

Abstract
Dopaminergic cells in the substantia nigra are highly vulnerable to the neurodegenerative process of Parkinson’s disease. Therefore, mechanisms that enhance their susceptibility to injury bear important implications for disease pathogenesis. Repeated injections with the herbicide paraquat cause oxidative stress and a selective loss of dopaminergic neurons in mice. In this model, the first paraquat exposure, though not sufficient to induce any neurodegeneration, predisposes neurons to damage by subsequent insults. The purpose of this study was to elucidate the mechanisms underlying this “priming” event. We found that a single paraquat exposure was followed by an increase in the number of cells with immunohistochemical, morphological and biochemical characteristics of activated microglia, including induction of NADPH-oxidase. If this microglial response was inhibited by the anti-inflammatory drug minocycline, subsequent exposures to the herbicide failed to cause oxidative stress and neurodegeneration. On the other hand, if microglial activation was induced by pre-treatment with lipopolysaccharide, a single paraquat exposure became capable of triggering a loss of dopaminergic neurons. Finally, mutant mice lacking functional NADPH-oxidase were spared from neurodegeneration caused by repeated paraquat exposures. Data indicate that microglial activation and consequent induction of NADPH-oxidase may act as risk factors for Parkinson’s disease by increasing the vulnerability of dopaminergic cells to toxic injury.
Keywords: Parkinson’s disease, Substantia nigra, Paraquat, Pesticide, Microglia, Inflammation, NADPH-oxidase, Minocycline
Introduction
Parkinson’s disease (PD) is a common neurodegenerative disorder, second only to Alzheimer’s disease in its incidence among the aging population (Mayeux, 2003; Van Den Eeden et al., 2003). The pathological features of PD include an abnormal accumulation of the protein α-synuclein throughout various brain regions and the degeneration of dopaminergic neurons in the nigrostriatal pathway (Forno, 1996; Spillantini et al., 1998). This latter degenerative process is largely responsible for the disabling motor abnormalities that characterize PD in the form of slowness in movements, muscle stiffness, resting tremor and postural instability (Fahn, 2003). The etiology of PD is likely to involve both environmental and genetic factors that would ultimately contribute to the progressive demise of specific neuronal cell populations (Di Monte, 2003). However, the exact mechanisms of neurodegeneration remain relatively unknown and, for this reason, no effective neuroprotective intervention is as yet available.
Several lines of clinical and experimental evidence support the hypothesis that glial activation and inflammatory processes play a role in the pathogenesis of PD. Epidemiological studies found a lower risk for PD associated with regular use of non-steroidal anti-inflammatory drugs (Chen et al., 2003 and 2005). Reactive microglial cells, which are important mediators of neuroinflammation, are abundant in the substantia nigra and striatum of patients with idiopathic PD (McGeer and McGeer, 2004; Ouchi et al., 2005). In humans who developed a parkinsonian syndrome after accidentally injecting themselves with the neurotoxicant 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), post-mortem analysis of the substantia nigra revealed clusters of reactive microglia around nerve cells (Langston et al., 1999). This finding was suggested to reflect an ongoing neurodegenerative process that persisted years after the initial toxic injury and could have been perpetuated, at least in part, by chronic neuroinflammation (Langston et al., 1999). Microglia have also been implicated in the production of reactive oxygen species (ROS) and the selective degeneration of nigrostriatal dopaminergic neurons caused by MPTP and other neurotoxicants in animal models and in vitro experimental paradigms (Gao et al., 2003a and b; Sherer et al., 2003; Wu et al., 2003; Ling et al., 2004).
Systemic exposure of rodents to the herbicide paraquat, alone or in combination with the fungicide maneb, reproduces pathological features of PD, including the intraneuronal deposition of α-synuclein and the selective degeneration of dopaminergic neurons in the substantia nigra (Manning-Bog et al., 2002; McCormack et al., 2002; Thiruchelvam et al., 2003; Peng et al., 2004). Multiple treatments with paraquat or paraquat/maneb have been associated with activation of microglia, although a causal relationship between this effect and neurodegeneration has yet to be demonstrated (McCormack et al., 2002; Saint-Pierre et al., 2006). An intriguing feature, which characterizes the development of neurodegeneration after sequential administrations of paraquat alone, is that the initial insult does not itself induce any neuronal loss but predisposes to the toxic consequences of subsequent challenges (McCormack et al., 2005). Mechanisms responsible for this “priming” effect are of likely relevance to PD since they could underlie, for example, the action of disease risk factors and help identify conditions that enhance the vulnerability of dopaminergic cells to degenerative processes.
The purpose of this study was twofold. Experiments were designed to provide evidence in favor of or against a direct contribution of microglia to paraquat-induced neurodegeneration. More in particular, however, we tested the possibility that a single paraquat administration triggers microglial activation and that a relationship exists between this inflammatory response and the loss of dopaminergic neurons observed upon a subsequent exposure to the herbicide. Biochemical and histological analyses were used to document paraquat-induced microglial response, and pharmacological and genetic tools were employed to manipulate this response and thus enhance or mitigate neurodegeneration.
Materials and methods
Animals and treatment
Experiments were carried out in ten- to twelve-week old male C57BL/6 (Charles River, Hollister, CA; Jackson Laboratory, Bar Harbor, ME) and gp91phox-deficient (B6.129S6-Cybbtm1din, Jackson Laboratory) mice. All chemicals administered to the animals were purchased from Sigma (St. Louis, MO), dissolved in saline and injected intraperitoneally. In all experiments, control groups of mice received saline injections in lieu of the treatment agents. Paraquat was given at a dose of 10 mg/kg once a week for one or two consecutive weeks, and animals were sacrificed at different time points following the first or second injection. In experiments aimed at assessing the effects of minocycline, mice received one or two injections of paraquat one-week apart. Animals treated with minocycline (45 mg/kg) received an injection of this anti-inflammatory drug 30 min prior to the first paraquat exposure. Minocycline was then administered every 12 hours for three or seven days, and mice were sacrificed at different time points during the first or second week of treatment. In experiments in which lipopolysaccharide (LPS) was used to induce microglial activation, LPS (from Escherichia coli 055:B5, Sigma) was administered once at a dose of 2 or 4 mg/kg. Then, mice received a single injection of paraquat at two days post LPS and were killed seven days later. Experimental protocols were in accordance with the NIH guidelines for use of live animals and were approved by the Animal Care and Use Committee at The Parkinson’s Institute.
Immunohistochemistry
After removal of the brains, midbrain blocks were either (i) snap frozen in cold isopentane for single and double staining with antibodies against macrophage antigen complex 1 (Mac-1), tyrosine hydroxylase (TH), and gp91phox, or (ii) immersion fixed in 4% paraformaldehyde (PFA) and cryoprotected in sucrose for TH or 4-hydroxynonenal (4-HNE) immunostaining.
Fresh frozen tissues were cryostat-cut into 12 μm serial coronal sections and used for brightfield microscopy. Sections were fixed in 4% PFA, and endogenous background was reduced by treatment with hydrogen peroxide, an avidin-biotin blocking kit (Vector Labs, Burlingame, CA) and blocking serum. Double-labeling for Mac-1 and TH involved sequential incubations with: (i) rat anti-Mac-1 antibody (1:400, Chemicon, Temecula, CA), (ii) biotinylated anti-rat IgG (mouse adsorbed) and ABC reagents (Vector Labs), (iii) 3,3′-diaminobenzidine (DAB), (iv) rabbit blocking serum, (v) sheep anti-TH antibody (1:1,200, Pel-Freez Biologicals, Rogers, AR), (vi) biotinylated anti-sheep IgG and ABC reagents (Vector Labs), and (vii) Vector SG chromagen (Vector Labs). Sections labeled for gp91phox were incubated overnight at 4°C in mouse anti-gp91phox (1:200, BD-Pharmingen, San Diego, CA). Immunostaining was visualized using DAB as the chromagen after incubation with the biotinylated secondary antibody and ABC reagents supplied in the Mouse on Mouse (M.O.M) kit (Vector Labs).
Immersion-fixed midbrain blocks were cryostat-cut into 40 μm serial coronal sections. For TH immunoreactivity and Nissl counterstaining, sections were incubated with a rabbit anti-TH antibody (1:600, Pel-Freez Biologicals) and 0.5% Cresyl violet as previously described (McCormack et al., 2002). 4-HNE-positive neurons were immunostained using an antibody that recognizes chemically reduced amino acid-4-HNE adducts as previously described (McCormack et al., 2005). Briefly, sections were reduced using sodium borohydride and incubated overnight at 4°C with rabbit anti-4-HNE (1:15,000, Calbiochem, San Diego, CA). After immersion in biotinylated goat anti-rabbit IgG, immunostaining was visualized using an ABC kit (Vector Labs) with DAB as the chromagen. Sections were lightly counterstained with Cresyl violet. In all immunohistochemical procedures, negative (unstained) control sections were incubated with appropriate IgG in lieu of the primary antibody.
Cell counting
Activated microglial cells were quantified in midbrain sections double-labeled for Mac-1 and TH. They were counted strictly within the substantia nigra pars compacta in sections immediately anterior to the 3rd nerve and were identified based on their robust Mac-1 immunoreactivity as well as typical morphology (e.g. enlarged cell body with swollen processes). Nigral 4-HNE-immunoreactive neurons were visualized and counted in midbrain sections at the level of the 3rd nerve as previously described (McCormack et al., 2005). A series containing every 6th section throughout the substantia nigra that had been immunostained for TH and counterstained with Cresyl violet was used for stereological cell counting. After delineation of the substantia nigra pars compacta at low magnification, sections were sampled at high magnification using the optical fractionator method (StereoInvestigator, MicroBrightfield, Williston, VT) (McCormack et al., 2002).
Immunoblotting
The ventral mesencephalon was dissected on ice and sonicated in lysis buffer with protease inhibitors. After centrifugation (14,000 x g), the supernatant was decanted and separated by Tris-Glycine gel electrophoresis under non-reducing conditions. Proteins were transferred to nitrocellulose membranes, and blots were blocked with 5% nonfat milk in 25 mM Tris-saline solution containing 0.05% Tween. Membranes were then incubated with either rat anti-F4/80 (1:200, Abcam, Cambridge, MA) or mouse anti-α-tubulin (1:7,000, Abcam). Secondary anti-rat or anti-mouse IgG conjugated to horseradish peroxidase (1:3,000, Pierce, Rockford, IL) was applied, and blots were incubated with a chemiluminescent substrate (Pierce) and exposed to Hyperfilm™ ECL™ (Amersham Biosciences, Piscataway, NJ). Non-reducing conditions for recognition of the F4/80 epitope were used based on manufacturer’s recommendations. Control blots were incubated with the appropriate IgG instead of the primary antibody, resulting in blank films.
RNA extraction and reverse transcriptase-polymerase chain reactions (RT-PCR)
Total RNA from ventral mesencephalon samples was extracted using RNeasy lipid tissue mini kit (Qiagen, Valencia, CA) as per the protocol provided by the manufacturer, and cDNAs were prepared by reverse transcription (Superscript III, Invitrogen, Carlsbad, CA). PCR was performed using PCR Express Thermal Cycler (Hybaid, Franklin, MA). The primer sequences were as follows: mouse Mac-1 (Itgam) forward primer 5′-TTC TCA TGG TCA CCT CCT GC-3′ and reverse primer 5′-GGT CTG ACC ATC TGA ACC TG-3′, mouse gp91phox (cybb) forward primer 5′-CAG GAG TTC CAA GAT GCC TG-3′ and reverse primer 5′-GAT TGG CCT GAG ATT CAT CC-3′, mouse glyceraldehyde 3-phosphate dehydrogenase (GAPDH) forward primer 5′-TGA TGA CAT CAA GAA GGT GGT GAA G-3′ and reverse primer 5′-TCC TTG AGG CCA TGT AGG CCA T-3′. Amplification was carried out for 27 cycles for Mac-1, 29 cycles for gp91phox, and 20 cycles for GAPDH. Amplification products were electrophoresed on 4% agarose gels containing ethidium bromide. Gels were placed on an ultraviolet box, and images were captured using Kodak Gel Logic 100 (Eastman Kodak Company, Rochester, NY).
Statistical analysis
For each experiment, at least five animals per group were studied. Data were analyzed by one-way analysis of variance (ANOVA). Fisher’s protected LSD post-hoc analysis was employed when differences were observed in ANOVA testing (P < 0.05).
Results
Microglial response after paraquat exposure
In agreement with data previously reported (McCormack et al., 2005), a single intraperitoneal injection of 10 mg/kg paraquat did not cause any loss of dopaminergic neurons in the mouse substantia nigra pars compacta (data not shown). We tested the possibility however that, under the same experimental conditions, paraquat was capable of inducing microglial activation. When midbrain sections were immunostained with an antibody against Mac-1, cells with amoeboid-shaped cytoplasm and thick, short ramifications typical of activated microglia were rarely observed in saline-treated control mice (Figure 1A). They became relatively abundant, however, in animals injected once with paraquat, after which Mac-1-immunoreactive cells were intermingled with TH-positive neurons in the substantia nigra pars compacta (Figure 1B). A significant number of cells with morphological and immunocytochemical characteristics of activated microglia was already present at one day after paraquat administration. Counts of these cells further increased between one and two days and reached their maximum at four and seven days post exposure (Figure 1C). Biochemical assays confirmed changes in microglial markers induced by a single paraquat injection. Mac-1 mRNA expression was significantly enhanced at two and seven days in homogenates from the ventral mesencephalon of mice treated with the herbicide as compared to control animals (Figure 2A). Furthermore, western blots of mesencephalic homogenates assayed with an antibody against mouse F4/80 antigen, another microglial marker, showed denser immunoreactive bands in paraquat-exposed versus saline-injected mice (Figure 2B).
Fig. 1.
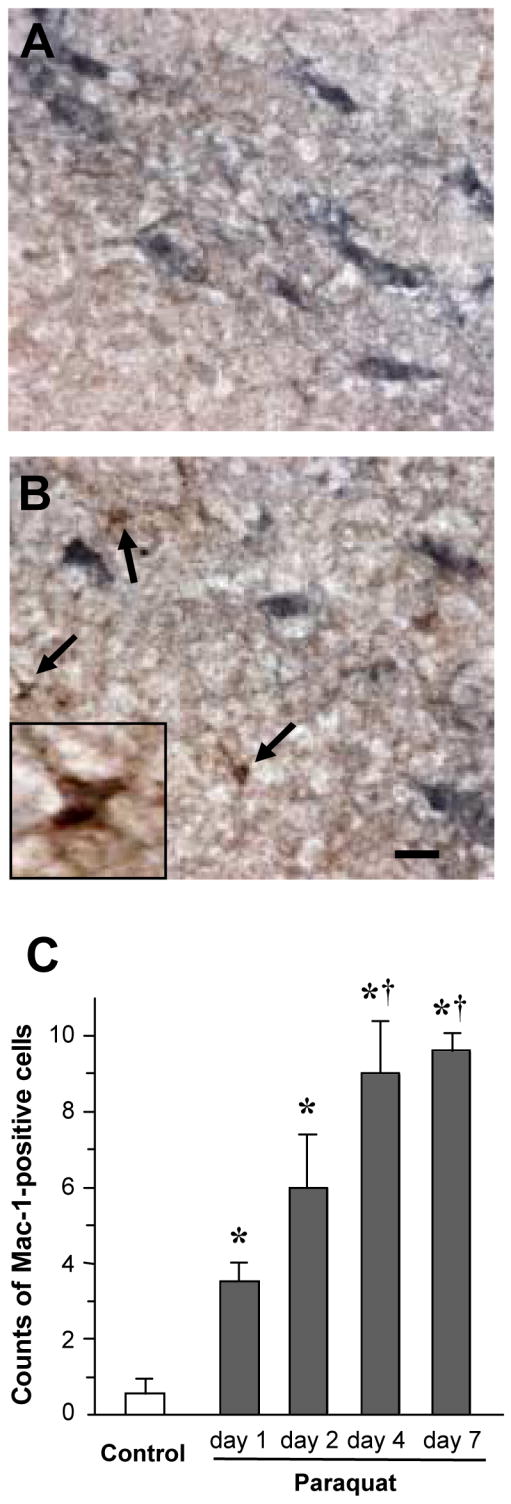
Paraquat exposure causes a time-dependent increase in the number of cells with immunocytochemical and morphological features of activated microglia. Mice received a single intraperitoneal injection of either saline or 10 mg/kg paraquat. A and B, representative images of midbrain sections from mice killed seven days after treatment with saline (A) or paraquat (B). Sections were double-labeled with antibodies against TH (blue) and Mac-1 (brown). Arrows indicate cells with morphological characteristics typical of activated microglia in the substantia nigra pars compacta of the paraquat-treated mouse. One of these cells is shown at higher magnification in the inset of panel B. Scale bar = 20μm. C, the number of Mac-1-positive cells with morphological features of activated microglia was counted in the substantia nigra pars compacta of midbrain sections anterior to the 3rd nerve. Sections were obtained from mice sacrificed at different time points after saline or paraquat treatment. Control values from saline-treated mice were similar at the different time points and were therefore pooled. Values are the mean of bilateral counts ± SEM. P < 0.05 compared with *the saline control value, and †the values at one and two days after paraquat exposure.
Fig. 2.
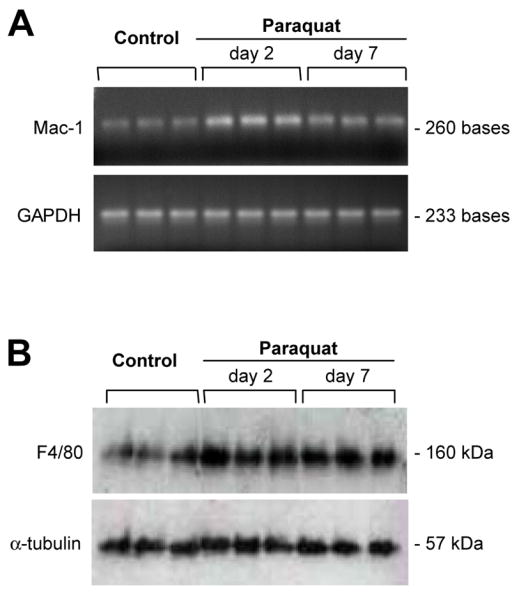
Microglial markers are increased at both the mRNA and protein levels in the ventral mesencephalon of mice injected with paraquat. Mice received a single intraperitoneal injection of either saline or 10 mg/kg paraquat. A, agarose gels showing Mac-1 and GAPDH RT-PCR amplification products in samples from the ventral mesencephalon of control mice injected with saline or mice treated with paraquat and sacrificed two or seven days later. Mac-1 but not GAPDH mRNA expression was markedly enhanced after paraquat exposure. B, homogenates from the ventral mesencephalon of saline- (control) or paraquat-treated mice were separated on a Tris-Glycine (native) gel and probed with antibodies against F4/80 (a microglial marker) or α-tubulin (loading control). Non-reducing conditions for recognition of the F4/80 epitope were used based on manufacturer’s recommendations. F4/80 but not α-tubulin immunoreactivity was increased in all samples (at day two and day seven post exposure) from animals injected with paraquat.
Induction of NADPH-oxidase in paraquat-treated mice
NADPH-oxidase is a complex enzyme composed of three cytosolic and two membrane-bound subunits (Babior, 2004). Upon microglial activation, the cytosolic components migrate to the membrane where they bind to the other subunits to assemble the active oxidase, which becomes capable of catalyzing the one-electron reduction of oxygen to superoxide. Because changes in NADPH-oxidase are not only a reflection of microglial response but also markers of potentially toxic events (e.g. ROS formation), the expression of the NADPH-oxidase main subunit gp91phox was compared in mice injected once with either saline or paraquat. Expression of gp91phox mRNA was low in homogenates from the ventral mesencephalon of control animals, whereas it was significantly enhanced at both two and seven days after paraquat administration (Figure 3A). Staining of midbrain sections with an antibody against gp91phox revealed minimal immunoreactivity in saline-injected mice (Figure 3B). In contrast, gp91phox-immunoreactive cells with morphological features of activated microglia were observed in the substantia nigra pars compacta of mice challenged with paraquat (Figure 3C).
Fig. 3.
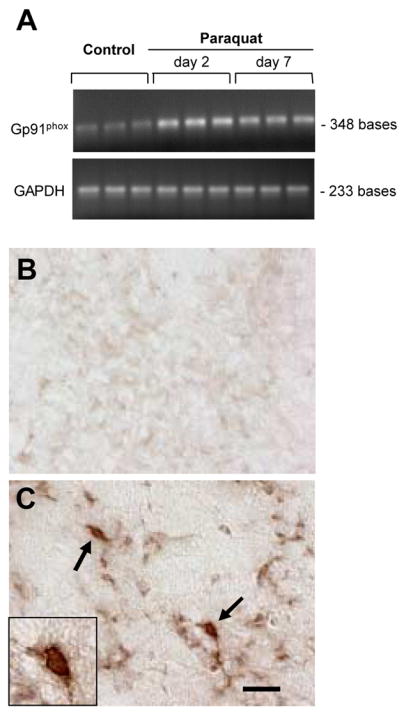
Induction of NADPH-oxidase in paraquat-exposed mice. Mice received a single intraperitoneal injection of either saline or 10 mg/kg paraquat. A, agarose gels showing gp91phox (a subunit of NADPH-oxidase) and GAPDH RT-PCR amplification products in samples from the ventral mesencephalon of control mice injected with saline or mice treated with paraquat and sacrificed two or seven days later. Gp91phox but not GAPDH mRNA expression was increased after paraquat exposure. B and C, representative images of midbrain sections from mice killed seven days after treatment with saline (B) or paraquat (C). Sections were stained with an antibody against gp91phox. Arrows indicate two brown cells with morphological characteristics typical of activated microglia in the substantia nigra pars compacta of the paraquat-treated mouse. One of these cells is shown at higher magnification in the inset of panel C. Scale bar = 20μm.
Pharmacological manipulations of paraquat-induced microglial response and neurodegeneration
Having demonstrated that a single toxicant administration triggers a robust microglial response, we next carried out a series of experiments to test the hypothesis that this initial effect may predispose to damage after subsequent paraquat exposures. Minocycline is a second-generation semisynthetic tetracycline that possesses potent anti-inflammatory properties, including attenuation of microglial response, distinct from its anti-microbial activity (Tikka et al., 2001; Wu et al., 2002). When mice were given minocycline after receiving a single injection of paraquat, the number of cells resembling activated microglia did not rise as it did in animals treated with paraquat alone (Figure 4A). Interestingly, the extent of inhibition of microglial activation was similar regardless of whether paraquat-exposed animals received minocycline for three or seven days (Figure 4A). In the next set of experiments, mice received two injections of paraquat separated by a one-week interval. Two groups of animals were also administered minocycline for three or seven days after the first paraquat exposure. A significant loss of nigral dopaminergic neurons was triggered by the second injection of paraquat in mice that had not received minocycline. In contrast, when animals received both paraquat and minocycline, the number of TH-positive cells remained unchanged as compared to control values (Figure 4B). Similar results were obtained when nigral neurons were counted based on their Nissl staining: treatment with minocycline fully protected against the degeneration of neurons caused by two paraquat injections (Table 1).
Fig. 4.

Effects of minocycline on paraquat-induced microglial activation and neurotoxicity. A, mice received a single injection of either saline or paraquat and were sacrificed at seven days after treatment. Sets of animals were also given minocycline. The first injection of this anti-inflammatory drug was administered 30 min prior to saline or paraquat, with subsequent treatments given every 12 hours for three or seven days. The number of Mac-1-positive cells with morphological features of activated microglia was counted in the substantia nigra pars compacta of midbrain sections anterior to the 3rd nerve. B, mice received two injections of saline or paraquat one week apart and were sacrificed seven days after the second injection. Sets of animals were also administered minocycline for three or seven days following the initial saline or paraquat exposure. The number of TH-positive neurons was estimated in the substantia nigra pars compacta using stereological techniques. C, mice received two injections of saline or paraquat one week apart and were sacrificed at two days after the second injection. Sets of animals were also administered minocycline for three or seven days following the initial saline or paraquat exposure. The number of neurons immunoreactive for 4-HNE (a lipid peroxidation marker) was counted in the substantia nigra pars compacta of midbrain sections at the level of the 3rd nerve. Data in mice treated with saline/minocycline were similar to control values in saline/saline-injected animals and were therefore omitted from the graphs. Values are the mean of bilateral counts ± SEM. * P < 0.05 compared with the other treatment groups.
Table 1.
Minocycline protects against paraquat-induced loss of nigral Nissl-stained neurons.
| Treatment | Number of Nissl-stained neurons |
|---|---|
| Saline | 14,438 ± 250 |
| Minocycline (7 days) | 13,974 ± 332 |
| Paraquat | 11,576 ± 378* |
| Paraquat + Minocyline (3 days) | 14,620 ± 256 |
| Paraquat + Minocyline (7 days) | 15,022 ± 404 |
All mice received two injections of saline or paraquat one week apart and were sacrificed seven days after the second injection. Sets of animals were also administered minocycline for three or seven days following the first saline or paraquat exposure. The number of Nissl-stained neurons was estimated in the substantia nigra pars compacta using stereological techniques. Values are the mean of bilateral counts ± SEM.
P < 0.01 as compared to the other treatment groups.
In a previous study, dopaminergic cell death caused by two weekly administrations of paraquat was associated with oxidative stress in the form of an enhanced number of nigral neurons immunoreactive for 4-HNE, a lipid peroxidation marker (McCormack et al., 2005 and 2006). Maximum increase in 4-HNE-immunoreactive neurons occurred at one, two and four days after the second paraquat treatment (McCormack et al., 2005). We therefore determined if neuroprotection by minocycline was paralleled by changes in oxidative stress. Mice received two injections of paraquat one week apart with or without treatment with minocycline after the first toxicant administration. The number of 4-HNE-positive neurons was then counted in the substantia nigra pars compacta at two days following the second paraquat injection. A significant increase in 4-HNE-positive neurons was caused by paraquat but only in mice that had not received minocycline (Fig. 4C).
If an initial paraquat injection enhances neuronal vulnerability to subsequent toxicant exposure by triggering microglial activation, other pharmacological inducers of CNS inflammation would be expected to cause a similar “priming” effect. Systemic administration of LPS is capable of inducing microglial activation in mice (Kitazawa et al., 2005). We confirmed that a single intraperitoneal injection of LPS caused a robust increase in the number of reactive microglia in the mouse substantia nigra pars compacta that was evident at two days post treatment. Using 4 mg/kg LPS, the number of Mac-1-immunoreactive cells rose to 26.0 ± 2.0 from 0.5 ± 0.4 in saline-injected control mice. Counts of nigral dopaminergic neurons were then compared in animals that received a single injection of paraquat preceded or not by LPS administration (2 or 4 mg/kg). LPS itself had no effect on nigral neuronal counts. Similarly, no change in neuronal number was found between saline-injected control mice and animals treated with paraquat alone. However, if 2 or 4 mg/kg LPS was administered two days prior to the herbicide, a significant loss of dopaminergic neurons occurred, consistent with the ability of LPS to enhance neuronal vulnerability to paraquat toxicity (Figure 5).
Fig. 5.

Pre-treatment with LPS predisposes to paraquat-induced neurodegeneration. Mice received a single injection of 2 or 4 mg/kg LPS and, after a two-day interval, were treated once with saline or paraquat. They were then sacrificed seven days later. The number of TH-positive (A) and Nissl-stained (B) neurons was estimated in the substantia nigra pars compacta using stereological techniques. Values are the mean of bilateral counts ± SEM. * P < 0.05 compared with the values in saline-injected control mice as well as animals treated with LPS or paraquat alone. † P < 0.05 compared with the value in mice treated with both 2 mg/kg LPS and paraquat.
Lack of paraquat-induced neurodegeneration in NADPH-oxidase deficient mice
To further assess the role of microglial activation and NADPH-oxidase-dependent reactions in neurodegeneration caused by paraquat, a final set of experiments was carried out in mutant mice lacking the gp91phox subunit of NADPH-oxidase. The absence of this subunit prevents assembly of the enzyme at the plasma membrane, thus abolishing its catalytic activity (Wu et al., 2003; Babior, 2004). Gp91phox-deficient mice and control wild-type littermates received two injections of either saline or paraquat one week apart. Microscopic examination of midbrain sections stained with an antibody against TH revealed an apparent reduction in density of TH-positive cells in non-mutant animals treated with paraquat versus saline (Figure 6A, B). No differences were evident, however, between gp91phox-deficient mice injected with saline or paraquat (Figure 6C, D). To confirm and quantitate these observations, the number of dopaminergic neurons was estimated stereologically in the substantia nigra pars compacta. Paraquat administration caused a 25% reduction of neurons in wild-type animals. In contrast, gp91phox-deficient mice were completely resistant to neurodegeneration since the number of TH-positive and Nissl-stained neurons was unchanged between mutant animals injected with saline or paraquat (Figure 6E, F).
Fig. 6.
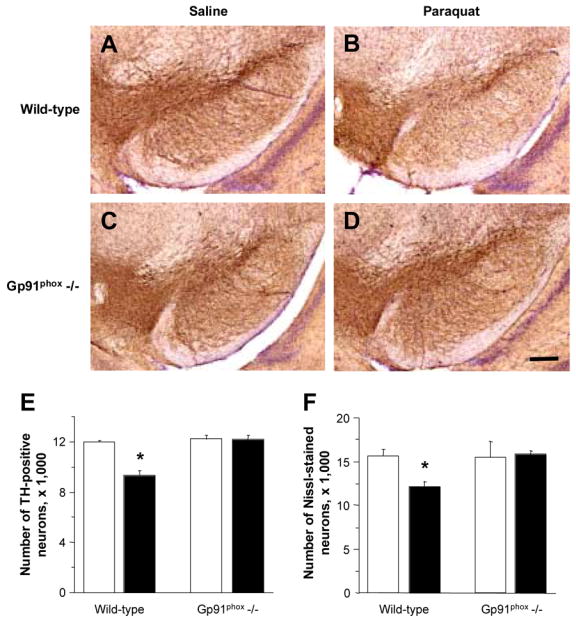
Mice lacking functional NADPH-oxidase are resistant to paraquat-induced neurodegeneration. Mutant mice lacking the gp91phox subunit of NADPH-oxidase and wild-type littermates received two injections of either saline or paraquat one week apart and were sacrificed seven days after the second injection. A–D, representative images of midbrain sections from wild-type littermates (A, saline and B, paraquat) and gp91phox-deficient mice (C, saline and D, paraquat) were labeled with an antibody against TH (brown) and counterstained with cresyl violet (purple). Scale bar = 200μm. E and F, the number of TH-immunoreactive (E) and Nissl-stained (F) neurons was estimated in the substantia nigra pars compacta using stereological techniques. Open (saline) and filled (paraquat) bars represent the mean of bilateral counts ± SEM. * P < 0.01 compared with the corresponding saline control group.
Discussion
Microglial cells have been implicated in the pathogenesis of neurodegeneration in PD and experimental models of PD (Langston et al., 1999; Gao et al., 2003a and b; Sherer et al., 2003; Wu et al., 2003; McGeer and McGeer, 2004; Ling et al., 2004). The present study not only supports this involvement but elucidates a new mechanism by which microglial activation could act as a risk factor for dopaminergic cell death. In the paraquat model, an initial insult does not kill neurons but predisposes dopaminergic cells to degeneration once subjected to a subsequent challenge (McCormack et al., 2005). Our current findings are consistent with the interpretation that microglia play a critical role in this “priming” effect. We observed a time-dependent increase in the number of cells with morphological and immunocytochemical characteristics typical of activated microglia in the substantia nigra pars compacta of mice injected once with paraquat. Substantial activation was achieved at seven days post exposure and coincided with the time when, if a second administration of paraquat was given, it triggered dopaminergic cell death. Additional support in favor of a relationship between microglial activation and enhanced susceptibility to neurodegeneration derives from experiments with minocycline. This anti-inflammatory drug completely blocked the microglial response elicited by an initial paraquat exposure and, as a likely consequence of this inhibition, no dopaminergic cell death was observed even when mice received a second injection with the herbicide. Neuroprotection by minocycline was achieved regardless of whether the drug was administered for three or seven days after the first paraquat treatment. Because the half-life of minocycline is relatively short in mice (a few hours, van Ogtrop et al., 2000), these findings emphasize that protection against neurodegeneration caused by two injections of paraquat one week apart was directly related to the ability of minocycline to counteract events that immediately followed the first toxicant administration. We were also able to show that enhanced vulnerability to paraquat-induced dopaminergic cell death could be achieved through treatment with LPS. When mice were injected with LPS and then exposed to paraquat, this resulted in a clear synergistic toxicity: no degeneration was caused by LPS or paraquat alone, whereas significant nigral cell loss followed the sequential administration of both compounds. Treatment with LPS induced microglial activation and interestingly, in LPS-primed animals, neurodegeneration was observed even after a single paraquat administration. Thus, two different mechanisms of microglial activation, i.e. pre-treatment with either paraquat or LPS, both seem to underlie an enhanced susceptibility of dopaminergic neurons to subsequent toxic insults.
Microglial activation in the paraquat model described in this study was accompanied by an induction of NADPH-oxidase. Indeed, the main membrane-bound subunit of this enzyme complex, gp91phox, was enhanced at both the mRNA and protein levels in the substantia nigra of mice injected once with paraquat. Furthermore, immunoreactivity for gp91phox in paraquat-treated animals stained cells with morphological features of activated microglia. Most importantly, however, data support a role for NADPH-oxidase in paraquat neurotoxicity. In the absence of functional NADPH-oxidase, i.e. in mutant mice lacking gp91phox, sequential paraquat injections failed to cause any significant loss of nigral dopaminergic neurons. Taken together, these data suggest that induction of microglial NADPH-oxidase represents a critical event in the process that sensitizes neurons to toxic injury and ultimately leads to dopaminergic cell degeneration after exposures to paraquat.
At least two mechanisms, not mutually exclusive, could underlie the role of microglia in enhancing the susceptibility of dopaminergic neurons to toxic injury. Microglial response after the initial paraquat administration could contribute to inflict a sublethal injury that, upon a subsequent toxicant exposure, would lead to the death of vulnerable neurons. A second mechanism by which microglial activation could predispose neurons to damage by paraquat relates to the specific chemical properties of this herbicide. A variety of cellular diaphorases are capable of catalyzing the one-electron reduction of paraquat that generates a cation radical (Bus and Gibson, 1984; Clejan and Cederbaum, 1989; Day et al., 1999). The subsequent transfer of one electron from this radical onto molecular oxygen readily forms superoxide anion and regenerates the parent herbicide, thus completing a cycle of redox reactions. Reiteration of this cycle generates considerable amounts of superoxide, which can in turn cause oxidative stress and cytotoxicity. A recent study has shown that paraquat reduction and consequent superoxide formation are markedly promoted by microglia (Bonneh-Barkay et al., 2005), raising the possibility of a relationship between microglial activation, induction of redox cycling and injury of dopaminergic neurons. Under this paradigm, once microglia are activated by an initial exposure to paraquat or through other mechanisms (e.g. LPS administration), a second challenge with the herbicide would trigger a robust redox cycling process and consequently cause enhanced ROS formation and significant neuronal damage. The one-electron reduction of paraquat in the presence of microglia is catalyzed by NADPH-oxidase (Bonneh-Barkay et al., 2005). Therefore, a role of redox cycling in the priming effect of microglial activation is consistent with our findings that NADPH-oxidase is induced after a single paraquat exposure and that mice with defective NADPH-oxidase are resistant to neurodegeneration caused by the herbicide. Redox cycling of paraquat as a consequence of microglial activation is also in line with the effects of minocycline. Once the microglial response elicited by an initial toxicant administration is blocked by treatment with minocycline, the redox cycling of any paraquat that is subsequently administered would be substantially inhibited. This inhibition could in turn prevent oxidative damage (lipid peroxidation) and result in neuroprotection.
In summary, findings of this study describe mechanisms of dopaminergic cell degeneration of potential relevance to human parkinsonism. They emphasize the role of neuroinflammation and pesticide exposure and illustrate how the interplay between toxic events can ultimately affect the neurodegenerative outcome. In our paraquat model, microglial activation and pesticide exposure act synergistically, and the susceptibility of dopaminergic neurons to toxic injury is dramatically exacerbated by an underlying inflammatory process. This priming effect is likely dependent upon the induction of NADPH-oxidase and the toxic consequences of NADPH-oxidase-catalyzed reactions such as the redox cycling of paraquat with molecular oxygen. Besides paraquat and other bipyridyl pesticides (e.g. diquat), a variety of naturally occurring and synthetic molecules (e.g. quinones) share the ability to redox cycle (Frank et al., 1987; O’Brien, 1991). This ability could therefore characterize an entire class of toxic agents of particular relevance to neurodegenerative processes targeting dopaminergic neurons.
Acknowledgments
This work was supported by grants from the National Institute of Environmental Health Sciences (ES10806 and ES12077, DAD) and the Backus Foundation. We thank Ms. Jennifer Atienza for her excellent assistance with the experiments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Babior BM. NADPH oxidase. Curr Opin Immunol. 2004;16:42–7. doi: 10.1016/j.coi.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Bonneh-Barkay D, Reaney SH, Langston WJ, Di Monte DA. Redox cycling of the herbicide paraquat in microglial cultures. Brain Res Mol Brain Res. 2005;134:52–56. doi: 10.1016/j.molbrainres.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Bus JS, Gibson JE. Paraquat: model from oxidant-initiated toxicity. Environ Health Perspect. 1984;55:37–46. doi: 10.1289/ehp.845537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Jacobs E, Schwarzschild MA, McCullogh ML, Calle EE, Thun MJ, Acherio A. Nonsteroidal anti-inflammatory drug use and the risk for Parkinson’s disease. Ann Neurol. 2005;58:963–967. doi: 10.1002/ana.20682. [DOI] [PubMed] [Google Scholar]
- Chen H, Zhang SM, Hernan MA, Schwarzschild MA, Willett WC, Colditz GA, Speizer FE, Ascherio A. Nonsteroidal anti-inflammatory drugs and the risk of Parkinson’s disease. Arch Neurol. 2003;60:1059–1064. doi: 10.1001/archneur.60.8.1059. [DOI] [PubMed] [Google Scholar]
- Clejan L, Cederbaum AI. Synergistic interactions between NADPH-cytochrome P-450 reductase, paraquat, and iron in the generation of active oxygen radicals. Biochem Pharmacol. 1989;38:1779–1786. doi: 10.1016/0006-2952(89)90412-7. [DOI] [PubMed] [Google Scholar]
- Day BJ, Patel M, Calavetta L, Chang LY, Stamler JS. A mechanism of paraquat toxicity involving nitric oxide synthase. Proc Natl Acad Sci. 1999;96:12760–12765. doi: 10.1073/pnas.96.22.12760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Monte DA. The environment and Parkinson’s disease: is the nigrostriatal system preferentially targeted by neurotoxins? Lancet Neurol. 2003;2:531–538. doi: 10.1016/s1474-4422(03)00501-5. [DOI] [PubMed] [Google Scholar]
- Fahn S. Description of Parkinson’s disease as a clinical syndrome. Ann N Y Acad Sci. 2003;991:1–14. doi: 10.1111/j.1749-6632.2003.tb07458.x. [DOI] [PubMed] [Google Scholar]
- Forno LS. Neuropathology of Parkinson’s disease. J Neuropathol Exp Neurol. 1996;55:259–272. doi: 10.1097/00005072-199603000-00001. [DOI] [PubMed] [Google Scholar]
- Frank DM, Arora PK, Blumer JL, Sayre LM. Model study on the bioreduction of paraquat, MPP+, and analogs. Evidence against a “redox cycling” mechanism in MPTP neurotoxicity. Biochem Biophys Res Commun. 1987;147:1095–1104. doi: 10.1016/s0006-291x(87)80183-3. [DOI] [PubMed] [Google Scholar]
- Gao HM, Liu B, Hong JS. Critical role for microglial NADPH oxidase in rotenone-induced degeneration of dopaminergic neurons. J Neurosci. 2003a;23:6181–6187. doi: 10.1523/JNEUROSCI.23-15-06181.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao HM, Liu B, Zhang W, Hong JS. Synergistic dopaminergic neurotoxicity of MPTP and inflammogen lipopolysaccharide: relevance to the etiology of Parkinson’s disease. FASEB J. 2003b;17:1957–1959. doi: 10.1096/fj.03-0203fje. [DOI] [PubMed] [Google Scholar]
- Kitazawa M, Oddo S, Yamasaki TR, Green KN, LaFerla FM. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J Neurosci. 2005;25:8843–8853. doi: 10.1523/JNEUROSCI.2868-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langston JW, Forno LS, Tetrud J, Reeves AG, Kaplan JA, Karluk D. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann Neurol. 1999;46:598–605. doi: 10.1002/1531-8249(199910)46:4<598::aid-ana7>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Ling Z, Chang QA, Tong CW, Leurgans SE, Lipton JW, Carvey PM. Rotenone potentiates dopamine neuron loss in animals exposed to lipopolysaccharide prenatally. Exp Neurol. 2004;190:373–383. doi: 10.1016/j.expneurol.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Manning-Boğ AB, McCormack AL, Li J, Uversky VN, Fink AL, Di Monte DA. The herbicide paraquat causes up-regulation and aggregation of alpha-synuclein in mice: paraquat and alpha-synuclein. J Biol Chem. 2002;277:1641–1644. doi: 10.1074/jbc.C100560200. [DOI] [PubMed] [Google Scholar]
- Mayeux R. Epidemiology of neurodegeneration. Annu Rev Neurosci. 2003;26:81–104. doi: 10.1146/annurev.neuro.26.043002.094919. [DOI] [PubMed] [Google Scholar]
- McCormack AL, Atienza JG, Johnston LC, Andersen JK, Vu S, Di Monte DA. Role of oxidative stress in paraquat-induced dopaminergic cell degeneration. J Neurochem. 2005;93:1030–1037. doi: 10.1111/j.1471-4159.2005.03088.x. [DOI] [PubMed] [Google Scholar]
- McCormack AL, Atienza JG, Langston JW, Di Monte DA. Decreased susceptibility to oxidative stress underlies the resistance of specific dopaminergic cell populations to paraquat-induced degeneration. Neuroscience. 2006;141:929–937. doi: 10.1016/j.neuroscience.2006.03.069. [DOI] [PubMed] [Google Scholar]
- McCormack AL, Thiruchelvam M, Manning-Boğ AB, Thiffault C, Langston JW, Cory-Slechta DA, Di Monte DA. Environmental risk factors and Parkinson’s disease: selective degeneration of nigral dopaminergic neurons caused by the herbicide paraquat. Neurobiol Dis. 2002;10:119–127. doi: 10.1006/nbdi.2002.0507. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. Inflammation and neurodegeneration in Parkinson’s disease. Parkinsonism Relat Disord. 2004;10 (Suppl 1):S3–7. doi: 10.1016/j.parkreldis.2004.01.005. [DOI] [PubMed] [Google Scholar]
- O’Brien PJ. Molecular mechanisms of quinone cytotoxicity. Chem Biol Interact. 1991;80:1–41. doi: 10.1016/0009-2797(91)90029-7. [DOI] [PubMed] [Google Scholar]
- Ouchi Y, Yoshikawa E, Sekine Y, Futatsubashi M, Kanno T, Ogusu T, Torizuka T. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann Neurol. 2005;57:168–175. doi: 10.1002/ana.20338. [DOI] [PubMed] [Google Scholar]
- Peng J, Mao XO, Stevenson FF, Hsu M, Andersen JK. The herbicide paraquat induces dopaminergic nigral apoptosis through sustained activation of the JNK pathway. J Biol Chem. 2004;279:32626–32632. doi: 10.1074/jbc.M404596200. [DOI] [PubMed] [Google Scholar]
- Saint-Pierre M, Tremblay ME, Sik A, Gross RE, Cicchetti F. Temporal effects of paraquat/maneb on microglial activation and dopamine neuronal loss in older rats. J Neurochem. 2006;98:760–772. doi: 10.1111/j.1471-4159.2006.03923.x. [DOI] [PubMed] [Google Scholar]
- Sherer TB, Betarbet R, Kim JH, Greenamyre JT. Selective microglial activation in the rat rotenone model of Parkinson’s disease. Neurosci Lett. 2003;341:87–90. doi: 10.1016/s0304-3940(03)00172-1. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci U S A. 1998;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiruchelvam M, McCormack A, Richfield EK, Baggs RB, Tank AW, Di Monte DA, Cory-Slechta DA. Age-related irreversible progressive nigrostriatal dopaminergic neurotoxicity in the paraquat and maneb model of the Parkinson’s disease phenotype. Eur J Neurosci. 2003;18:589–600. doi: 10.1046/j.1460-9568.2003.02781.x. [DOI] [PubMed] [Google Scholar]
- Tikka T, Fiebich BL, Goldsteins G, Keinanen R, Koistinaho J. Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J Neurosci. 2001;21:2580–2588. doi: 10.1523/JNEUROSCI.21-08-02580.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Den Eeden SK, Tanner CM, Bernstein AL, Fross RD, Leimpeter A, Bloch DA, Nelson LM. Incidence of Parkinson’s disease: variation by age, gender, and race/ethnicity. Am J Epidemiol. 2003;157:1015–1022. doi: 10.1093/aje/kwg068. [DOI] [PubMed] [Google Scholar]
- van Ogtrop ML, Andes D, Stamstad TJ, Conklin B, Weiss WJ, Craig WA, Vesga O. In vivo pharmacodynamic activities of two glycylcyclines (GAR-936 and WAY 152,288) against various gram-positive and gram-negative bacteria. Antimicrob Agents Chemother. 2000;44:943–949. doi: 10.1128/aac.44.4.943-949.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, Choi DK, Ischiropoulos H, Przedborski S. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. J Neurosci. 2002;22:1763–1771. doi: 10.1523/JNEUROSCI.22-05-01763.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DC, Teismann P, Tieu K, Vila M, Jackson-Lewis V, Ischiropoulos H, Przedborski S. NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2003;100:6145–6150. doi: 10.1073/pnas.0937239100. [DOI] [PMC free article] [PubMed] [Google Scholar]