity was visualized by ECL with GAR-HRP (1:10,000). A) Each calpain inhibitor was used at 15μM. B) Results with 25μM and 50μM calpeptin. C) Results with 25μM and 50μM calpain inhibitor III. D) Results with 25μM and 50μM PD150606. E) The level of AHR protein was normalized to the level of actin as detailed [29, 30]. Three independent experiments for each calpain inhibitor were then averaged and plotted as the mean +/- SE with the DMSO treated cells in each experiment set to 100%. * = statistically different from DMSO treated cells (p<0.001).
Calpeptin, calpian inhibitor III, and PD150606 have distinct Ki values for the inhibition of calpain I or calpain II, so it was of interest to assess whether the compounds would have an affect on AHR-mediated signaling when they were used in combination. In the first study, Hepa-1 cells were pre-incubated with 15μM or 25μM of each inhibitor for an effective total concentration of 45μM or 75μM. Cells were then exposed to TCDD for an additional 4 hrs. Figure 2A shows that using the inhibitors in combination did not reduce the TCDD-induced degradation of the AHR or the induction of CYP1A1, however, as shown in Figs. 1B-D, exposure to the inhibitors actually elevated the basal level of CYP1A1 above controls. Studies next focused on whether longer pre-treatment times with the combination of inhibitors would impact AHR degradation or CYP1A1 induction. Hepa-1 cells were pretreated with the inhibitors for up to 3 hrs prior to exposure to TCDD. Figure 2B shows that the TCDD induced degradation of the AHR or the induction of CYP1A1 was not reduced in comparison to DMSO treated cells even when cells are pretreated with the inhibitors for 3 hrs.
Figure 2. Effect of combinations calpain inhibitors on TCDD-induced degradation of the AHR and induction of CYP1A1.
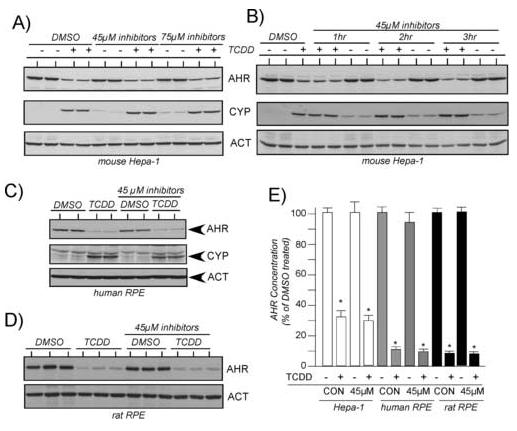
A) Duplicate plates of mouse Hepa-1 cells were treated with 0.05% DMSO or a combination of calpeptin (15μM or 25μM), calpain inhibitor III (15μM or 25μM) and PD15606 (15μM or 25μM) for 1 hr at 37°C. Cells were then exposed to TCDD (2nM) for an additional 4 hrs and total cell lysates prepared. Equal amounts of total cell lysates were resolved by SDS-PAGE, blotted and stained with A-1A anti-AHR IgG (1.0μg/ml), anti-ß-actin IgG (1:1000) or anti-CYP1A1 IgG (1:1000). Reactivity was visualized by ECL with GAR-HRP (1:10,000). B) Hepa-1 cells were treated and analyzed as described in A, except the incubation with the calpain inhibitors was carried out for 1, 2 or 3 hrs prior to the exposure of TCDD. C) Human RPE cells were treated and analyzed as described in A. D) Rat RPE cells were treated and analyzed as described in A. E) The level of AHR protein was normalized to the level of actin as detailed [29, 30]. Three independent experiments for each cell line were then averaged and plotted as the mean +/- SE with the DMSO treated cells in each experiment set to 100%. * = statistically different from DMSO treated cells (p<0.001).
To validate that the observed results were not specific to the Ahb-1 receptor expressed in the Hepa-1 line, human or rat RPE cells were treated with 45μM effective concentration of the three inhibitors and then exposed to TCDD as detailed above. Representative Western blot results are presented in Figures 2C-D and the quantified results from three independent studies are presented in Figure 2E. Consistent with the results observed in the Hepa-1 line, the combination of calpain inhibitors did not impact the TCDD-induced degradation of the AHR in either cell line and did not reduce the level of CYP1A1 induced by TCDD in the human RPE line. The induction of endogenous CYP1A1 protein could not be evaluated in the rat RPE line since it does not appear to induce levels that can be detected with the commercial CYP1A1 antibody. Collectively, these results support the hypothesis that calpain proteases are not involved in ligand-induced degradation of the AHR. In addition, the results also show that treatment of the mouse and human lines with calpain inhibitors does not impact the level of TCDD-mediated induction of endogenous CYP1A1.
It was next pertinent to assess the TCDD-induced nuclear translocation of the AHR since previous studies suggest that calpain inhibitors block this event [25]. For these studies, Hepa-1 cells were evaluated since they exhibit a predominant cytoplasmic AHR in the absence of ligand exposure and this allows the nuclear translocation event to be prominently observed [21, 22, 24, 28-30, 35]. Hepa-1 cells were pre-incubated with 15μM of each inhibitor for an effective total concentration of 45μM and then exposed to TCDD for an additional hour. Cells were then fixed and stained with AHR antibodies and representative micrographs are presented in Figure 3. It can be observed that the AHR shows a predominant nuclear localization in TCDD treated cells regardless of whether they were incubated with calpain inhibitors. This finding is consistent with the induction of CYP1A1 as shown in Figures 1 and 2 and validate that the calpain inhibitors are having a minimal impact on all aspects of AHR signal transduction.
Figure. 3. In vivo calpain assay in the presence and absence of caplain inhibitors.
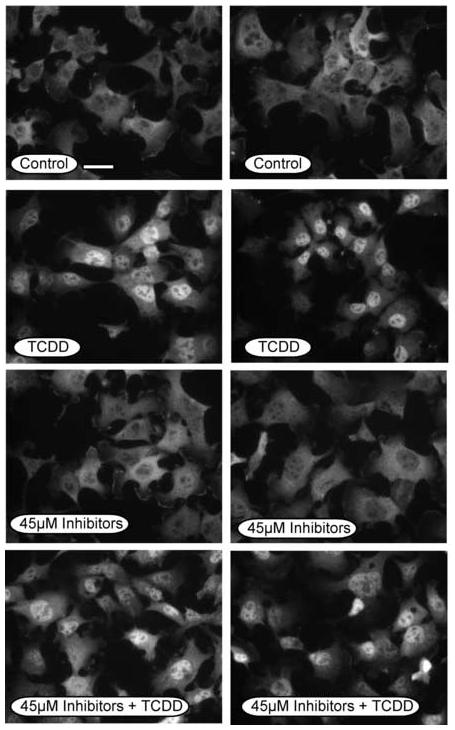
Human RPE or Hepa-1 cells were grown on glass coverslips and treated with 0.05% DMSO (0.05%) or a combination of calpeptin (15μM), calpain inhibitor III (15μM) and PD15606 (15μM) for 1 hr at 37°C. Cells were then incubated with t-BOC-L-leucyl-L-methionine (10μM) for 20 minutes and wet mounted on slides in phosphate buffered saline. Fluorescence was observed at 405nm and individual fields photographed for identical times. Bar in the control panels = 5μm.
Since the various calpain inhibitors had no measurable impact on the degradation of the AHR or the induction of CYP1A1, it was essential to verify that the calpain activity in the cells was actually reduced. Thus, Hepa-1 and RPE cells were exposed to a combination of 45μM calpeptin, calpian inhibitor III, and PD150606 (15μM of each inhibitor) for 1 hr and then incubated with the calpain substrate, t-BOC-L-leucyl-L-methionine (t-BOC-leu-met). t-BOC-leu-met is metabolized by calpains and emits a fluorescent signal at 400-440nm that can be evaluated in the living cells by fluorescence microscopy [36, 37]. These experiments were carried out six times and a representative study is presented in Figure 4. It can be observed that control-treated Hepa-1 and RPE cells exhibit a fluorescent signal that is indicative of basal levels of calpain activity. Importantly, when the Hepa-1 and human RPE cells were exposed to the calpain inhibitors, the level of fluorescence was dramatically reduced. Thus, these studies validate that the calpain inhibitors were effective in reducing calpain activity in vivo and that their lack of effect on AHR degradation or CYP1A1 induction was not due to loss of activity of the inhibitor.
Figure. 4.
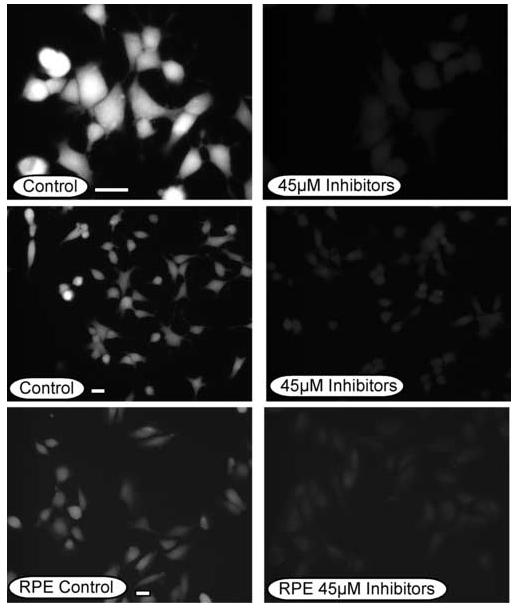
Subcellular localization of the AHR in Hepa-1 cells exposed to calpain inhibitors. Hepa-1 cells were grown on glass coverslips and treated with DMSO (0.05%), or a combination of calpeptin (15μM), calpain inhibitor III (15μM) and PD15606 (15μM) for 1 hr at 37°C. Cells were then incubated with TCDD (2nM) for and additional hour at 37°C, fixed and then incubated with A-1 anti-AHR IgG (1.0μg/ml) and visualized with GAR-RHO IgG (1:400). All panels were photographed with identical exposures. Bar in the control panel = 5μm.
3.2 Effect of proteasome inhibitors on TCDD-induced degradation of the AHR and induction of endogenous CYP1A1
Previous studies have implicated the 26S proteasome in the ligand-induced degradation of the AHR [21, 23, 33-35]. In many of these studies, the proteasome was implicated because ligand induced degradation of the AHR was blocked by using the peptidyl aldehyde of di-leucine, MG-132 [38]. However, since MG-132 is also reported to inhibit calpain activity [38], it was pertinent to assess ligand-induced degradation of the AHR in the presence of other proteasome inhibitors that show greater specificity. In the first study, Hepa-1 cells were treated with proteasome inhibitor 1 (PSI-1; 10μM, 20μM) or lactacystin (1μM, 15μM) for 1 hr and then exposed to TCDD for an additional 4 hrs. Total cell lysates were evaluated for the level of AHR, actin and CYP1A1 by quantitative Western blotting. A representative experiment is shown in Figure 5. In contrast to the results obtained with the calpain inhibitors, treatment of Hepa-1 cells with 10μM PSI-1 or 15μM lactacystin resulted in a complete block of TCDD-induced degradation of the AHR and did not affect the induction of CYP1A1. Identical results were observed in both rat and human RPE cell lines.1
Figure 5. Effect of individual proteasome inhibitors on TCDD-induced degradation of the AHR and induction of CYP1A1.
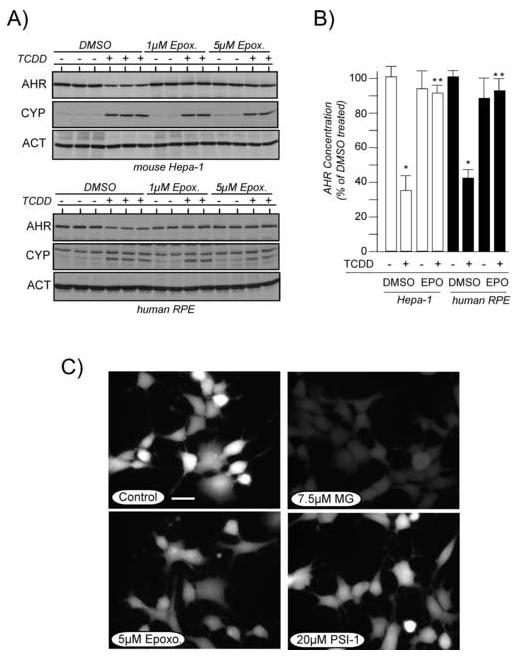
A) Duplicate plates of Hepa-1 cells were treated with 0.05% DMSO or the indicated proteasome inhibitors for 1 hr at 37°C. Cells were then exposed to TCDD (2nM) for an additional 4 hrs and total cell lysates prepared. Equal amounts of total cell lysates were resolved by SDS-PAGE, blotted and stained with A-1A anti-AHR IgG (1.0μg/ml), anti-ß-actin IgG (1:1000) or anti-CYP1A1 IgG (1:1000). Reactivity was visualized by ECL with GAR-HRP (1:10,000). The level of AHR protein at each time point was divided by the corresponding level of actin and the average +/-SD plotted with DMSO treated cells set to 100%. PSI-1 = proteasome inhibitor 1; LAC = lactacystin. B) Hepa-1 cells were treated with MG-132 (1μM or 7.5μM) for 1 hr and then exposed to TCDD (2nM) for an additional 4 hrs. AHR and CYP1A1 protein was evaluated as detailed in A.
To further validate that the proteasome was involved in TCDD-induced degradation of the AHR, studies were repeated using epoxomicin, a highly specific irreversible inhibitor of the proteasome [39, 40]. It is important to note that epoxomicin has limited stability in aqueous solution, thus, the compound was purchased in DMSO under inert gas and was opened and used immediately. For these studies, Hepa-1 or human RPE cells were exposed to 1μM or 5μM epoxomicin for 1hr and then treated for an additional 4 hrs with TCDD. Total cell lysates were generated and evaluated for the level of AHR, actin and CYP1A1 by Western blotting. Representative Western blots experiments are presented in Figure 6A and the quantified results from three independent studies is presented in Figure 6B. It can be observed that treatment of either cell line with as little as 1μM epoxomicin was sufficient to block the TCDD-induced degradation of the AHR without affecting the induction of CYP1A1. To confirm that the specific proteasome inhibitors did not reduce calpain activity, Hepa-1 cells were exposed to MG-132, PSI-1, or epoxomicin for 1 hr and then incubated with t-BOC-leu-met as detailed in Materials and Methods. Figure 6C shows that PSI-1 and epoxomicin do not reduce the level of fluorescence (calpain activity) in the Hepa-1 line, while treatment of cells with MG-132 lead to a reduction in cellular fluorescence. Collectively, these results implicate the 26S proteasome in the TCDD-induced degradation of the AHR and conclusively show that blocking the ligand-induced degradation of the AHR does not inhibit the TCDD-mediated induction of endogenous CYP1A1.
Figure 6. Effect of epoxomicin on TCDD-induced degradation of the AHR and induction of CYP1A1.
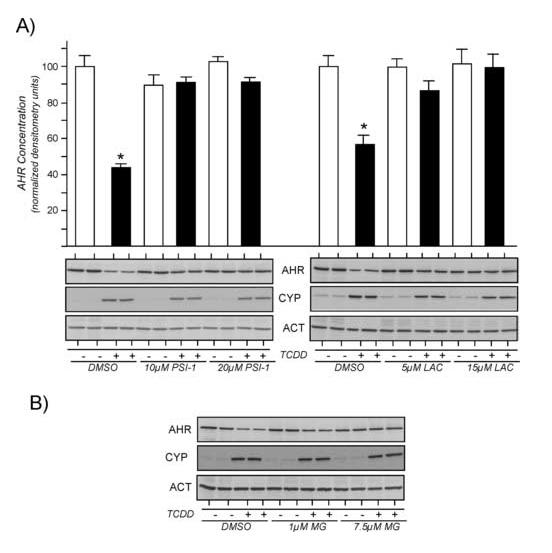
A) Hepa-1 or human RPE cells were treated with 0.05% DMSO or epoxomicin (1μM or 5μM) for 1 hr at 37°C. Cells were then exposed to TCDD (2nM) for an additional 4 hrs and total cell lysates prepared. Equal amounts of total cell lysates were resolved by SDS-PAGE, blotted and stained with A-1A anti-AHR IgG (1.0μg/ml), anti-ß-actin IgG (1:1000) or anti-CYP1A1 IgG (1:1000). Reactivity was visualized by ECL with GAR-HRP (1:10,000). B) The level of AHR protein was normalized to the level of actin as detailed [29, 30]. Three independent experiments for each cell line were then averaged and plotted as the mean +/- SE with the DMSO treated cells in each experiment set to 100%. * = statistically different from DMSO treated cells (p<0.001). ** = statistically different from TCDD treated control cells (p<0.001). C) Hepa-1 cells were grown on glass coverslips and treated with DMSO (0.05%), MG-132 (7.5μM), epoxomicin (5μM), or proteasome inhibitor 1 (20μM) for 1 hr at 37°C. Cells were then incubated with t-BOC-L-leucyl-L-methionine (10μM) for 20 minutes and wet mounted on slides in phosphate buffered saline. Fluorescence was observed at 405nm and individual fields photographed for identical times. Bar in the control panel = 5μm.
Treatment with proteasome inhibitors causes in the nuclear localization of the endogenous AHR
It has been hypothesized that calpains cleave the AHR in the C-terminal transactivation domain and this event is required for the AHR to become translocated to the nucleus and bind DNA [25]. Thus, it would be expected that if the degradation pathway were inhibited, the AHR would exhibit a predominantly cytoplasmic localization even in the presence of ligand. Since previous studies suggest that MG-132 can cause the accumulation of endogenous AHR in the nucleus of cells in the absence of ligand exposure [34, 41, 42], it was pertinent to assess whether other proteasome inhibitors would also cause the AHR to become nuclear. For these studies, Hepa-1 cells were exposed to MG-132 (15μM), lactacystin (10μM), PSI-1 (15μM), or epoxomicin (2.5μM) for 2 hrs and the cells fixed and stained for the AHR as detailed in Methods. Hepa-1 cells were used since the endogenous AHR is predominantly cytoplasmic in the absence of ligand exposure in this line [21, 24, 25, 28, 30]. Fields of cells from a representative experiment and the relative nuclear fluorescence intensities are presented in Figure 7. It is first important to note that the AHR is predominantly cytoplasmic in control treated cells but is readily accumulated in the nucleus following TCDD exposure for 1 hr (nuclear fluorescence intensity increases >3-fold over control treated cells). Importantly, when the cells were exposed to any of the proteasome inhibitors, there was a statistically significant accumulation of the AHR in the nucleus even though the proteasome had been inhibited. The mechanism underlying this event is currently undefined, but these results are in agreement with previous studies that have used MG-132 to assess the localization of the AHR [34, 41, 42].
Figure 7. Subcellular localization of the AHR in Hepa-1 cells exposed to proteasome inhibitors.
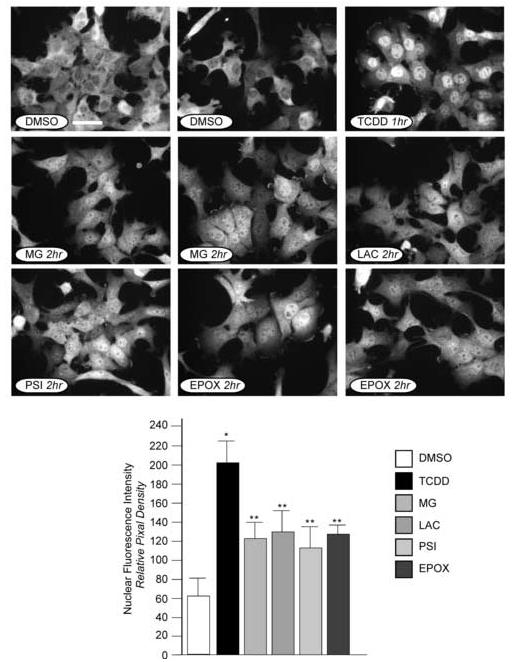
Hepa-1 cells were grown on glass coverslips and treated with DMSO (0.05%), MG-132 (7.5μM), lactacystin (15μM), proteasome inhibitor 1 (20μM) or epoxomicin (5μM), or for 2 hrs at 37°C. A sample was also treated with TCDD (2nM) for 1 hr at 37°C. Cells were then fixed and incubated with A-1 anti-AHR IgG (1.0μg/ml) and visualized with GAR-RHO IgG (1:400). All panels were photographed with identical exposures. Bar in the control panel = 5μm. Nuclear fluorescence intensities were determined for each of the treatments. 50-75 cells in 3-4 distinct fields of view were quantified using MicroSuite image analysis software and the average +/-SE plotted as relative pixel density. * = statistically different from DMSO treated cells (p<0.001). ** = statistically different from DMSO treated cells and TCDD treated cells (p<0.001).
3.3 The DNA bound AHR does not show a reduction in molecular mass
Since the results presented in the current study suggest that the AHR is readily transloacted to the nucleus in the presence of proteasome or calpain inhibitors, it was pertinent to evaluate whether the DNA bound form of the AHR showed any changes in molecular mass. For these studies, Hepa-1, rat RPE and human RPE cells were exposed to TCDD for 30 minutes and the cytoplasmic and nuclear fractions isolated and evaluated for AHR by Western blotting. Since each of the cells express an AHR with a different molecular mass, the proteins were evaluated on a 7% SDS polyacrylamide gel so that the different sized AHRs could be resolved. The Ahb-1 AHR is 805 amino acids while the rat and human AHRs are approximately 847 amino acids. Using this method, it would be expected that any cleavage event that removed as few as 10 amino acids (approximately 1.3kDa) could be resolved. The results in Figure 8A show that TCDD exposure results in a dramatic elevation in the level of AHR detected in the nuclear fraction in the Hepa-1 and rat RPE lines while the nuclear fraction isolated from the human RPE cells showed a modest elevation in AHR. Importantly, the molecular mass of the nuclear AHR in all of the cell lines did not exhibit a difference in molecular mass when compared to the AHR found in the cytoplasmic fractions and there were no smaller molecular mass bands that were present in a TCDD-dependent manner. Since nuclear lysates fractions represent the total complement of proteins associated with nuclear structures, it was pertinent to repeat the studies using proteins that had been extracted from the DNA. Thus, nuclear extracts were prepared from control and TCDD-treated Hepa-1 and rat RPE cells and the AHR evaluated by Western blotting. The results in Figure 8B shows that there is a dramatic elevation in the level of nuclear AHR detected in the TCDD treated samples of both lines and that the molecular mass is not reduced when compared to untreated samples. In addition there are no smaller immuno-reactive bands that appear in the fractions in a TCDD-dependent manner (i.e., the smaller bands that are observed in the nuclear fractions are present at the same level in both the control and TCDD-treated samples). It is important to note, the lack of detection of smaller molecular mass AHRs is not due to the antibody being used in the studies, since it is polyclonal, was generated against amino acids 1-416 of the mAHR, and has been used to detect AHRs truncated in the C-terminal domain (24, 43). Therefore, these studies provide no evidence that ligand treatment results in a DNA bound form of the AHR that has a reduced molecular mass.
Figure 8. Analysis of nuclear AHR following TCDD exposure.
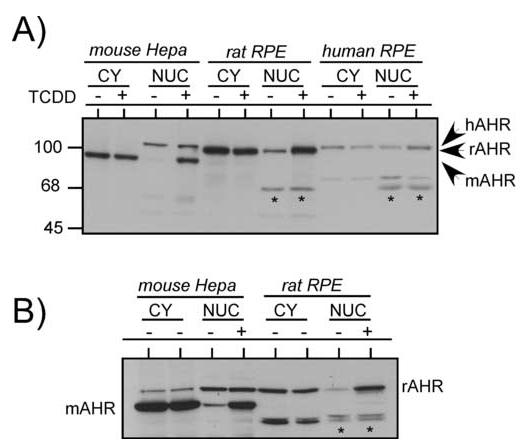
A) Hepa-1, rat RPE, and human RPE cells were treated with TCDD (5nM) for 1 hr and nuclear and cytoplasmic lysates prepared as detailed in Materials and Methods. Equal amounts of cytosol, or nuclear lysates were resolved by SDS-PAGE, blotted and stained with A-1A anti-AHR IgG (1.0μg/ml). Reactivity was visualized by ECL with GAR-HRP (1:10,000). Note the ability to resolve the different species of AHR and lack of lower molecular mass bands that show changes between the control and TCDD treated samples (asterisk). B) Hepa-1 or rat RPE cells were treated with TCDD as in A and nuclear extracts prepared as detailed in Materials and Methods. Equal amount of cytosol or nuclear extracts were resolved by SDS-PAGE and evaluated as detailed in A. As in (A), asterisks mark lower molecular mass bands that show no changes between control and TCDD treated samples.
4.0 DISCUSSION
The concentration of AHR protein is a critical component in the response of cells to toxicologically relevant ligands, thus it is important to define the pathways involved in maintaining AHR levels. Numerous studies from this and other laboratories have clearly established that the AHR is subject to degradation following ligand binding both in vitro and in vivo [5, 6]. Although the precise enzymes involved in the degradation of the AHR have not been identified, it has recently been hypothesized that cleavage of the C-terminus of the AHR by calpain proteases is required for not only the translocation of the AHR to the nucleus but also for its ligand-induced degradation [25]. This hypothesis is based in part on a study by Poland and Glover from 1988 showing that there was a Ca++ dependent cleavage of the murine AHR from a 95kDa species to a 70kDa species when the AHR was purified from tissues and cells [44]. The authors concluded that the likely protease was calpain II (m-calpain) and that the cleavage of the 95kDa species was an artifact of the sample preparation (as is often observed when isolating native proteins from tissue or cell extracts). Thus, the importance of this observation in the mechanism of AHR-mediated signaling was not pursued. However, due to the recent hypotheses concerning calpains in AHR signaling and the targeting of proteases by novel pharmacologicals in a number of disease states [26, 27], it was pertinent to reassess whether calpain proteases were involved in AHR-mediated signal transduction.
The results of the current study do not support a role of calpain proteases in either the nuclear translocation or ligand-induced degradation of the mouse, human or rat AHR in vivo. First, none of the three different calpain inhibitors used in any of the studies had an affect on the ligand-induced degradation of the AHR when used independently or in combination and did not block the nuclear localization of the AHR after ligand exposure. Second, ligand-induced degradation was blocked by three different inhibitors that were specific to the proteasome and these inhibitors all caused accumulation of the latent AHR in the nucleus in the absence of ligand. Finally, the AHR extracted from the nucleus following ligand exposure was the same molecular mass as control AHR.
It is important to note that the conclusions of the current study are consistent with other reports have also evaluated the role of calpains in AHR signaling. Ma and Baldwin [23] for example, previously showed that exposure of Hepa-1 cells to the calpain inhibitors calpastatin or PD150606 had no impact on TCDD-mediated degradation of the AHR or induction of CYP1A1 and other studies have shown that the calpain inhibitor ALLM did not block AHR degradation in a rat cell line [21]. Consistent with these findings, amino acid sequence analysis of all mammalian AHRs using PESTfind (https://emb1.bcc.univie.ac.at/toolbox/pestfind/pestfind-analysis-webtool.htm) indicates that the AHR does not contain defined “PEST sequences” that have been identified in many proteins that are targets for calpain proteases [45, 46]. It is also important to note that if the latent 95kDa Ahb-1 AHR required calpain cleavage for a nuclear translocation event, this likely have a substantial impact on the transactivation potential of the AHR. For example, truncation of 165 amino acids from the C-terminus of the Ahb-1 receptor (approximately 21kDa) has no affect on the cytoplasmic localization of the AHR, but reduces the ability of the AHR to induce expression of endogenous CYP1A1 by 50% [24]. In addition, removal of 305 amino acids from the C-terminus of the AHR (approximately 40kDa) causes the latent AHR to become nuclear but completely abolishes the ability of the AHR to induce endogenous CYP1A1 [24]. A cleavage from 95kDa to 70kDa (as suggested by Poland and Glover [44]) would be a loss of approximately 270 amino acids. It is also difficult to reconcile a cleavage event with the finding that the rat and the mouse Ahb-2 receptors are undergoing dynamic nucleocytoplasmic shuttling in the absence of ligand stimulation [47]. In these cells, one molecular mass of AHR is detected [47]. There are also numerous studies in which the AHR has been tagged on the C-terminus with a GFP tag. In all these instances the AHR appears to be capable of TCDD-mediated nuclear translocation and gene regulation and retains the GFP tag [48, 49]. Thus, taken collectively, these results are not consistent with a calpain cleavage event in the initiation of the AHR signal transduction pathway or in the ligand-induced degradation of the AHR.
So what is the pathway that mediates ligand-induced AHR degradation? Currently, nearly all studies point to degradation through the 26S proteasome although there may be alternate pathways to target the AHR to the proteasome depending on whether it is ligand bound [24]. This conclusion is based on the ability to block ligand-induced degradation of he AHR with specific proteasome inhibitors (the current study) as well as previous studies showing that degradation is blocked in cells containing a temperature sensitive mutation in the E1 ubiquitin conjugating enzyme [23]. Importantly, since inhibition of the proteasome blocks the normal process of AHR degradation and the proteasome in now being targeted by various novel pharmacologics, it will be critical to determine the impact of the reduced degradation on the magnitude and duration of gene regulation. The current challenge also continues to be the identification of target residues of the AHR that confer degradation, the identification on the enzymes involved and the isolation of an AHR that has been ubiquitylated in vivo.
ACKNOWLEDGEMENTS
This work was supported by a grant from the National Institutes of Health to RSP (ES 10991).
ABBREVIATIONS
- TCDD
2,3,7,8-tetrachlorodibenzo-p-dioxin
- AHR
aryl hydrocarbon receptor
- ARNT
Ah receptor nuclear translocator
- GAR-HRP
goat anti-rabbit antibodies conjugated to horseradish peroxidase
- GAR-RHO
goat anti-rabbit antibodies conjugated to rhodamine
- t-BOC-leu-met
t-BOC-L-leucyl-L-methionine amide
Footnotes
RS Pollenz, unpublished results
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1].Kewley RJ, Whitelaw ML, Chapman-Smith A. The mammalian basic helix-loop-helix/PAS family of transcriptional regulators. Int J Biochem Cell Biol. 2004;36:189–204. doi: 10.1016/s1357-2725(03)00211-5. [DOI] [PubMed] [Google Scholar]
- 2].Pandini A, Bonati L. Conservation and specialization in PAS domain dynamics. Protein Eng Des Sel. 2005;18:127–37. doi: 10.1093/protein/gzi017. [DOI] [PubMed] [Google Scholar]
- 3].Carney SA, Prasch AL, Heideman W, Peterson RE. Understanding dioxin developmental toxicity using the zebrafish model. Birth Defects Res A Clin Mol Teratol. 2006;76:7–18. doi: 10.1002/bdra.20216. [DOI] [PubMed] [Google Scholar]
- 4].Mandal PK. Dioxin: a review of its environmental effects and its aryl hydrocarbon receptor biology. J Comp Physiol. 2005;175:221–30. doi: 10.1007/s00360-005-0483-3. [DOI] [PubMed] [Google Scholar]
- 5].Pollenz RS. Mechanism of Ah receptor down regulation (degradation) and its impact on AHR-mediated gene regulation. Chemico Biological Inter. 2002;141:41–61. doi: 10.1016/s0009-2797(02)00065-0. [DOI] [PubMed] [Google Scholar]
- 6].Harper PA, Riddick DS, Okey AB. Regulating the regulator: factors that control levels and activity of the aryl hydrocarbon receptor. Biochem Pharmacol. 2006;72:267–79. doi: 10.1016/j.bcp.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 7].Pollenz RS, Santostefano MJ, Klett E, Richardson VM, Necela B, Birnbaum LS. Female Sprague Dawley rats exposed to a single oral dose of 2,3,7,8-tetrachlorodibenzo-p-dioxin exhibit sustained depletion of aryl hydrocarbon receptor protein in liver, spleen, thymus and lung. Toxicol Sci. 1998;42:117–28. doi: 10.1006/toxs.1998.2439. [DOI] [PubMed] [Google Scholar]
- 8].Ma Q, Whitlock JP., Jr The aromatic hydrocarbon receptor modulates the Hepa 1c1c7 cell cycle and differentiated state independently of dioxin. Mol Cell Biol. 1996;16:2144–50. doi: 10.1128/mcb.16.5.2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9].Andersson P, McGuire J, Rubio C, Gradin K, Whitelaw ML, Pettersson S, Hanberg A, Poellinger L. A constitutively active dioxin/aryl hydrocarbon receptor induces stomach tumors. Proc Natl Acad Sci U S A. 2002;99:9990–9995. doi: 10.1073/pnas.152706299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10].McGuire J, Okamoto K, Whitelaw ML, Tanaka H, Poellinger L. Definition of a dioxin receptor mutant that is a constitutive activator of transcription: delineation of overlapping repression and ligand binding functions within the PAS domain. J Biol Chem. 2001;276:41841–41849. doi: 10.1074/jbc.M105607200. [DOI] [PubMed] [Google Scholar]
- 11].Fernandez-Salguero P, Pineau T, Hilbert DM, McPhail T, Lee SS, Kimura S, Nebert DW, Rudikoff S, Ward JM, Gonzalez FJ. Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor. Science. 1995;268:722–726. doi: 10.1126/science.7732381. [DOI] [PubMed] [Google Scholar]
- 12].Schmidt JV, Su GH, Reddy JK, Simon MC, Bradfield CA. Characterization of a murine Ahr null allele: involvement of the Ah receptor in hepatic growth and development. Proc Natl Acad Sci U S A. 1996;93:6731–6. doi: 10.1073/pnas.93.13.6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13].Hushka LJ, Williams JS, Greenlee WF. Characterization of 2,3,7,8-tetrachlorodibenzofuran-dependent suppression and AH receptor pathway gene expression in the developing mouse mammary gland. Toxicol Appl Pharmacol. 1998;152:200–210. doi: 10.1006/taap.1998.8508. [DOI] [PubMed] [Google Scholar]
- 14].Peters JM, Narotsky MG, Elizondo G, Fernandez-Salguero PM, Gonzalez FJ, Abbott BD. Amelioration of TCDD-Induced Teratogenesis in Aryl Hydrocarbon Receptor (AhR)-Null Mice. Toxicological Sciences. 1999;47:86–92. doi: 10.1093/toxsci/47.1.86. [DOI] [PubMed] [Google Scholar]
- 15].Walisser JA, Glover E, Pande K, Liss AL, Bradfield CA. Aryl hydrocarbon receptor-dependent liver development and hepatotoxicity are mediated by different cell types. Proc Natl Acad Sci U S A. 2005;102:17858–6. doi: 10.1073/pnas.0504757102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16].Walisser JA, Bunger MK, Glover E, Harstad EB, Bradfield CA. Patent ductus venosus and dioxin resistance in mice harboring a hypomorphic Arnt allele. J Biol Chem. 2004;279:16326–31. doi: 10.1074/jbc.M400784200. [DOI] [PubMed] [Google Scholar]
- 17].Abbott BD, Schmid JE, Pitt JA, Buckalew AR, Wood CR, Held GA, Diliberto JJ. Adverse Reproductive Outcomes in the Transgenic Ah Receptor-Deficient Mouse. Tox.and App. Pharm. 1999;155:62–70. doi: 10.1006/taap.1998.8601. [DOI] [PubMed] [Google Scholar]
- 18].Heimler I, Trewin AL, Chaffin CL, Rawlins RG, Hutz RJ. Modulation of ovarian follicle maturation and effects on apoptotic cell death in Holtzman rats exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in utero and lactationally. Reprod. Toxicol. 1998;12:69–73. doi: 10.1016/s0890-6238(97)00101-9. [DOI] [PubMed] [Google Scholar]
- 19].Wolf CJ, Ostby JS, Gray LE. gestantional exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) severely alters reproductive function of female hamster offspring. Toxicol. Sci. 1999;51:259–264. doi: 10.1093/toxsci/51.2.259. [DOI] [PubMed] [Google Scholar]
- 20].Benedict JC, Lin TM, Loeffler IK, Peterson RE, Flaws JA. Physiological role of the aryl hydrocarbon receptor in mouse ovary development. Toxicol. Sci. 2000;56:382–388. doi: 10.1093/toxsci/56.2.382. [DOI] [PubMed] [Google Scholar]
- 21].Davarinos NA, Pollenz RS. Aryl hydrocarbon receptor imported into the nucleus following ligand binding is rapidly degraded via the cytosplasmic proteasome following nuclear export. J Biol Chem. 1999;274:28708–15. doi: 10.1074/jbc.274.40.28708. [DOI] [PubMed] [Google Scholar]
- 22].Pollenz RS, Barbour ER. Analysis of the complex relationship between nuclear export and Ah receptor-mediated gene regulation. Mol. Cell. Biol. 2000;20:6094–6105. doi: 10.1128/mcb.20.16.6095-6104.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23].Ma Q, Baldwin KT. 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced degradation of aryl hydrocarbon receptor (AhR) by the ubiquitin-proteasome pathway. Role of the transcription activaton and DNA binding of AhR. J Biol Chem. 2000;275:8432–8. doi: 10.1074/jbc.275.12.8432. [DOI] [PubMed] [Google Scholar]
- 24].Pollenz RS, Popet J, Dougherty EJ. Role of the carboxy-terminal transactivation domain and active transcription in the ligand-induced degradation of the mouse Ahb-1 receptor. Biochemical Pharmacology. 2005;70:1623–1633. doi: 10.1016/j.bcp.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 25].Dale YR, Eltom SE. Calpain mediates the dioxin-induced activation and down-regulation of the aryl hydrocarbon receptor. Mol Pharmacol. 2006;70:1481–7. doi: 10.1124/mol.106.027474. [DOI] [PubMed] [Google Scholar]
- 26].Ciechanover A. Intracellular Protein Degradation: From a Vague Idea thru the Lysosome and the Ubiquitin-Proteasome System and onto Human Diseases and Drug Targeting. Hematology Am Soc Hematol Educ Program. 2006:1–12. doi: 10.1182/asheducation-2006.1.1. [DOI] [PubMed] [Google Scholar]
- 27].Joazeiro CA, Anderson KC, Hunter T. Proteasome inhibitor drugs on the rise. Cancer Res. 2006;66:7840–2. doi: 10.1158/0008-5472.CAN-06-2033. [DOI] [PubMed] [Google Scholar]
- 28].Pollenz RS, Sattler CA, Poland A. The aryl hydrocrabon receptor and aryl hydrocarbon receptor nuclear translocator protein show distinct subcellular localization in Hepa 1c1c7 cell by immunofluorescence microscopy. Mol Pharmacol. 1994;45:428–38. [PubMed] [Google Scholar]
- 29].Holmes JL, Pollenz RS. Determination of aryl hydocarbon receptor nuclear translocator protein concentration and subcellular localization in hepatic and nonhepatic cell culture lines: Development of quantitative western blotting protocols for calculation of aryl hydrocarbon receptor and aryl hydrocarbon receptor nuclear translocator protein in total cell lysates. Mol Pharmacol. 1997;52:202–11. doi: 10.1124/mol.52.2.202. [DOI] [PubMed] [Google Scholar]
- 30].Pollenz RS. The aryl hydrocarbon receptor but not Arnt protein is rapidly depleted in hepatic and non hepatic culture cells exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Mol Pharmacol. 1996;49:391–8. [PubMed] [Google Scholar]
- 31].Pollenz RS, Santostefano MJ, Klett E, Richardson V, Necela B, Birnbaum LS. A single oral dose of TCDD results in sustained depletion of AHR protein in female Sprague-Dawley rats. Toxicological Sciences. 1998;42:117–128. doi: 10.1006/toxs.1998.2439. [DOI] [PubMed] [Google Scholar]
- 32].Roman BL, Pollenz RS, Peterson RE. Responsiveness of the adult male rat reproductive tract to 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure: Ah receptor and ARNT expression, CYP1A1 induction, and Ah receptor downregulation. Toxicol. Applied Phramacol. 1998;150:228–239. doi: 10.1006/taap.1998.8388. [DOI] [PubMed] [Google Scholar]
- 33].Sommer RJ, Sojka K, Pollenz RS, Cooke P, Peterson RE. AHR and ARNT protein and mRNA concentrations in rat prostate: Effects of stage of development and TCDD. Toxicol. Appl. Pharmacol. 1999;155:177–189. doi: 10.1006/taap.1998.8597. [DOI] [PubMed] [Google Scholar]
- 34].Wormke M, Stoner M, Saville B, Walker K, Abdelrahim M, Burghardt R, Safe S. The aryl hydrocarbon receptor mediates degradation of estrogen receptor alpha through activation of proteasomes. Mol Cell Biol. 2003;23:1843–55. doi: 10.1128/MCB.23.6.1843-1855.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35].Song Z, Pollenz RS. Ligand-dependent and independent modulation of aryl hydrocarbon receptor localization, degradation, and gene regulation. Mol Pharmacol. 2002;62:806–16. doi: 10.1124/mol.62.4.806. [DOI] [PubMed] [Google Scholar]
- 36].Roberts BJ, Whitelaw ML. Degradation of the basic helix-loop-helix/Per-ARNT-Sim homology domain dioxin receptor via the ubiquitin/proteasome pathway. J Biol Chem. 1999:27436351–6. doi: 10.1074/jbc.274.51.36351. [DOI] [PubMed] [Google Scholar]
- 37].Glading A, Uberall F, Keyse SM, Lauffenburger DA, Wells A. Membrane proximal ERK signaling is required for M-calpain activation downstream of epidermal growth factor receptor signaling. J Biol Chem. 2001;276:23341–8. doi: 10.1074/jbc.M008847200. [DOI] [PubMed] [Google Scholar]
- 38].Carragher NO, Fonseca BD, Frame MC. Calpain activity is generally elevated during transformation but has oncogene-specific biological functions. Neoplasia. 2004;6:53–73. doi: 10.1016/s1476-5586(04)80053-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39].Tsubuki S, Saito Y, Tomioka M, Ito H, Kawashima S. Differential inhibition of calpain and proteasome activities by peptidyl aldehydes of di-leucine and tri-leucine. J Biochem. 1996;119:572–6. doi: 10.1093/oxfordjournals.jbchem.a021280. [DOI] [PubMed] [Google Scholar]
- 39].Kim KB, Myung J, Sin N, Crews CM. Proteasome inhibition by the natural products epoxomicin and dihydroeponemycin: insights into specificity and potency. Bioorg Med Chem Lett. 1999;9:3335–40. doi: 10.1016/s0960-894x(99)00612-5. [DOI] [PubMed] [Google Scholar]
- 40].Sin N, Kim KB, Elofsson M, Meng L, Auth H, Kwok BH, Crews CM. Total synthesis of the potent proteasome inhibitor epoxomicin: a useful tool for understanding proteasome biology. Bioorg Med Chem Lett. 1999;9:2283–8. doi: 10.1016/s0960-894x(99)00376-5. [DOI] [PubMed] [Google Scholar]
- 41].Santiago-Josefat B, Fernandez-Salguero PM. Proteasome inhibition induces nuclear translocation of the dioxin receptor through an Sp1 and protein kinase C-dependent pathway. J Mol Biol. 2003;333:249–60. doi: 10.1016/j.jmb.2003.08.020. [DOI] [PubMed] [Google Scholar]
- 42].Santiago-Josefat B, Pozo-Guisado E, Mulero-Navarro S, Fernandez-Salguero PM. Proteasome inhibition induces nuclear translocation and transcriptional activation of the dioxin receptor in mouse embryo primary fibroblasts in the absence of xenobiotics. Mol Cell Biol. 2001;21:1700–9. doi: 10.1128/MCB.21.5.1700-1709.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43].Pollenz RS, Wilson SE, Dougherty EJ. Role of the endogenous XAP2 protein on the localization and nucleocytoplasmic shuttling of the endogenous mouse Ah b-1 receptor in the presence and absence of ligand. Molecular Pharmacol. 2006;70:1369–1376. doi: 10.1124/mol.106.027672. [DOI] [PubMed] [Google Scholar]
- 44].Poland A, Glover E. Ca2+-dependent proteolysis of the Ah receptor. Arch Biochem Biophys. 1988;261:103–11. doi: 10.1016/0003-9861(88)90109-9. [DOI] [PubMed] [Google Scholar]
- 45].Rechsteiner M, Rogers SW. PEST sequences and regulation by proteolysis. Trends Biochem Sci. 1996;21:267–71. [PubMed] [Google Scholar]
- 46].Sandoval A, Oviedo N, Tadmouri A, Avila T, De Waard M, Felix R. Two PEST-like motifs regulate Ca2+/calpain-mediated cleavage of the CaVbeta3 subunit and provide important determinants for neuronal Ca2+ channel activity. Eur J Neurosci. 2006;23:2311–20. doi: 10.1111/j.1460-9568.2006.04749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47].Pollenz RS, Dougherty EJ. Redefining the role of XAP2 and CHIP in the degradation of endogenous AHR in cell culture models. J. Biol. Chem. 2005;280:33346–33356. doi: 10.1074/jbc.M506619200. [DOI] [PubMed] [Google Scholar]
- 48].Petrulis JR, Hord NG, Perdew GH. Subcellular localization of the aryl hydrocarbon receptor is modulated by the immunophilin homolog hepatitis B virus X-associated protein 2. J Biol Chem. 2000;275:37448–37453. doi: 10.1074/jbc.M006873200. [DOI] [PubMed] [Google Scholar]
- 49].Ramadoss P, Petrulis JR, Hollingshead BD, Kusnadi A, Perdew GH. Divergent roles of hepatitis B virus X-associated protein 2 (XAP2) in human versus mouse Ah receptor complexes. Biochemistry. 2004;43:700–709. doi: 10.1021/bi035827v. [DOI] [PubMed] [Google Scholar]