

Summary
Phosphodiesterases (PDEs) are key enzymes that control the cellular concentrations of the second messengers cAMP and cGMP. The mechanism for selective recognition of substrates cAMP and cGMP by individual PDE families remains a puzzle. To understand the mechanism for substrate recognition by PDE enzymes, the crystal structure of the catalytic domain of an inactive D201N mutant of PDE4D2 in complex with substrate cAMP has been determined at 1.56 Å resolution. The structure shows that Gln369 forms only one hydrogen bond with the adenine of cAMP. This finding provides experimental evidence against the hypothesis of two hydrogen bonds between the invariant glutamine and the substrate cAMP in PDE4, and thus suggests that the widely circulated “glutamine switch” model is unlikely the mechanism for substrate recognition by PDEs. A structure comparison between PDE4D2-cAMP and PDE10A2-cAMP reveals an anti configuration of cAMP in PDE4D2 but syn in PDE10A2, in addition to different contact patterns of cAMP in these two structures. These observations imply that individual PDE families have their characteristic mechanisms for substrate recognition.
Keywords: PDE4, cAMP/cGMP, substrate specificity
Cyclic nucleotide phosphodiesterases (PDEs) regulate the cellular concentrations of the second messengers cAMP and cGMP and play important roles in many biological processes (1-7). The human genome encodes twenty-one PDE genes that are categorized into eleven families. Alternative mRNA splicing of the PDE genes produces about one hundred proteins (1,3). Selective inhibitors against individual PDE families have been widely studied as therapeutics for treatment of various human diseases (8-15). For example, the PDE5 selective inhibitor sildenafil has been approved for treatment of male erectile dysfunction and pulmonary hypertension (16,17).
Individual PDE families have different specificities in hydrolysis of substrates cAMP and cGMP. Thus, PDE families of 4, 7, and 8 prefer to hydrolyze cAMP while PDE5, 6, and 9 are cGMP specific. The remaining PDE families 1, 2, 3, 10, and 11 utilize both cAMP and cGMP as substrates, but have slightly different catalytic efficacies. Understanding substrate specificity is important not only for illustration of the catalytic mechanism, but also for development of PDE inhibitors for treatment of human diseases. On the basis of the crystal structures of PDE4 and PDE5 in complex with products AMP and GMP, a “glutamine switch” mechanism was proposed for substrate specificity (18) and is now widely circulated in the field of PDE research. However, a couple of lines of evidence suggest that the invariant glutamine may not be the key for the substrate recognition. First, the invariant glutamine forms a hydrogen bond with a tyrosine in the crystal structures of dual-substrate specific PDE2 (19) and PDE10 (20) and is thus unlikely to freely switch its conformation for recognition of the different substrates. Second, the recent PDE10-substrate structures show that cAMP and cGMP have the same syn configuration, but different orientations and interactions (20), suggesting that cAMP and cGMP are recognized by the different contact patterns in PDE10. Thus, it is of interest to know if cAMP in PDE4 has the same contact pattern and configuration as that in PDE10 and whether PDE4 and PDE10 have a similar mechanism of substrate recognition. Also, it is not known if product AMP simulates binding of substrate cAMP in PDE4 as what the glutamine switch mechanism suggested. To address these questions, we constructed an inactive D201N mutant of PDE4D2 (D201N) and determined the crystal structure of the catalytic domain of the mutant in complex with substrate cAMP.
Protein expression, crystallization and structure determination
The cDNA for expression of the catalytic domain of the wild type PDE4D2 was purchased from ATCC (American Type Culture Collection, ATCC #5654749) and subcloned into pET15b, as described previously (21). The plasmid pET-PDE4D for expression of the wild type PDE4D2 (residues 86-413) was mutated to pET-D201N with the QuickChange site-directed mutagenesis kit (Stratagene) using the PCR primers of 5′-GCCAGTGCAATACATAATGTAGATCATCCTGG-3′ and 5′-CCAGGATGATCTACATTATGTATTGCACTGGC-3′. Protein overexpression and purification of the D201N mutant followed the protocol for the wild type PDE4D2 (22). Briefly, the plasmid pET-D201N was introduced into E. coli BL21-CodonPlus (Invitrogen). The E coli cell was grown in LB medium at 37°C to absorption A600 = 0.7 and then 0.1 mM isopropyl β-Dthiogalactopyranoside was added to induce overexpression at 15°C for 40 hours. To purify the recombinant protein of D201N, the cell lysate was passed through a Ni-NTA affinity (Qiagen) column. The eluted protein was cleaved with thrombin and further purified by passing through the columns of Q-sepharose (Amersham Biosciences) and Sephacryl S300 (Amersham Biosciences). A typical purification yielded about 50 mg PDE4D2 D201N mutant from 2 liters of cell culture. The purified D201N protein showed a single band in SDS-PAGE and was estimated to be > 95% pure.
The 30mg/ml unliganded D201N mutant was stored in a buffer of 20 mM Tris.base, pH 7.5, 50 mM NaCl, 1 mM β-mercaptoethanol, 1 mM EDTA and crystallized at 4°C by hanging drop against a well buffer of 0.1 M HEPES pH 7.5, 0.1 M MgCl2, 12% PEG3350, 30% ethylene glycol, and 10% isopropanol. The D201N-cAMP complex was prepared by soaking the unliganded crystals in the crystallization buffer plus 20 mM cAMP for one hour and then flash-frozen in liquid nitrogen. Diffraction data were collected on beamline X29 at Brookhaven National Laboratory and processed by program HKL (Table 1) (23). The D201N mutant structure was solved by molecular replacement, using the wild type PDE4D2 as the initial model. The atomic model was rebuilt by program O (24) and refined by program CNS (Table 1) (25).
Table 1.
Statistics on diffraction data and structure refinement
| Data collection | D201N-cAMP |
| Space group | P212121 |
| Unit cell (a, b, c, Å) | 57.8, 81.0, 163.6 |
| Resolution (Å) | 30-1.56 |
| Total measurements | 1,429,939 |
| Unique reflections | 108,849 |
| Completeness (%) | 98.9 (90.0)* |
| Average I/σ | 10.5 (2.4)* |
| Rmerge | 0.072 (0.43)* |
| Structure Refinement | |
| R-factor/R-free | 0.212/0.231(10%)‡ |
| Reflections | 104,214 |
| RMS deviation for | |
| Bond (Å) | 0.0043 |
| Angle | 1.07° |
| Average B-factor (Å2) | |
| Protein | 22.4 (5280)§ |
| cAMP | 23.0 |
| Waters | 31.5 (528)§ |
| Zn | 22.3 (2)§ |
The numbers in parentheses are those at the highest resolution shell.
The percentage of reflections omitted for calculation of R-free.
The number of atoms in the crystallographic asymmetric unit.
Enzymatic activity and structure of the D201N mutant
The D201N mutant has a KM of 1.8 μM, comparable to that of the wild type PDE4D2, but its kcat of 6.8 × 10−4 s−1 is 4 magnitudes lower than 3.9 s−1 of the wild type enzyme (22). The structure of the PDE4D2 D201N mutant contains sixteen α-helices (Fig. 1) that are folded into an entity as other PDE families (4). Superposition of the D201N mutant over the wild type PDE4D2 yielded a root-mean-squared deviation (RMSD) of 0.47 Å for Cα atoms in the entire catalytic domain (residues 87-410), indicating no significant conformational changes caused by the mutation. In addition, the graphic display of the superimposed structures shows no substantial conformational changes of the active site residues upon cAMP binding (Fig 1B). However, the D201N mutation causes loss of the magnesium ion binding and thus loss of catalytic activity. This loss is understandable because Asp201 is the only residue that the magnesium ion binds in the wild type PDE4D2.
Fig. 1.
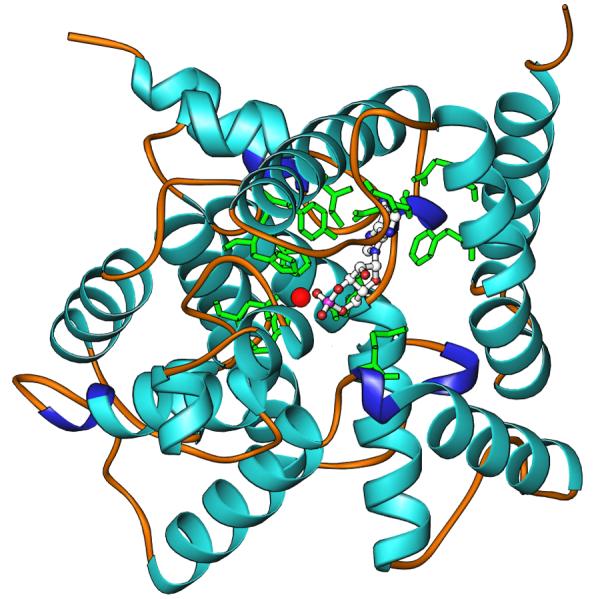
Structure of the PDE4D D201N catalytic domain. (A) Ribbons diagram. The large red ball represents a zinc ion. The green sticks are the residues at the active site of the PDE4D2 D201N mutant. (B) Superposition of the active site residues of the wild type PDE4D2 (blue sticks) over those of the D201N mutant. The zinc ion has the same position in the wild type and D201N structures. The magnesium ion is from the wild type PDE4D2.
Cyclic AMP binds PDE4D2 in anti configuration
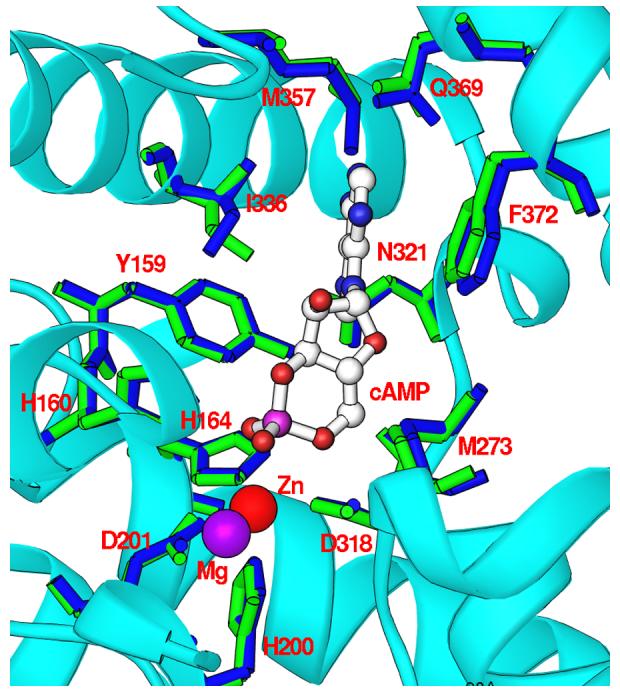
The adenine of cAMP binds to the substrate recognition pocket or so called S-pocket (20). The cyclic phosphate group of cAMP orients toward the metal binding pocket, but neither directly contacts the zinc ion in this structure nor the second metal site generated from the structural superposition (Fig. 2). Substrate cAMP forms only two hydrogen bonds with PDE4D2, which occur between N1 of the adenine of cAMP and Ne2 of Gln369 and between a phosphate oxygen of cAMP and Ne2 of His160 (Fig. 2). The D201N-cAMP structure shows that N6 of adenine of cAMP is 3.78 Å away from Oe1 of Gln369. This distance represents a weak van der Waals' interaction and is much longer than the ideal length of 2.9 Å for a hydrogen bond. Four water molecules bind the phosphate oxygens and ribose O2' of cAMP. For the hydrophobic interactions, adenine of cAMP stacks against the highly conserved Phe372 in the PDE families (except tryptophan in PDE11) on one side and contacts Ile336 and Phe340 on another side. In addition, cAMP forms van der Waals' interactions with residues Tyr159, Met273, Leu319, Asn321, Thr333, and Met357.
Fig. 2.
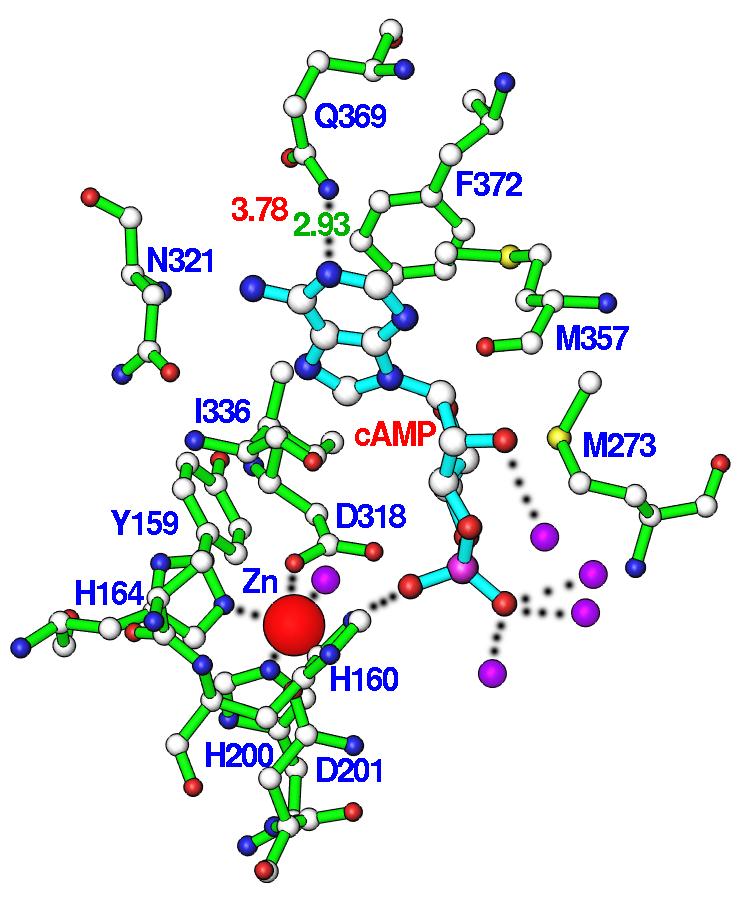
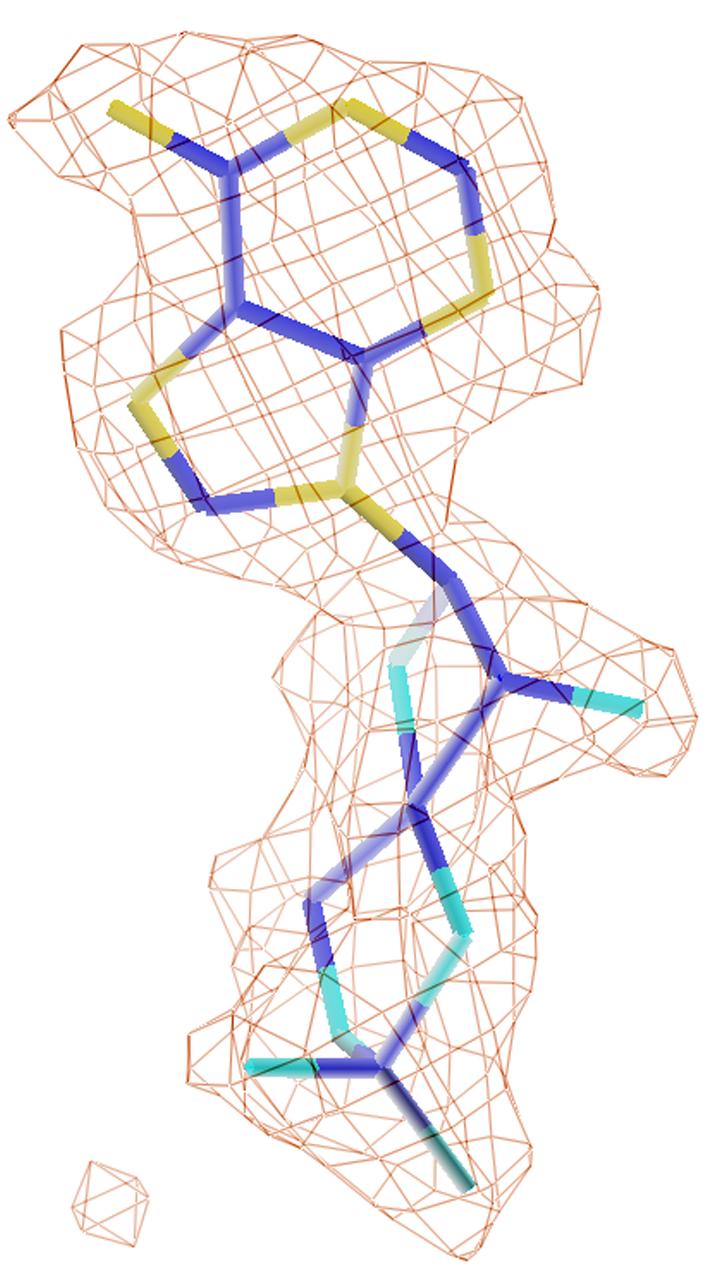
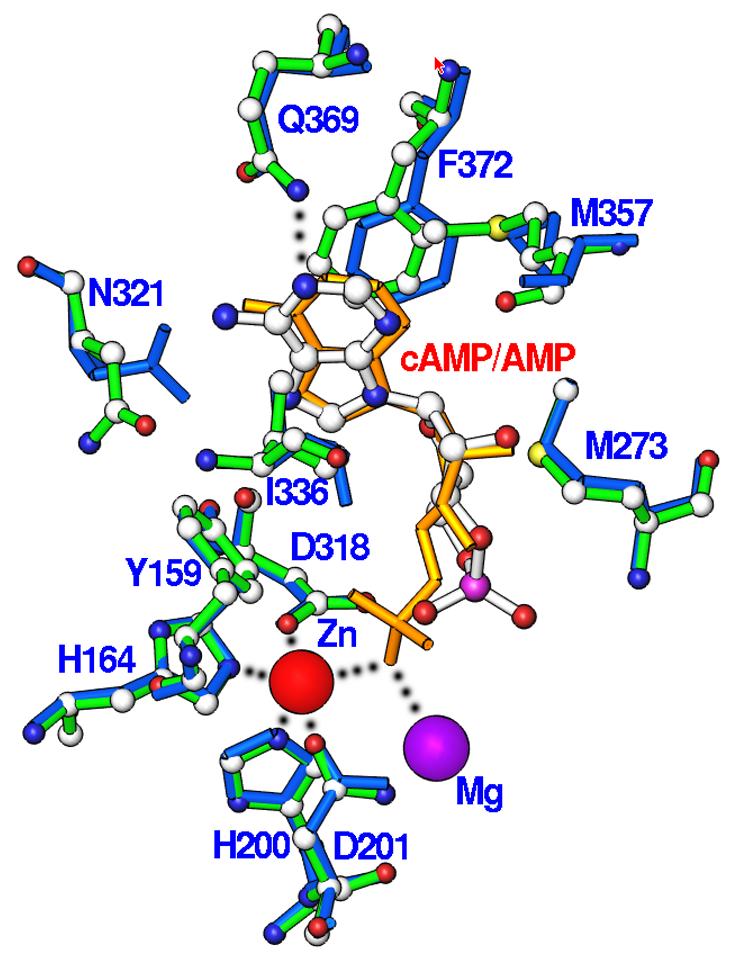
Nucleotide binding. (A) Interactions of cAMP with residues of PDE4D2. Dotted lines represent hydrogen bonds or metal coordinations. Isolated purple balls are bound water molecules. (B) The electron density for cAMP, which was calculated from the omitted structure and contoured at 3 sigmas. (C) Structural superposition between PDE4D2-cAMP and PDE4D2-AMP. The color codes for bonds are green for residues of PDE4D2-cAMP, white for cAMP, gold for AMP, and blue for residues of PDE4D2-AMP.
The bound cAMP has an anti configuration for the adenine and a 3'-endo puckering for the ribose (Fig. 2a), thus suggesting that the anti conformer of cAMP is the genuine substrate of PDE4, although cAMP has a syn/anti equilibrium of 30:70 in free solution (26). Our observation is consistent with the topological mapping experiments, in which the anti conformer of cAMP was the preferable substrate for PDE4 (27). However, the topological mapping predicted a hydrogen bond of the invariant glutamine with N7 of cAMP (27), in contrast to N1 in our crystal structure.
Product AMP does not simulate binding of substrate cAMP
The superposition of PDE4D2-cAMP over PDE4D-AMP and PDE4B-AMP (18,21,28) yielded the small RMSDs of 0.26-0.73 Å, indicating overall similarity of the structures. In addition, the structural comparison shows some features shared by AMP and cAMP. For example, both substrate and product have the same anti configuration and interact with a similar set of amino acids. However, the hydrogen bonding patterns of cAMP and AMP in the structures of PDE4D2-cAMP and PDE4-AMP are totally different (Fig. 2c). First, a phosphate oxygen of AMP bridges two divalent metal ions, but cAMP does not directly contact the metal ions. Second, the side chain of Asn321 in the PDE4-product complexes (18,21,28) changes its conformation to form two hydrogen bonds with N6 and N7 of AMP. In comparison, Asn321 in the PDE4D2-cAMP complex retains its conformation in the unliganded state and does not form hydrogen bonds with cAMP. Finally, two hydrogen bonds are formed between N1 and N6 of AMP and Ne2 and Oe1 of Gln369 in the PDE4-product complexes (18,21,28). In contrast, Gln369 in the PDE4D2-cAMP structure forms only one hydrogen bond with N1 of cAMP, and the distance of 3.78 Å between Oe1 of Gln369 and N6 of cAMP indicates their weak van der Waals' interaction. Therefore, the structural comparison suggests that the product does not simulate substrate binding in PDE4.
For the same chemical formulae, the nucleoside portions of the products AMP and GMP have been assumed to have the same configuration and interaction as those of the substrates cAMP and cGMP. This belief has led to the widely circulated “glutamine switch” mechanism for substrate specificity of PDEs (18). On the basis of the crystal structures of PDE4-AMP and PDE5-GMP, the invariant glutamine (Gln369 in PDE4D2) was assumed to have two hydrogen bonds with cAMP but only one with cGMP in the cAMP-specific PDE families and vice versa in the cGMP-specific families. For dual-substrate specific PDE families, the invariant glutamine is expected to switch its side chain conformation to form two hydrogen bonds with cAMP and cGMP (18).
Several lines of evidence suggest that the “glutamine switch” is unlikely the mechanism for the recognition of substrates. First, a hydrogen bond between the invariant glutamine and a scaffolding tyrosine in the structures of dual substrate specific PDE2 and PDE10 (19,20) will block free rotation of the glutamine side chain and thus prohibit gain of two hydrogen bonds with cAMP or cGMP. In fact, the structures of PDE10A2 in complex with both substrates show that the invariant Gln726 forms two hydrogen bonds with cAMP, but one with cGMP (20). A structural model shows that Gln726 will retain one hydrogen bond with cGMP even after switching the side chain conformation of the invariant glutamine (20). Secondly, the Q817A mutation in PDE5A1 reduced the cGMP affinity by 60-fold, but did not significantly impact cAMP affinity, suggesting that the glutamine is not a key residue to differentiate substrates (29). Finally, only one hydrogen bond between Gln369 and cAMP in our D201N-cAMP structure provides experimental evidence against the glutamine switch mechanism that hypothesizes two hydrogen bonds between the glutamine and cAMP in the cAMP-specific PDE families (18). Thus, the invariant glutamine appears to be important for substrate affinity, but less critical for differentiation of substrates. Most likely, the substrate differentiation is achieved through variation of amino acids in the substrate recognition pocket (20).
Different mechanisms for substrate recognition in PDE4 and PDE10
The binding of cAMP in PDE4D2 is significantly different from that in PDE10A2 (Fig. 3). First, the adenine rings of cAMP in the two structures have about 180o orientation difference and show positional difference of about 2 Å between their N9 atoms, although both adenines stack against a phenylalanine. Second, cAMP has an anti configuration in PDE4D2, but both cAMP and cGMP in PDE10A2 have a syn configuration (20). Finally, cAMP forms one hydrogen bond with Gln369 of PDE4D2, but two with the invariant glutamine in PDE10A2. These differences suggest that individual PDE families possess characteristic recognition mechanisms for substrate specificity.
Fig. 3.
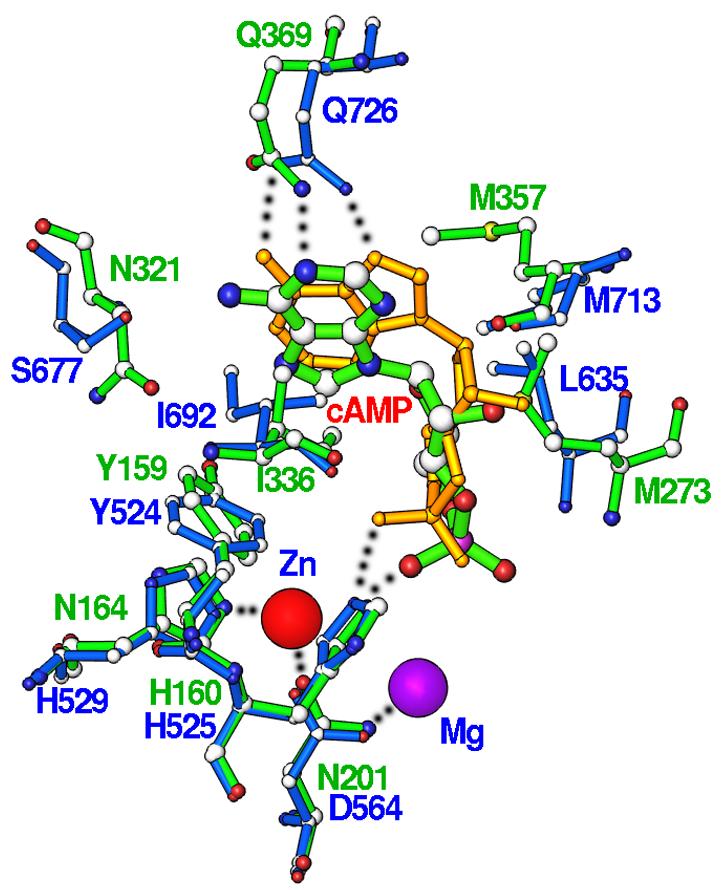
Structural superposition of PDE4D2-cAMP (green bonds and labels) over PDE10A2-cAMP (blue bonds and labels, and gold for cAMP). The bound cAMP has the anti configuration in PDE4D2 and syn in PDE10A2.
In the dual-substrate specific PDE10, the substrates are distinguished through different orientations and interactions of cAMP and cGMP (20). One extra hydrogen bond of cAMP with Gln726 of PDE10A2 in comparison to cGMP may contribute to the 70-fold affinity difference, as shown by KM of 56 nM for cAMP and 4.4 μM for cGMP (20). However, explanation for the preference of cAMP over cGMP by PDE4 is not obvious. To observe the interaction of cGMP with PDE4, we collected a data set from a crystal of the PDE4D2 D201N mutant, which had been soaked in 30 mM cGMP and the same buffer for preparation of the D201N-cAMP complex. However, no cGMP binding was observed in the structure. This result is consistent with the enzymatic data that PDE4 has a KM of 1 mM for cGMP, in comparison to 1.5 μM for cAMP (22). If the position and conformation of cAMP is taken as the model of cGMP, N2 of cGMP will be about 2.5 Å away from Cε of Met357. This distance is too close for the van der Waals' interaction and will push the guanine away so as to eliminate the hydrogen bond between the invariant glutamine and cGMP. Thus, the substrate specificity in PDE4 appears to be the consequence of a poor fit of cGMP to the active site.
In general, we believe that the substrate specificity is determined by multiple elements and individual PDE families have characteristic mechanisms. The amino acid variation in the nucleotide binding pocket across PDE families and the shape/size of the pocket are two factors determining the binding affinity. Substrates cAMP and cGMP may take similar or different orientations to fit the pocket with different affinity in dual substrate specific PDE families. Meanwhile, PDE families with unique substrate specificity may have a perfect match with one substrate such as cAMP in PDE4, but unfavorable interactions with another substrate.
Acknowledgements
We thank beamline X29 at NSLS for collection of diffraction data, Dr. Howard Fried for proofreading of the manuscript and Xiuyan Qin for assistance on cell culture and protein expression. This work was supported by NIH GM59791 to HK.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Accession codes
The coordinates and structural factors have been deposited into the Protein Data Bank with accession code of 2PW3.
References
- 1.Mehats C, Andersen CB, Filopanti M, Jin SL, Conti M. Cyclic nucleotide phosphodiesterases and their role in endocrine cell signaling. Trends in Endocrinol. Metabolism. 2002;13:29–35. doi: 10.1016/s1043-2760(01)00523-9. [DOI] [PubMed] [Google Scholar]
- 2.Houslay MD, Adams DR. PDE4 cAMP phosphodiesterases: modular enzymes that orchestrate signalling cross-talk, desensitization and compartmentalization. Biochem. J. 2003;370:1–18. doi: 10.1042/BJ20021698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bender AT, Beavo JA. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol. Rev. 2006;58:488–520. doi: 10.1124/pr.58.3.5. [DOI] [PubMed] [Google Scholar]
- 4.Ke H, Wang H. Crystal structures of phosphodiesterases and implications on substrate specificity and inhibitor selectivity. Curr. Top. Med. Chem. 2007;7:391–403. doi: 10.2174/156802607779941242. [DOI] [PubMed] [Google Scholar]
- 5.Omori K, Kotera J. Overview of PDEs and their regulation. Circ. Res. 2007;100:309–327. doi: 10.1161/01.RES.0000256354.95791.f1. [DOI] [PubMed] [Google Scholar]
- 6.Conti M, Beavo J. Biochemistry and Physiology of Cyclic Nucleotide Phosphodiesterases: Essential Components in Cyclic Nucleotide Signaling. Annu Rev Biochem. 2007 doi: 10.1146/annurev.biochem.76.060305.150444. Published online March 21, 2007. [DOI] [PubMed] [Google Scholar]
- 7.Lugnier C. Cyclic nucleotide phosphodiesterase (PDE) superfamily: a new target for the development of specific therapeutic agents. Pharmacol Ther. 2006;109:366–398. doi: 10.1016/j.pharmthera.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 8.Truss MC, Stief CG, Uckert S, Becker AJ, Wafer J, Schultheiss D, Jonas U. Phosphodiesterase 1 inhibition in the treatment of lower ruinary tract dysfunction: from bench to bedside. World J. Urol. 2001;19:344–350. doi: 10.1007/s003450100221. [DOI] [PubMed] [Google Scholar]
- 9.Liu Y, Shakur Y, Yoshitake M, Kambayashi JJ. Cilostazol (pletal): a dual inhibitor of cyclic nucleotide phosphodiesterase type 3 and adenosine uptake. Cardiovascular Drug Rev. 2001;19:369–386. doi: 10.1111/j.1527-3466.2001.tb00076.x. [DOI] [PubMed] [Google Scholar]
- 10.Lipworth BJ. Phosphodiesterase-4 inhibitors for asthma and chronic obstructive pulmonary disease. Lancet. 2005;365:167–175. doi: 10.1016/S0140-6736(05)17708-3. [DOI] [PubMed] [Google Scholar]
- 11.Lerner A, Epstein PM. Cyclic nucleotide phosphodiesterases as targets for treatment of haematological malignancies. Biochem. J. 2006;393:21–41. doi: 10.1042/BJ20051368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Menniti FS, Faraci WS, Schmidt CJ. Phosphodiesterases in the CNS: targets for drug development. Nat. Rev. Drug Discov. 2006;5:660–670. doi: 10.1038/nrd2058. [DOI] [PubMed] [Google Scholar]
- 13.Giembycz MA, Smith SJ. Phosphodiesterase 7A: a new therapeutic target for alleviating chronic inflammation? Curr. Pharm. Des. 2006;12:3207–3220. doi: 10.2174/138161206778194123. [DOI] [PubMed] [Google Scholar]
- 14.Reffelmann T, Kloner RA. Cardiovascular effects of phosphodiesterase 5 inhibitors. Curr Pharm Des. 2006;12:3485–3494. doi: 10.2174/138161206778343073. [DOI] [PubMed] [Google Scholar]
- 15.Palmer MJ, Bell AS, Fox DN, Brown DG. Design of second generation phosphodiesterase 5 inhibitors. Curr. Top. Med. Chem. 2007;7:405–419. doi: 10.2174/156802607779941288. [DOI] [PubMed] [Google Scholar]
- 16.Corbin JD, Francis SH. Pharmacology of phosphodiesterase-5 inhibitors. Int. J. Clin. Pract. 2002;56:453–459. [PubMed] [Google Scholar]
- 17.Galie N, Ghofrani HA, Torbicki A, Barst RJ, Rubin LJ, Badesch D, Fleming T, Parpia T, Burgess G, Branzi A, Grimminger F, Kurzyna M, Simonneau G. Sildenafil use in pulmonary arterial hypertension (SUPER) study group. N. Engl. J. Med. 2005;353:2148–2157. doi: 10.1056/NEJMoa050010. [DOI] [PubMed] [Google Scholar]
- 18.Zhang KY, Card GL, Suzuki Y, Artis DR, Fong D, Gillette S, Hsieh D, Neiman J, West BL, Zhang C, Milbum MV, Kim SH, Schlessinger J, Bollag G. A glutamine switch mechanism for nucleotide selectivity by phosphodiesterases. Mol. Cell. 2004;15:279–286. doi: 10.1016/j.molcel.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 19.Iffland A, Kohls D, Low S, Luan J, Zhang Y, Kothe M, Cao Q, Kamath AV, Ding YH, Ellenberger T. Structural determinants for inhibitor specificity and selectivity in PDE2A using the wheat germ in vitro translation system. Biochemistry. 2005;44:8312–8325. doi: 10.1021/bi047313h. [DOI] [PubMed] [Google Scholar]
- 20.Wang H, Liu Y, Hou J, Zheng M, Robinson H, Ke H. Structural insight into substrate specificity of phosphodiesterase 10. Proc. Natl. Acad. Sci., USA. 2007;104:5782–5787. doi: 10.1073/pnas.0700279104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huai Q, Colicelli J, Ke H. The crystal structure of AMP-bound PDE4 suggests a mechanism for phosphodiesterase catalysis. Biochemistry. 2003;42:13220–13226. doi: 10.1021/bi034653e. [DOI] [PubMed] [Google Scholar]
- 22.Wang H, Liu Y, Chen Y, Robinson H, Ke H. Multiple elements jointly determine inhibitor selectivity of cyclic nucleotide phosphodiesterases 4 and 7. J. Biol. Chem. 2005;280:30949–30955. doi: 10.1074/jbc.M504398200. [DOI] [PubMed] [Google Scholar]
- 23.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 24.Jones TA, Zou J-Y, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Cryst. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 25.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR system: A new software suite for macromolecular structure determination. Acta Cryst. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 26.Yathindra N, Sunderalingam M. Conformations of cyclic 3',5'-nucleotides. Effect of the base on the Syn-Anti conformer distribution. Biochem. Biophys. Res. Commun. 1974;56:119–126. doi: 10.1016/s0006-291x(74)80323-2. [DOI] [PubMed] [Google Scholar]
- 27.Butt E, Beltman J, Becker DE, Jensen GS, Rybalkin SD, Jastorff B, Beavo JA. Characterization of cyclic nucleotide phosphodiesterases with cyclic AMP analogs: topology of the catalytic sites and comparison with other cyclic AMP-binding proteins. Mol. Pharmacol. 1995;47:340–347. [PubMed] [Google Scholar]
- 28.Xu RX, Rocque WJ, Lambert MH, Vanderwall DE, Luther MA, Nolte RT. Crystal structures of the catalytic domain of phosphodiesterase 4B complexed with AMP, 8-Br-AMP, and rolipram. J. Mol. Biol. 2004;337:355–365. doi: 10.1016/j.jmb.2004.01.040. [DOI] [PubMed] [Google Scholar]
- 29.Zoraghi R, Corbin JD, Francis SH. Phosphodiesterase-5 Gln817 is critical for cGMP, vardenafil, or sildenafil affinity; Its orientation impacts cGMP but not cAMP affinity. J. Biol. Chem. 2006;281:5553–5558. doi: 10.1074/jbc.M510372200. [DOI] [PubMed] [Google Scholar]