

Synopsis
A serious disorder with characteristic microvascular thrombosis involving the brain and other organs, TTP typically presents with thrombocytopenia, hemolysis with schistocytes on blood smears and mental changes or seizures and may rapidly progresses to a fatal end if the patients are not immediately treated with plasma. Recent advances have shown that TTP is caused by deficiency of a circulating, von Willebrand factor cleaving metalloprotease, ADAMTS13. This new knowledge will provide clues to improve the diagnosis and management of this intriguing disease.
Keywords: Thrombotic thrombocytopenic purpura, Von Willebrand factor, ADAMTS13, Metalloprotease, Microangiopathic hemolysis, Thrombocytopenia
First reported in 1924 by Moschcowitz [1], thrombotic thrombocytopenic purpura (TTP) usually refers to the disorder of thrombocytopenia, hemolysis with schistocytes on blood smears, and neurologic abnormalities such as headache, confusion, focal deficits, seizures or coma. These manifestations are due to widespread microvascular thrombosis involving the capillaries and arterioles of the brain and other organs. Thrombocytopenia results from consumption of platelets, while erythrocyte fragmentation and hemolysis may be due to mechanical injury as the red cells encounter the intravascular thrombi.
TTP typically affects previously healthy individuals in their adolescent or adult ages and almost invariably undertakes a rapid course of deterioration and death unless plasma infusion or exchange therapy is immediately instituted. Plasma therapy, whose effectiveness in the treatment of TTP was discovered through astute clinical observations and has been validated in randomized control investigation [2], is technically demanding, frequently complicated with transfusion-related complications, and quite costly. When plasma exchange does not lead to remission, clinicians often resort to immunosuppressive drugs or splenectomy, although the effectiveness of these modalities remains unclear.
Advances in recent years have transformed TTP from a complete mystery to a relatively explicable disease. This article reviews the recent advances and explains how the knowledge of its molecular defect has changed our diagnostic and therapeutic approaches to TTP. It also discusses current controversies and future directions of investigation.
Is TTP a syndrome or a disease?
There is controversy whether TTP is a syndrome or a disease. The constellation of thrombocytopenia and microangiopathic hemolysis, often accompanied by acute renal failure (the hemolytic uremic syndrome, or HUS), occurs in up to 15% of the patients after infection with shiga toxin producing E. coli or Sh. dysenteriae. Previously, typical or post-diarrhea HUS was distinguished from TTP primarily based on the presence of a diarrhea prodrome, severe renal involvement and absent neurologic complications. However, the basis of this distinction is not entirely reliable. In some patients with documented E. coli O157:H7 infection, the prodrome of hemorrhagic diarrhea may be absent or not obvious and renal function impairment may be mild. Such cases have been reported under the diagnosis of TTP. Conversely, neurologic complications such as mental changes, strokes and seizures affect approximately 20% of patient with the HUS [3], raising doubt of whether TTP and HUS were indeed different disorders.
The syndrome of thrombocytopenia and microangiopathic hemolysis, with variable combination of neurologic and renal manifestations, may also occur in association with conditions such as lupus or related autoimmune connective tissue disorders, various drugs or infections, metastasizing cancers and bone marrow transplantation. Occasionally patients may present with thrombocytopenia, microangiopathic hemolysis and renal function impairment without evidence of preceding infections or other plausible causes. These patients of atypical HUS have high risk of relapses and many evolve to end stage renal failure with or without relapses of HUS. Nevertheless, because the renal function impairment may be mild and reversible, a clear distinction between atypical HUS and TTP is impossible in some cases. In the absence of direct pathogenetic basis to support a clear distinction of TTP, typical HUS, atypical HUS and secondary HUS, some investigators proposed that these entities were one disease process affecting different target organs.
This ambivalence has led to two common schemes for classifying patients with thrombotic microangiopathy. In one scheme, all patients presenting with thrombocytopenia and microangiopathic hemolysis are grouped under the generic syndrome of TTP -HUS or simply TTP. In a modified version of this scheme, patients with shiga toxin associated HUS were excluded [4]. In the other scheme, the diagnosis of TTP was applied to patients with specific neurologic signs, without or without acute renal failure, while the diagnosis of HUS is reserved for patients with evidence of acute renal function impairment but no specific neurologic signs [5]. Neither classification has proven satisfactory. Both schemes include under one diagnosis patients with entirely different disorders. This obscure the importance that these disorders require different therapeutic and prognostic considerations. In the second scheme, some patients present with specific neurologic signs on some occasions but no such signs on other occasions. Such patients have ended up shifting between the diagnoses TTP and HUS during the course of their illness, although their really had relapses of the same disease.
To address the uncertainty resulting from lack of knowledge in the underlying pathogenesis, Bukowski proposed in 1982 a set of strict criteria for diagnosis of idiopathic TTP (Table 1). This definition was based on the view that patients presenting with neurologic deficits but no or minimal renal function impairment most likely had TTP, provided there were no alternative plausible explanations. It was hoped that a strict definition of TTP would enhance the uniformity of the cases in clinical studies and facilitate comparison among different series. However, this strict definition might exclude some patients with TTP. In practice, since the consequence of not treating a patient of TTP with plasma is likely to be grave, physicians often offer plasma exchange therapy to patients even though the diagnosis of TTP was uncertain or unlikely. Consequently, Bukowski’s proposal was not widely heeded. Investigators continued to define TTP and HUS using widely different criteria (Table 2). This heterogeneity in diagnosis contributes to some of the discrepancy among various series of TTP but surprisingly is often ignored in literature reviews.
Table 1.
Common clinical and laboratory features of TTP.
| Thrombocytopenia (platelet count <20×109/L in acute cases) |
| Microangiopathic hemolytic anemia |
| Neurologic abnormality or complaint |
| Renal abnormalities |
| Proteinuria and microscopic hematuria |
| Peak BUN ≤ 0.4 g/L or Cr ≤ 0.03 g/L |
| Fever ≥ 38.3°C |
| Microthrombi on tissue biopsy |
| Exclusion |
| Evidence of intravascular coagulation |
| Evidence of underlying condition associated with or producing microangiopathic syndrome |
| Positive antinuclear antibody, anti-DNA antibody, or LE preparation |
| Oliguria or anuria |
Modified from Bukowski [66].
Table 2.
Heterogeneity in diagnosis of TTP
| Authors | Year | No. | Exclusion of renal failure | Exclusion of secondary cases | Severe deficiency | ADAMTS13 |
|---|---|---|---|---|---|---|
| Furlan, et al | 1998 | 24 | Yesa | No | 83% | |
| Tsai, et al | 1998 | 39 | Yesb | Yesc | 100% | |
| Veyradier, et al | 2001 | 111 | Yesa | No | 71% | |
| Rick, et al | 2002 | 50 | Not stated | Not stated | 78% | |
| Bohm, et al | 2002 | 22 | Not stated | No | 91% | |
| Vesely, et al | 2003 | 48 | No | Yesd | 33% | |
| Hovinga, et al | 2004 | 396 | Not stated | Not stated | 57% | |
| Peyvandi, et al | 2004 | 100 | No | No | 48% | |
| Zhou, et al | 2004 | 34 | Yesb | Yesc | 100% | |
| Coppo, et al | 2004 | 46 | No | Yes | 67% | |
| Zheng et al | 2004 | 37 | No | No | 43% | |
| Kokame, et al | 2005 | 41 | Not stated | Not stated | 80% | |
| Terrel, et al | 2005 | 70 | No | Yesd | 34% | |
| Matsumoto, et al | 2005 | 108 | Not stated | Yesa | 52% |
Based on referral diagnosis
Crmax >4.0 g/dL
Based on overall course
Based on initial assessment
In recent years, it has become clear that defective regulation of von Willebrand factor activity by a circulating metalloprotease, ADAMTS13, is found in most patients with classic TTP, while defective regulation of complement activation may be detected in many patients with atypical HUS. These advances, along with well-known association between shiga toxins and the post-diarrhea HUS, provide a basis to classify microthrombotic disorders based on their pathogenesis and etiology (Table 3). Deficiency of ADAMTS13, exposure to shiga toxins, and dysregulation of complement activation may all lead to microvascular thrombosis. Consequently, these disorders share the common features of thrombocytopenia and microangiopathic hemolysis in the peripheral blood. Nevertheless, the pathways leading to microvascular thrombosis, the patterns of organ involvement, and the strategies of treatment are quite different (Table 4). Microvascular thrombosis tends to involve the brain and heart in ADAMTS13 deficient patients and the kidney in shiga toxin and complement dysreguation cases. Consequently, patients with ADAMTS13 deficiency often present with neurological deficits while shiga toxin and complement dysregulation is more likely to be complicated with prominent renal failure. Nevertheless, it is impossible to completely distinguish these entities based on clinical features because of overlapping in the pattern of organ involvement. Interestingly, another disorder of complement dysregulation, paroxysmal nocturnal hemoglobinuria (PNH), may also cause thrombosis. Rarely, widespread thrombosis may involve the mesenteric microcirculation in a patient with PNH, resulting in microangiopathic hemolysis and thrombocytopenia. This difference between atypical HUS and PNH in predilection of organ involvement suggests that the endothelial cells use tissue-specific regulatory proteins to control complement activation.
Table 3.
A classification of microvascular thrombosis based on pathogenesis and etiology
| Clinical entity | Molecule | Cause | Etiology |
|---|---|---|---|
| TTP | ADAMTS13 | ADAMTS13 Ab or rarely ADAMTS13 mutations | Ticlopidine, HIV, or idiopathic (most cases) |
| Autosomal recessive | |||
| Atypical HUS | |||
| CFH | CFH mutation | Autosomal dominant, variable penetrance | |
| Ab | Idiopathic | ||
| MCP | MCP mutation | Autosomal dominant, variable penetrance | |
| IF | IF mutation | Autosomal dominant, variable penetrance | |
| BF | BF mutation (gain of function) | Autosomal dominant | |
| Unknown | Unknown | Unknown (50%-70% of atypical HUS) | |
| Secondary HUS | |||
| Stx-HUS | Shiga toxins | Bacterial infection | Stx+ E. coli or Sh. dysenteriae |
| SP-HUS | TA-antigen | Bacterial infection | Neuraminidase (e.g. S. pneumoniae) |
| Other secondary HUS or TMA | Unknown | Unknown | Lupus and related disorders, BMT, neoplastic diseases, drugs, surgery, infection, pregnancy, pancreatitis |
| PNH | CSRF | PIG-A gene mutation | Somatic mutation |
Abbreviations. BF: complement B factor; BTM: bone marrow transplantation; CFH: complement factor H; CRSF: complement regulating surface proteins (e.g. CD55, CD59); IF: complement factor I; MCP: membrane cofactor protein; PNH: paroxysmal nocturnal hemoglobinuria; SP: S. pneumoniae associated HUS; Stx: shiga toxins; TMA: thrombotic microangiopathy
Table 4.
Comparison of microvascular thrombosis due to various molecular defects
| Molecule | Mechanism | Organ* | Presentation* | Course* | Treatment* |
|---|---|---|---|---|---|
| ADAMTS13 | VWF-platelet aggregation | Brain, heart | Neurologic change | Relapse | Plasma, rituximab, splenectomy |
| Shiga toxins | Endothelial injury | Kidney | Renal function impairment | Relapse is uncommon | Supportive |
| Complement dysregulation | Endothelial injury | Kidney | Renal function impairment | Relapse, ESRD | - |
| CFH, IF, or BF | - | - | - | - | Plasma, liver-kidney graft? |
| MCP | - | - | - | - | Kidney graft |
| PIG-A | Endothelial injury? | Mesenteric vasculature | Abdominal pain, bloody diarrhea | PNH | PNH |
Only the most common features are listed.
Abbreviations. CFH: complement factor H; ESRD: end stage renal disease; IF: complement factor I; MCP: membrane cofactor protein; PIG-A: phosphatidylinositol glycan anchor biosynthesis, class A; VWF: von Willebrand factor; PNH: paroxysmal nocturnal hemoglobinuria
In hindsight, TTP as defined by Bukowski would have included essentially only patients with severe ADAMTS13 deficiency. In clinical practice, this set of criteria may be used to assess the probability of TTP. Nevertheless, it does not exclude the diagnosis of TTP.
ADAMTS13 - structure and function
ADAMTS13 is a circulating metalloprotease of the “a disintegrin and metalloprotease with thrombospond type 1 motif” enzyme family that cleaves plasma von Willebrand factor (VWF) at its Y1605-M1606 peptidyl bond in a shear dependent manner.
The ADAMTS13 gene contains 29 exons spanning approximately 37 kb on chromosome 9q34 [6-8]. ADAMTS13 encodes a 4.7-kb transcript that is detectable in the liver, and a 2.4-kb transcript detectable in placenta, skeletal muscle, and certain tumor cell lines. In the liver, ADAMTS13 is expressed primarily in the vitamin A-enriched stellate cells [9;10]. ADAMTS13 may also be expressed in platelets and endothelial cells [11-13].
The full-length ADAMTS13 transcript encodes a precursor protein of 1427 amino acid residues. The protein undergoes extensive glycosylation and other post-translation modifications before secretion. The sequence of ADAMTS13 exhibits a multi-domain structure that is common among proteases of the ADAMTS family and but also contains two unique CUB domains (Figure 1).
Figure 1.
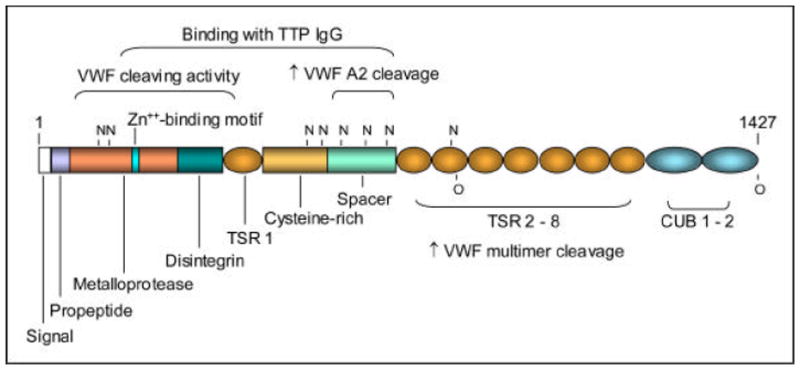
Domain structure of ADAMTS13. The consensus sequence for zinc binding, potential N- and O-glycosylation sites, and the regions sufficient for VWF cleaving activity and for interaction with IgG antibodies of TTP are marked. Truncation upstream of the spacer domain markedly decreases the cleaving activity against either VWF multimers or VWF fragments of the A2 domain, while truncation downstream of the spacer domain decreases the cleaving activity against VWF multimers.
ADAMTS13 is inactivated by disulfide bond-reducing agents, tetracyclines, or cation chelators such as phenanthroline and EGTA [14]. Although ADAMTS13 is stable in normal plasma, its activity may deteriorate rapidly in plasma samples from patients with liver diseases, disseminated intravascular coagulopathy or other pathological conditions. In vitro, thrombin, plasmin, and perhaps hemoglobin may inactivate ADAMTS13 [15;16]. Thrombospondin may also protect VWF from cleavage by ADAMTS13 [17]. Based on observations following infusion of normal plasma in patients with genetic deficiency of ADAMTS13, the elimination half-life of ADAMTS13 is approximately 2 days.
ADAMTS13 diverts early from other members of the ADAMTS family of proteases [18;19]. In particular, ADAMTS13 contains an unusually short (41 amino acid residues) propeptide whose cleavage does not appear to be necessary for expression of proteolytic activity [20]. Enzymatic analysis of recombinant proteins expressed in cultured mammalian cells reveals that the VWF cleaving activity is markedly decreased [21] but not abolished as previously reported [22;23] when ADAMTS13 is truncated upstream of its spacer domain. The spacer domain is required for binding to a short fragment in the VWF A2 domain downstream of the cleavage site [24] and also for recognition by TTP IgG [21;25]. One study also detected interaction of TTP IgG with other domain fragments [26], although this observation remains controversial. The TSR 2-8 region is required for effective cleavage of VWF multimers but not of the VWF A2 fragment.
ADAMTS13 prevents microvascular platelet thrombosis
VWF, a glycoprotein secreted from vascular endothelial cells in very large polymeric forms, supports platelet adhesion and aggregation at sites of vessel injury. High levels of shear stress promote platelet adhesion to VWF, presumably because it causes conformational change of VWF. ADAMTS13, a plasma metalloprotease derived primarily from hepatic stellate cells, cleaves von Willebrand factor (VWF) when the flexible macromolecule is partially unfolded by high levels of shear stress. By cleaving VWF before it is fully activated by shear stress, ADAMTS13 prevents accumulation of super active forms of VWF and VWF-platelet aggregation. This explains why a severe deficiency of ADAMTS13 causes microvascular thrombosis characteristic of TTP [27].
This scheme provides a conceptual basis for understanding why ADAMTS13 deficiency leads to microvascular thrombosis of TTP. It is supported by in-vitro studies demonstrating that brief (<15 seconds) exposure to 70 – 120 dynes/cm2 of shear stress results in increased platelet aggregating capacity of VWF [28]. However, VWF-platelet aggregation is observed only after exposure to extremely high levels (400 dynes/cm2) of shear stress [29], a level not known to exist in the normal circulation. On the other hand, levels of wall shear stress, in the range encountered in normal arterioles and capillaries [30], is sufficient to enhance platelet adhesion to immobilized VWF. This discrepancy in shear stress levels necessary for enhancing VWF-platelet binding most likely relates to the fact that shear stress in the vascular lumen is inversely proportional to the distance from the luminal surface. Since VWF is unlikely to be exposed to wall shear stress for sustained durations after it is released in the circulation, how could VWF-platelet aggregation ever occur to cause TTP?
In vitro and in vivo studies have observed that some VWF molecules may be anchored to endothelial surface upon secretion from endothelial cells [31-33]. This anchoring appears fragile but may be sufficient to allow conformational unfolding of VWF by shear stress. ADAMTS13 quickly cleaves VWF before it is fully unfolded. by wall shear stress. In the absence of ADAMTS13, VWF become fully unfolded and activated, providing a ligand to support platelet adhesion, leading to platelet aggregation and microvascular thrombosis. In this scheme, VWF-platelet thrombosis may develop even if VWF does not remain anchored to endothelial surface (Figure 2).
Figure 2.
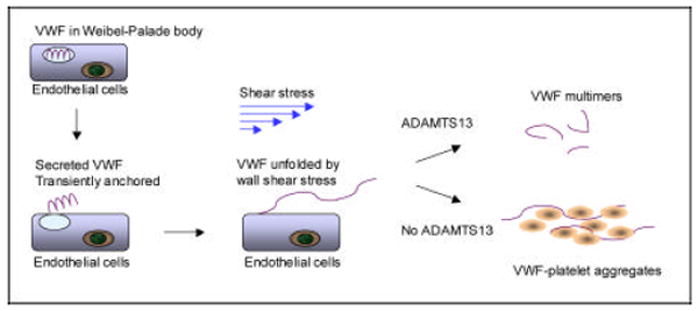
A scheme depicting how ADAMTS13 deficiency may lead to VWF-platelet aggregation and microvascular thrombosis. Some of the VWF molecules secreted from endothelial cells may remain transiently anchored to endothelial surface. After exposure to wall shear stress, VWF is conformationally unfolded and is immediately cleaved ADAMTS13 to smaller forms. In the absence of ADAMTS13, VWF becomes activated, leading to intravascular VWF-platelet binding and microvascular thrombosis.
Severe ADAMTS13 deficiency cause thrombotic thrombocytopenic purpura
Severe deficiency of ADAMTS13 was first demonstrated in relapsing TTP and acute acquired TTP [28;34;35]. These and subsequent studies demonstrate that ADAMTS13 deficiency is associated with inhibitory autoantibodies of ADAMTS13 in patients with acquired TTP [36]. Separately, another study has linked hereditary TTP to mutations of the ADAMTS13 gene on chromosome 9q34 [37]. Taken together, these observations demonstrate that ADAMTS13 deficiency causes TTP.
Does severe ADAMTS13 deficiency account for all cases of TTP? Severe ADAMTS13 deficiency can be detected in essentially every adolescent or adult case of thrombocytopenia and microangiopathic hemolysis without renal failure or plausible causes [28;38]. In other series, severe deficiency of ADAMTS13 is detected in 34%-91% of the cases (Table 2) [35;39-47]. This lower prevalence of ADAMTS13 deficiency was not unexpected because patients with renal failure or underlying disorders were not rigorously excluded in these series. As discussed earlier, patients without ADAMTS13 deficiency should no longer be included under the diagnosis of TTP because they may have complement dysregulation or other mechanisms that require separate therapeutic considerations.
Is ADAMTS13 deficiency sufficient for diagnosis of TTP? ADAMTS13 deficiency may be detected in patients without thrombocytopenia or microangiopathic hemolysis. This is observed most commonly in patients with a history of TTP and has been interpreted as evidence that ADAMTS13 deficiency alone is not sufficient to cause TTP. This is equivalent to proposing that von Willebrand factor mutation or deficiency is insufficient for diagnosis of von Willebrand factor disease if the patient is not bleeding. The propensity to develop thrombotic complications in a patient with ADAMTS13 deficiency is affected by the activity levels of ADAMTS13 as well as environmental and genetic factors. Such patient may develop complications of TTP either spontaneously or following exposure to stresses of infection, surgery, or pregnancy.
Some studies have reported that ADAMTS13 is also decreased in conditions such as sepsis, disseminated intravascular coagulopathy and liver diseases, occasionally to very low levels [48-50]. However, these observations remain controversial [51;52], and the significance of decreased ADAMTS13 levels remains uncertain if they are not accompanied by evidence of decreased VWF proteolysis. As will be further elaborated later, a low ADAMTS13 assay value requires further confirmatory investigation before it can be used to support the diagnosis of TTP.
Mouse models of ADAMTS13 deficiency
Animal models of ADAMTS13 deficiency have been generated in mice by using homologous recombination technologies [33;53]. Initially, the ADAMTS13-null mice exhibited no phenotypic abnormalities except more severe thrombocytopenia following epinephrine and collagen injection. The size of VWF multimers was also not affected. Transfer of the ADAMTS13-null allele to a different genetic background produced mice that developed spontaneous thrombocytopenia and premature death with microvascular thrombosis quite similar to human TTP. Intriguingly, this TTP-like phenotype can also be precipitated by injection of shiga toxin to the ADAMTS13-null mice before they developed a complete form of the disease. Thus ADAMTS13 appears to be less critical in regulating the activity of VWF in mice, and the genetic background of the mice affects the phenotype of ADAMTS13 deficiency. Further investigation of the mouse models may elucidate the modifiers that affect the phenotypic severity of ADAMTS13 deficiency.
Clinical features
The incidence of TTP has been estimated using a variety of approaches and case definitions to be 2-7 per million person-years [44;54-56]. The incidence is likely to be affected by race, gender, prevalence of HIV infection, accuracy of diagnosis and other as yet unknown factors. Acquired TTP affects adolescents and adults, with a peak incidence between the ages of 30 and 40 years. TTP occurs more frequently among blacks, particularly black females. The female/male ratio is estimated to be 2-3:1. It has been proposed that the incidence of TTP is increasing; however this has not been consistently observed.
HIV was present in approximately 50% of the TTP cases at a major urban center [57], in 39% of our non-referral cases, and in 4% of the Oklahoma registry cases [44]. The use of ticlopidine may increase the risk of TTP by 50-200-folds [58;59]. In contrast, clopidogrel has not been definitively associated with ADAMTS13 deficiency [60;61]. With the exception of HIV infection and ticlopidine, most cases of TTP do not have an obvious cause.
TTP commonly presents in previously healthy individuals with a constellation of vague, non-specific symptoms of weakness, dizziness, and headache before advancing to more obvious neurological manifestations such as visual symptoms, ataxia, syncope, confusion, paresthesias, paresis, dysarthria, aphasia, seizures, stupor, and coma. Fever is present at admission in approximately 50% or less of the patients but may develop in additional cases during the course. Purpura is common at presentation but serious external or internal bleeding is rare except toward the terminal stage. TTP may present with abdominal pain or other gastrointestinal symptoms, presumably due to visceral ischemia or pancreatitis. Visual abnormalities due to retinal hemorrhage or thrombosis have been described in the literature. However, most of those patients had features suggestive of typical or atypical HUS and in no case has severe ADAMTS13 deficiency been documented. Patients of TTP may present with chest pain or arrhythmia. Nevertheless, clinically overt cardiopulmonary dysfunction is uncommon until the pre-terminal stage [62]. Rarely, TTP may present with sudden death due to widespread myocardial necrosis [63].
Diagnosis
A previously healthy adolescence or adult presenting with headache, confusion or focal neurologic deficits, accompanied by thrombocytopenia, microangiopathic hemolysis, and microscopic hematuria almost certainly has TTP, provided there are no alternative explanations and renal failure is absent or minimal throughout the course. The diagnosis of TTP becomes less certain or even unlikely if the patient has a prodrome of bloody diarrhea, develops significant renal failure or has underlying disorders such as disseminated intravascular coagulopathy, active autoimmune connective tissue diseases, malignancies, bone marrow transplantation, pneumococcal or other infections, or usage of certain drugs or chemicals (e.g gemcitabine, mitomycin, cyclosporin, quinine, or cocaine). Importantly, some of these underlying conditions may not be apparent initially and must be vigorously investigated as part of the differential diagnosis.
Sub-clinical or atypical forms of TTP also exist. Absence of thrombocytopenia or microangiopathic hemolysis is not inconsistent with the diagnosis of TTP. Some patients of TTP may be asymptomatic but are at risk of developing thrombotic complications when stressed with fever, infection, surgery, or pregnancy. Others may present with isolated thrombocytopenia and are mistaken to have ITP. Still others present with acute neurological deficits without thrombocytopenia or microangiopathic hemolysis [64]. Definitive diagnosis is difficult in atypical or complex cases without the aid of ADAMTS13 and VWF multimer analysis.
Thrombocytopenia, microangiopathic hemolysis and fleeting neurological deficits (triad), with fever and renal abnormalities (pentad) are common but not pathognomonic of TTP; these constellations of complications may also be present in some cases of systemic lupus erythematosus or various other disorders.
A prodrome of abdominal pain, diarrhea with bloody stools favors the diagnosis of shiga toxin associated HUS. Presence of advanced renal failure, oliguria, fluid overload or hypertension in a patient with favors the diagnosis of typical or atypical HUS. On the other hand, hypertension and renal failure may not be severe or irreversible in some patients with HUS. In such cases, a distinction from TTP may not be straightforward without specific laboratory tests.
ANA or other autoantibodies may be detectable in 10% - 50% of patients with TTP [43;65]. However, the titers are low and concurrent presentation of active lupus or other autoimmune diseases is uncommon with TTP [66]. ADAMTS13 activity level may be partially decreased in patients with systemic lupus erythematosus or related disorders [67]. Patients with positive serological studies for lupus or related disorders are more likely to have connective tissue disorders presenting as a TTP-like syndrome.
Patients with neoplastic diseases may present with microangiopathic hemolysis and thrombocytopenia due to embolism of tumor cells, chemotherapeutic agents or other unknown mechanisms [68;69]. Such cases are generally not associated with severe ADAMTS13 deficiency or inhibitors of the enzyme [70;71].
Paroxysmal nocturnal hemoglobinuria (PNH) may be complicated with microvascular thrombosis, particularly of the mesenteric circulation, presenting with abdominal pain, blood stools, microangiopathic hemolysis and mild or moderate thrombocytopenia. A prior history of PNH may not be elicited. A syndrome of thrombocytopenia, microangiopathic hemolysis, variable renal impairment, with or without acute respiratory syndrome, may occur after surgery or pancreatitis [72].
VWF multimers in TTP
Abnormalities in VWF multimers are common in TTP [73] but often misunderstood. Large VWF multimers are depleted in most patients presenting with acute TTP (Figure 3, panel A). Ultra large multimers frequently appeared during the early stage of recovery, and also during remission if the ADAMTS13 level remains decreased (Figure 3, panel B). These changes reflect evolving balance between endothelial secretion of VWF, VWF proteolysis by ADAMTS13, and VWF-platelet binding induced by high levels of shear stress.
Figure 3.
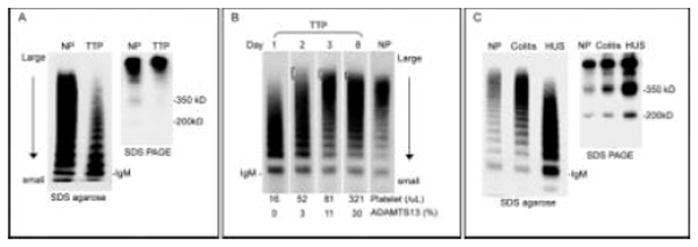
VWF multimer changes in TTP and HUS. A. VWF multimers and fragments in a representative case of acute TTP. Compared to normal plasma (NP), a gradient of decreased large multimers is noted in TTP. The decrease of large multimers is due to consumption as shear stress induces binding of large VWF multimers to platelets. In the absence of ADAMTS13, the 350kD and 200kD fragments of VWF are decreased. B. Serial changes of VWF multimers in TTP. A typical loss of large multimers is noted on day 1. As the patient improves with plasma exchange, ultra large multimers are present in subsequent days. The multimer pattern shows a gradually shift toward normal as the platelet count and ADAMTS13 activity level increase (bottom). C. VWF multimers and fragments in a representative case of shiga toxin associated HUS. VWF multimer is normal during the stage of colitis but shows a gradient of decreased large multimers at the onset of HUS. The VWF fragments are markedly increased, which are attributable to both increased VWF antigen level and increased proteolysis by ADAMTS13 [74]. VWF multimers are separated by SDS agarose gel electrophoresis and visualized. VWF fragments are separated by SDS agarose PAGE under non-reducing conditions. VWF is visualized with rabbit polyclonal anti-VWF and 125I labeled or horseradish peroxidase labeled goat anti-rabbit IgG.
Large VWF multimers are also decreased in patients with HUS, aortic stenosis or pulmonary arterial hypertension (Figure 3, panel C) [74-76]. In these conditions, ADAMTS13 levels are normal or mildly decreased. It is believed that large multimers are decreased because high levels of shear stress in these disorders enhance proteolysis of VWF by ADAMTS13. Consequently, the proteolytic fragments are increased. VWF multimer pattern normalizes when aortic stenosis corrected by surgery or pulmonary arterial hypertension is controlled with prostacyclin.
ADAMTS13 and its autoantibodies
ADAMTS13 activity levels are <10% (or <5%, depending on the assays used) of normal control in patients with acute TTP. During the course of plasma therapy, An ADAMTS13 activity level >10% is associated with clinical and hematological improvement. On the other hand, because ADAMTS13 assays are typically performed on samples obtained immediately prior to each session of plasma exchange, clinical response may be observed with ADAMTS13 activity level <10%. During remission of TTP, the ADAMTS13 levels remain decreased in many cases, occasionally <10% of normal control.
Several assays have been developed to measure ADAMTS13 activity. The assays use VWF multimers or abbreviated peptides of the VWF sequence for substrates. The detection methodology ranges from gel analysis to ELISA to FRET (fluorescnce resonance energy transfer). An international collaborative study find that not infrequently the assays produce variable results [77]. Thus, interpretation of ADAMTS13 activity assay results should be individualized according to the experience of the laboratories.
Enzyme immunoassays have been developed to measure ADAMTS13 antigen levels [78;79]. However, such assays also detect ADAMTS13-antibody complexes and based on early studies, are of limited diagnostic values.
Plasma mixing studies yield positive results for inhibitors in 50-90% of the cases [36]. When IgG molecules are isolated and tested at high concentrations, inhibitory activity is detectable in essentially every patient of TTP investigated. The inhibitors of ADAMTS13 are generally of very low levels (< 2 U/mL, using the Bethesda definition for factor VIII inhibitors) and are infrequently >10 U/mL [80].
ADAMTS13-binding IgG is detectable by ELISA in 97-100% of TTP cases [36;81]. The incidence declines substantially if the blood samples are obtained after the plasma therapy is initiated. The assay may yield false positive results in 5% - 15% of patients without TTP, unless a competitive binding procedure is also performed to confirm that the binding IgG is suppressible by ADAMTS13 [36]. The prevalence of non-inhibitory antibodies without evidence of inhibitors is probably quite low.
Overall, assays of ADAMTS13 activity, like most protease activity assays, are prone to be affected by multiple factors, including assay precision and accuracy, stability of the enzyme in plasma samples, and other ill-defined factors. Antigenic assays require further improvement before they can be used for diagnosis of TTP. ELISA for ADAMTS13 binding IgG is highly sensitive but requires a saturation step to improve its specificity. Because of these issues, ADAMTS13 analysis results should be correlated with clinical data. The experience of the laboratory is also critical. As yet clinical ADAMTS13 assays can not be used as the sole basis for diagnosis of TTP.
Clinical course
Without treatment, acute TTP is almost always fatal, often within 10-14 days. With plasma exchange, most patients are expected to survive the acute event. However, TTP may worsen after an initial response during plasma therapy. Occasionally it may be persistent, requiring long-term plasma exchange, especially after several relapses.
Relapses occur in 30% - 60% of the patients after an initial acute episode, contributing to further morbidity and mortality. Relapses occur most commonly during the first month after the acute episode but may also occur months or years thereafter, with or without obvious precipitating events.
Previously, because most cases die or achieved remission without the need of continuing plasma exchange, TTP was considered to be an acute disease. Serial measurements in patients that survive one or more episodes of TTP reveal that ADAMTS13 activity remains decreased in many patients, occasionally to <10% of normal control. Antibodies of ADAMTS13 may also remain detectable. The activity level tends to fluctuate, and a normal level is not necessarily an indication of complete immunological remission, as it may decrease subsequently. Thus, TTP is likely to be a chronic disease in many if not all patients.
A clinical relapse occurs when exacerbation of the autoimmune reaction suppresses the ADAMTS13 activity below a presumed threshold level. The threshold level is <10%, as relapse of TTP doe not occur with ADMTS13 activity >10%. However, since VWF-platelet interaction is affected by multiple factors, it is unlikely that a single threshold value exists for relapse.
Relapses often occur without apparent precipitating causes. On the other hand, patients with ADAMTS13 activity <10% of normal control are at risk of relapse when they are exposed to the stress of various conditions such as surgery, infection, or pregnancy.
When TTP relapses, it often begins with a decline of the platelet count without apparent hemolysis. The course evolves variably, ranging from rapid deterioration within a few days to smoldering over weeks or months. Occasionally, a relapse may present with focal neurological deficits such as dizziness, hemiparesis, slurred speech or aphasia [64]. Such neurological complications may pose a diagnostic challenge when they are not accompanied by thrombocytopenia or microangiopathic hemolysis.
Management
Approximately 80% of patients who are treated aggressively with exchange plasmapheresis survive the initial episode of TTP [2]. Death is frequently due to delay in diagnosis and treatment or ineffectiveness of plasma exchange. Platelet transfusion should be avoided because bleeding complications are uncommon in TTP and marked deterioration in neurological status has been reported in association with platelet transfusions.
Plasma therapy is often tapered and discontinued after the platelet count is normal for several days. However, the platelet count may not recover to normal if the patient has concurrent ITP or other causes of thrombocytopenia, as is commonly encountered in patients with HIV infection.
The therapeutic efficacy of plasma exchange therapy is believed to result from replenishment of the missing ADAMTS13. Although it may also remove the inhibitors, this process is not very effective and by itself is insufficient for therapeutic responses. Thus replacement with fluids lacking ADAMTS13 is not recommended. Limited by the risk of fluid overload, Infusion of fresh frozen plasma is less effective, inducing remission in approximately 60% of the cases. Plasma infusion is used primarily as a stand-in until plasmapheresis is available or during tapering.
It has been proposed that cryoprecipitate-depleted plasma is more effective than fresh frozen plasma because it lacks large von Willebrand factor multimers [82]. However, large VWF multimers may continue to derive from endothelial cells, and cryoprecipitate-depleted plasma is comparable to fresh frozen plasma in the ADAMTS13 activity level [83-85]. Two small randomized controlled trials have failed to demonstrate that the use of cryoprecipitate-depleted plasma is superior to fresh frozen plasma as initial replacement therapy [86;87].
The ADAMTS13 activity levels have been investigated in a variety of plasma products [83-85]. Levels comparable to that in fresh frozen plasma are present in various plasma fractions and some of its derived products. The ADAMTS13 activity exhibits negligible loss of activity after the plasma samples are stored at 4-6°C or even at room temperature for at least five days. During our experimentation to isolate ADAMTS13, the protease was not precipitated by the procedure of cryoprecipitation [88]. A formal investigation also found no difference between the cryoprecipitate and cryosupernatant preparations of fresh frozen plasma in their ADANTS13 activity levels [85]. However, in a recent study using FRETS -VWF73 as the substrate, the ADAMTS13 level is reported to be approximately 200% of pooled normal plasma in the cryoprecipitate fraction of plasma [84]. Whether this discrepancy relates to the difference in assay methodology remains unclear.
Patients are often treated with corticosteroids or antiplatelet drugs during plasmapheresis. The benefit of these therapies is unclear in patients receiving plasma exchange but probably minimal [66;89].
Refractory patients are often treated with cryoprecipitate-depleted plasma, vincristine, splenectomy, cyclophosphamide, azathioprine, splenectomy, high dose intravenous immunoglobulins or prostacyclin analogues, mostly in anecdotal case reports or with questionable responses [90]. More recently, rituximab, a chimeric monoclonal anti-CD20, has been used with presumed benefits in patients with protracted TTP. The role of rituximab in the treatment of acute TTP is will be investigated in a planned controlled trial [91]. The experience of calcineurin inhibitors as a treatment of TTP remains limited [92].
Relapsing TTP, which may occur either early in the first month after remission or later after months or years, is also treated with plasma therapy. The efficacy of anti-platelet drugs, corticosteroids, rituximab, or calcineurin inhibitors for prevention of relapse is unclear. Patients with two or three risk factors of age >40 years, fever >38.5°C and hemoglobin <90 g/L may have substantially worse prognosis [93].
If TTP is an autoimmune disease, why does it respond to plasma therapy? Based on experience in patients with inhibitors of factor VIII, replacement therapy is effective only in low responder patients whose inhibitor levels are no more than 5-10 Bethesda units/mL. A retrospective analysis of the participants in the Canadian Apheresis Group trial demonstrates that the levels of ADAMTS13 inhibitors are low in most patients of TTP [80]. This may be a critical reason that TTP responds to plasma exchange in most cases. Occasionally a patient may not respond to plasma exchange because they have very high levels of inhibitors either at presentation or during the course of therapy. Other patients may require prolonged courses of plasma exchange before a sustained clinical remission is achieved.
Since plasma therapy does not address the underlying immune abnormalities of TTP, why do most patients achieve a sustained remission after receiving treatment for their acute episodes? The answer for this question is not entirely clear. It is possible that the autoimmune reaction to ADAMTS13 develops in response to an otherwise innocuous infection. This may be analogous to the production of anti-red cell antibodies following certain bacterial or viral infections. Patients are able to remain in remission because after the acute phase, the antibody levels spontaneously decline to very low levels. In a subset of patients, the autoimmune B cell clones may persist or re-emerge, resulting in protracted or relapsing courses. In such cases, immunosuppressive drugs such as cyclophosphamide, azathioprine, or rituximab may deserve further investigation.
The cloning of ADAMTS13 has raised the expection that ADAMTS13 protein may eventually replace fresh frozen plasma as a more targeted treatment of TTP. However, the amount of ADAMTS13 protein might not be easy to determine for individual cases because patients’ antibody levels vary during the course of their disease. Truncated variants of ADAMTS13 lacking the space domain sequence necessary for recognition by the inhibitors are not suppressible and may circumvent the therapeutic difficulty of TTP [21].
Pregnancy and TTP
Some reports suggest that pregnancy is associated with TTP [94]. However, the evidence in support of this association remains circumstantial. TTP may appear to be associated with pregnancy because like many autoimmune disorders it is more likely to affect young and middle-age women. Thus TTP may occur co-incidentally during pregnancy. Pregnancy may also cause flaring of pre-existing subclinical hereditary or acquired TTP. Both de novo and relapsing TTP tends to occur in the first half of pregnancy. Additionally, the syndrome of microangiopathic hemolysis and thrombocytopenia may result from preeclampsia, disseminated intravascular coagulopathy, hemolysis with elevated liver enzyme and low platelet (HELLP) syndrome, or atypical HUS, with or without evidence of complement dysregulation. These disorders have different pathophysiology and prognosis and should be distinguished from TTP. TTP may also occur postpartum. Like acquired factor VIII inhibitors or other post autoimmune disorders, it may be related to the recovery of gestational immunotolerance.
Pregnancy counseling for women with a history of TTP continues to be a challenge. ADAMTS13 levels remain decreased in many patients of TTP during remission. Normal pregnancy per se is also associated with elevated VWF other procoagulant protein levels, decreased anticoagulant proteins, activation of platelets, and significant albeit modest decrease of ADAMTS13 levels [95;96]. A further decrease of ADAMTS13 may occur if pregnancy is complicated with preeclampsia or HELLP syndrome. Together, these changes of pregnancy may be sufficient to precipitate the complications of TTP if the baseline ADAMTS13 level of the patient is low. Thus, until more definitive data becomes available, pregnancy should be considered a high risk of TTP relapse, particularly if the patient’s baseline ADAMTS13 levels are less than 10% - 30% of normal control.
Hereditary Thrombotic Thrombocytopenic Purpura
Previously known as Schulman-Upshaw syndrome [97;98] or chronic relapsing TTP, hereditary TTP is a rare disorder, accounting for less than 1% of all cases of TTP. Frequently, hereditary TTP has its initial manifestations during the neonatal period but is often not recognized until later in life. Some of the cases have siblings of intrauterine or perinatal death due to presumed TTP.
DNA sequence analysis reveals that the patients have double heterozygous or, less commonly, homozygous mutations of the ADAMTS13 gene on chromosome 9q34. More than 65 mutations, including nonsense, missense, frame-shifting insertion or deletion and splicing mutations, have been detected in patients with hereditary TTP [99-103]. The mutations affect the process of protease synthesis, activity, or more commonly, secretion. At least three mutations have been detected in seemingly unrelated families. One mutation, 4143insA, has been detected in at least 15 patients that share a common haplotype in central-northern Europe [104]. More than 25 polymorphisms have also been detected in the coding sequence of ADAMTS13. Certain combinations of polymorphisms or mutation-polymorphism may result in severe ADAMTS13 deficiency [100].
In a typical case, the affected individual is born with evidence of neonatal stress or presents within a few hours after birth with severe jaundice and thrombocytopenia. Hemolysis with schistocytes on blood smears may be noted. Occasionally serious complications such as seizures and mental obtundation may occur. The symptoms typically improve immediately after blood transfusion or exchange transfusion, often performed unknowingly for anemia, thrombocytopenia, or hyperbilirubinemia. Consequently, the neonates may be discharged without a correct diagnosis, only to present with complications of TTP weeks or years later.
Like most genetic disorders, hereditary TTP varies substantially in its severity. Many patients of hereditary TTP invariably develop hematological or other complications if not treated regularly with plasma infusion every 2-4 weeks. Others may maintain normal or mildly subnormal platelet counts and develop more serious complications only during intermittent episodes of exacerbations. Such variability is also observed in siblings with the same ADAMTS13 mutations, indicating that extragenic or environmental factors are operative. The severity may also vary during the lifetime of individual cases, with or without apparent exacerbating conditions such as pregnancy. Because the clinical course may be mild, the hereditary form of TTP may be encountered in adults when thrombotic microangiopathy is precipitated by infection, diarrhea, pregnancy or surgery.
Occasionally the clinical course may affect specific organs, presenting with pancreatitis, focal neurological deficits, seizures, or acute renal failure. Acute renal failure may be severe but is reversible if plasma therapy is instituted promptly. Chronic renal failure is uncommon, but may occur if the patients are not treated with plasma therapy or have a concurrent mutation of atypical HUS [105].
Laboratory findings are similar to those of acquired TTP. Typically the ADAMTS13 activity levels are less than 10% of normal but may be slightly higher in milder cases. Inhibitory antibodies of ADAMTS13 are not detectable. Parents are partially deficient in ADAMTS13 activity but are asymptomatic. Ultra large von Willebrand factor multimers are detected during remission, while acute exacerbation is often accompanied by depletion of the large and ultra large multimers. Hereditary TTP responds to 10-15 mL fresh frozen plasma per kilogram of body weight administered every 2-4 weeks. Vascular access or transfusion related reactions might become problematic.
Summary
TTP is an important cause of microvascular thrombosis. Previously considered one of the most intriguing and mysterious disorders, TTP has been shown to be caused by deficiency of ADAMTS13 that may be hereditary or acquired. The onset and course of TTP are now explicable based on our knowledge of VWF-platelet physiology. Future challenges include improvement of the assays for reliable diagnosis of TTP, delineation of modifiers affecting the severity of the disease, and development of new therapeutic measures for more effective treatments. With heightened interest in the investigation of this disease, the prospect of meeting these challenges is encouraging.
Acknowledgments
This work was supported in part by Grant No. HL62136 from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Moschcowitz E. An acute febrile pleiochromic anemia with hyaline thrombosis of the terminal arterioles and capillaries: an undescribed disease. Arch Intern Med. 1925;36(1):89–93. [PubMed] [Google Scholar]
- 2.Rock GA, Shumak KH, Buskard NA, Blanchette VS, Kelton JG, Nair RC, Spasoff RA. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Canadian Apheresis Study Group. N Engl J Med. 1991;325(6):393–397. doi: 10.1056/NEJM199108083250604. [DOI] [PubMed] [Google Scholar]
- 3.Tapper D, Tarr P, Avner E, Brandt J, Waldhausen J. Lessons learned in the management of hemolytic uremic syndrome in children. J Pediatr Surg. 1995;30(2):158–163. doi: 10.1016/0022-3468(95)90554-5. [DOI] [PubMed] [Google Scholar]
- 4.Vesely SK, George JN, Lammle B, Studt JD, Alberio L, El-Harake MA, Raskob GE. ADAMTS13 activity in thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: relation to presenting features and clinical outcomes in a prospective cohort of 142 patients. Blood. 2003;102(1):60–68. doi: 10.1182/blood-2003-01-0193. [DOI] [PubMed] [Google Scholar]
- 5.Remuzzi G, Galbusera M, Noris M, Canciani MT, Daina E, Bresin E, Contaretti S, Caprioli J, Gamba S, Ruggenenti P, Perico N, Mannucci PM. von Willebrand factor cleaving protease (ADAMTS13) is deficient in recurrent and familial thrombotic thrombocytopenic purpura and hemolytic uremic syndrome. Blood. 2002;100(3):778–785. doi: 10.1182/blood-2001-12-0166. [DOI] [PubMed] [Google Scholar]
- 6.Levy GG, Nichols WC, Lian EC, Foroud T, McClintick JN, McGee BM, Yang AY, Siemieniak DR, Stark KR, Gruppo R, Sarode R, Shurin SB, Chandrasekaran V, Stabler SP, Sabio H, Bouhassira EE, Upshaw JD, Jr, Ginsburg D, Tsai HM. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature. 2001;413(6855):488–494. doi: 10.1038/35097008. [DOI] [PubMed] [Google Scholar]
- 7.Soejima K, Mimura N, Hirashima M, Maeda H, Hamamoto T, Nakagaki T, Nozaki C. A novel human metalloprotease synthesized in the liver and secreted into the blood: possibly, the von Willebrand factor-cleaving protease? J Biochem (Tokyo) 2001;130(4):475–480. doi: 10.1093/oxfordjournals.jbchem.a003009. [DOI] [PubMed] [Google Scholar]
- 8.Zheng X, Chung D, Takayama TK, Majerus EM, Sadler JE, Fujikawa K. Structure of von Willebrand factor-cleaving protease (ADAMTS13), a metalloprotease involved in thrombotic thrombocytopenic purpura. J Biol Chem. 2001;276(44):41059–41063. doi: 10.1074/jbc.C100515200. [DOI] [PubMed] [Google Scholar]
- 9.Zhou W, Inada M, Lee TP, Benten D, Lyubsky S, Bouhassira EE, Gupta S, Tsai HM. ADAMTS13 is expressed in hepatic stellate cells. Lab Invest. 2005;85(6):780–788. doi: 10.1038/labinvest.3700275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Uemura M, Tatsumi K, Matsumoto M, Fujimoto M, Matsuyama T, Ishikawa M, Iwamoto TA, Mori T, Wanaka A, Fukui H, Fujimura Y. Localization of ADAMTS13 to the stellate cells of human liver. Blood. 2005 doi: 10.1182/blood-2005-01-0152. [DOI] [PubMed] [Google Scholar]
- 11.Suzuki M, Murata M, Matsubara Y, Uchida T, Ishihara H, Shibano T, Ashida S, Soejima K, Okada Y, Ikeda Y. Detection of von Willebrand factor-cleaving protease (ADAMTS-13) in human platelets. Biochem Biophys Res Commun. 2004;313(1):212–216. doi: 10.1016/j.bbrc.2003.11.111. [DOI] [PubMed] [Google Scholar]
- 12.Liu L, Choi H, Bernardo A, Bergeron AL, Nolasco L, Ruan C, Moake JL, Dong JF. Platelet-derived VWF-cleaving metalloprotease ADAMTS-13. J Thromb Haemost. 2005;3(11):2536–2544. doi: 10.1111/j.1538-7836.2005.01561.x. [DOI] [PubMed] [Google Scholar]
- 13.Turner N, Nolasco L, Tao Z, Dong JF, Moake J. Human endothelial cells synthesize and release ADAMTS-13. J Thromb Haemost. 2006;4(6):1396–1404. doi: 10.1111/j.1538-7836.2006.01959.x. [DOI] [PubMed] [Google Scholar]
- 14.Tsai HM, Sussman II, Ginsburg D, Lankhof H, Sixma JJ, Nagel RL. Proteolytic cleavage of recombinant type 2A von Willebrand factor mutants R834W and R834Q: inhibition by doxycycline and by monoclonal antibody VP-1. Blood. 1997;89(6):1954–1962. [PubMed] [Google Scholar]
- 15.Crawley JT. Proteolytic inactivation of ADAMTS13 by thrombin and plasmin. 2005 doi: 10.1182/blood-2004-03-1101. [DOI] [PubMed] [Google Scholar]
- 16.Studt JD, Hovinga JA, Antoine G, Hermann M, Rieger M, Scheiflinger F, Lammle B. Fatal congenital thrombotic thrombocytopenic purpura with apparent ADAMTS13 inhibitor: in vitro inhibition of ADAMTS13 activity by hemoglobin. Blood. 2005;105(2):542–544. doi: 10.1182/blood-2004-06-2096. [DOI] [PubMed] [Google Scholar]
- 17.Bonnefoy A, Daenens K, Feys HB, De VR, Vandervoort P, Vermylen J, Lawler J, Hoylaerts MF. Thrombospondin-1 controls vascular platelet recruitment and thrombus adherence in mice by protecting (sub)endothelial VWF from cleavage by ADAMTS13. Blood. 2006;107(3):955–964. doi: 10.1182/blood-2004-12-4856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Apte SS. A disintegrin-like and metalloprotease (reprolysin type) with thrombospondin type 1 motifs: the ADAMTS family. Int J Biochem Cell Biol. 2004;36(6):981–985. doi: 10.1016/j.biocel.2004.01.014. [DOI] [PubMed] [Google Scholar]
- 19.Nicholson AC, Malik SB, Logsdon JM, Jr, Van Meir EG. Functional evolution of ADAMTS genes: evidence from analyses of phylogeny and gene organization. BMC Evol Biol. 2005;5(1):11. doi: 10.1186/1471-2148-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Majerus EM, Zheng X, Tuley EA, Sadler JE. Cleavage of the ADAMTS13 propeptide is not required for protease activity. J Biol Chem. 2003;278(47):46643–46648. doi: 10.1074/jbc.M309872200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou W, Dong L, Ginsburg D, Bouhassira EE, Tsai HM. Enzymatically active ADAMTS13 variants are not inhibited by anti-ADAMTS13 autoantibodies: a novel therapeutic strategy? J Biol Chem. 2005;280(48):39934–39941. doi: 10.1074/jbc.M504919200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng X, Nishio K, Majerus EM, Sadler JE. Cleavage of von Willebrand factor requires the spacer domain of the metalloprotease ADAMTS13. J Biol Chem. 2003;278(32):30136–30141. doi: 10.1074/jbc.M305331200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soejima K, Matsumoto M, Kokame K, Yagi H, Ishizashi H, Maeda H, Nozaki C, Miyata T, Fujimura Y, Nakagaki T. ADAMTS-13 cysteine-rich/spacer domains are functionally essential for von Willebrand factor cleavage. Blood. 2003;102(9):3232–3237. doi: 10.1182/blood-2003-03-0908. [DOI] [PubMed] [Google Scholar]
- 24.Gao W, Anderson PJ, Majerus EM, Tuley EA, Sadler JE. Exosite interactions contribute to tension-induced cleavage of von Willebrand factor by the antithrombotic ADAMTS13 metalloprotease. Proc Natl Acad Sci U S A. 2006;103(50):19099–19104. doi: 10.1073/pnas.0607264104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luken BM, Turenhout EA, Hulstein JJ, van Mourik JA, Fijnheer R, Voorberg J. The spacer domain of ADAMTS13 contains a major binding site for antibodies in patients with thrombotic thrombocytopenic purpura. Thromb Haemost. 2005;93(2):267–274. doi: 10.1160/TH04-05-0301. [DOI] [PubMed] [Google Scholar]
- 26.Klaus C, Plaimauer B, Studt JD, Dorner F, Lammle B, Mannucci PM, Scheiflinger F. Epitope mapping of ADAMTS13 autoantibodies in acquired thrombotic thrombocytopenic purpura. Blood. 2004;103(12):4514–4519. doi: 10.1182/blood-2003-12-4165. [DOI] [PubMed] [Google Scholar]
- 27.Tsai HM. Shear stress and von Willebrand factor in health and disease. Semin Thromb Hemost. 2003;29(5):479–488. doi: 10.1055/s-2003-44556. [DOI] [PubMed] [Google Scholar]
- 28.Tsai HM, Lian EC. Antibodies to von Willebrand factor-cleaving protease in acute thrombotic thrombocytopenic purpura. N Engl J Med. 1998;339(22):1585–1594. doi: 10.1056/NEJM199811263392203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruggeri ZM, Orje JN, Habermann R, Federici AB, Reininger AJ. Activation-independent platelet adhesion and aggregation under elevated shear stress. Blood. 2006;108(6):1903–1910. doi: 10.1182/blood-2006-04-011551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weiss HJ, Turitto VT, Baumgartner HR. Effect of shear rate on platelet interaction with subendothelium in citrated and native blood I Shear rate--dependent decrease of adhesion in von Willebrand's disease and the Bernard-Soulier syndrome. J Lab Clin Med. 1978;92(5):750–764. [PubMed] [Google Scholar]
- 31.Chauhan AK, Motto DG, Lamb CB, Bergmeier W, Dockal M, Plaimauer B, Scheiflinger F, Ginsburg D, Wagner DD. Systemic antithrombotic effects of ADAMTS13. J Exp Med. 2006;203(3):767–776. doi: 10.1084/jem.20051732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dong JF, Moake JL, Nolasco L, Bernardo A, Arceneaux W, Shrimpton CN, Schade AJ, McIntire LV, Fujikawa K, Lopez JA. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood. 2002;100(12):4033–4039. doi: 10.1182/blood-2002-05-1401. [DOI] [PubMed] [Google Scholar]
- 33.Motto DG, Chauhan AK, Zhu G, Homeister J, Lamb CB, Desch KC, Zhang W, Tsai HM, Wagner DD, Ginsburg D. Shigatoxin triggers thrombotic thrombocytopenic purpura in genetically susceptible ADAMTS13-deficient mice. J Clin Invest. 2005;115(10):2752–2761. doi: 10.1172/JCI26007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Furlan M, Robles R, Solenthaler M, Lammle B. Acquired deficiency of von Willebrand factor-cleaving protease in a patient with thrombotic thrombocytopenic purpura. Blood. 1998;91(8):2839–2846. [PubMed] [Google Scholar]
- 35.Furlan M, Robles R, Galbusera M, Remuzzi G, Kyrle PA, Brenner B, Krause M, Scharrer I, Aumann V, Mittler U, Solenthaler M, Lammle B. von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura and the hemolytic-uremic syndrome. N Engl J Med. 1998;339(22):1578–1584. doi: 10.1056/NEJM199811263392202. [DOI] [PubMed] [Google Scholar]
- 36.Tsai HM, Raoufi M, Zhou W, Guinto E, Grafos N, Ranzurmal S, Greenfield RS, Rand JH. ADAMTS13-binding IgG are present in patients with thrombotic thrombocytopenic purpura. Thromb Haemost. 2006;95(5):886–892. [PMC free article] [PubMed] [Google Scholar]
- 37.Levy GG, Nichols WC, Lian EC, Foroud T, McClintick JN, McGee BM, Yang AY, Siemieniak DR, Stark KR, Gruppo R, Sarode R, Shurin SB, Chandrasekaran V, Stabler SP, Sabio H, Bouhassira EE, Upshaw JD, Jr, Ginsburg D, Tsai HM. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature. 2001;413(6855):488–494. doi: 10.1038/35097008. [DOI] [PubMed] [Google Scholar]
- 38.Zhou W, Tsai HM. An enzyme immunoassay of ADAMTS13 distinguishes patients with thrombotic thrombocytopenic purpura from normal individuals and carriers of ADAMTS13 mutations. Thromb Haemost. 2004;91(4):806–811. doi: 10.1160/TH03-11-0675. [DOI] [PubMed] [Google Scholar]
- 39.Veyradier A, Obert B, Houllier A, Meyer D, Girma JP. Specific von Willebrand factor-cleaving protease in thrombotic microangiopathies: a study of 111 cases. Blood. 2001;98(6):1765–1772. doi: 10.1182/blood.v98.6.1765. [DOI] [PubMed] [Google Scholar]
- 40.Bohm M, Vigh T, Scharrer I. Evaluation and clinical application of a new method for measuring activity of von Willebrand factor-cleaving metalloprotease (ADAMTS13) Ann Hematol. 2002;81(8):430–435. doi: 10.1007/s00277-002-0502-3. [DOI] [PubMed] [Google Scholar]
- 41.Hovinga JA, Studt JD, Alberio L, Lammle B. von Willebrand factor-cleaving protease (ADAMTS-13) activity determination in the diagnosis of thrombotic microangiopathies: the Swiss experience. Semin Hematol. 2004;41(1):75–82. doi: 10.1053/j.seminhematol.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 42.Matsumoto M, Yagi H, Ishizashi H, Wada H, Fujimura Y. The Japanese experience with thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Semin Hematol. 2004;41(1):68–74. doi: 10.1053/j.seminhematol.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 43.Coppo P, Bengoufa D, Veyradier A, Wolf M, Bussel A, Millot GA, Malot S, Heshmati F, Mira JP, Boulanger E, Galicier L, Durey-Dragon MA, Fremeaux-Bacchi V, Ramakers M, Pruna A, Bordessoule D, Gouilleux V, Scrobohaci ML, Vernant JP, Moreau D, Azoulay E, Schlemmer B, Guillevin L, Lassoued K. Severe ADAMTS13 deficiency in adult idiopathic thrombotic microangiopathies defines a subset of patients characterized by various autoimmune manifestations, lower platelet count, and mild renal involvement. Medicine (Baltimore) 2004;83(4):233–244. doi: 10.1097/01.md.0000133622.03370.07. [DOI] [PubMed] [Google Scholar]
- 44.Terrell DR, Williams LA, Vesely SK, Lammle B, Hovinga JA, George JN. The incidence of thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: all patients, idiopathic patients, and patients with severe ADAMTS -13 deficiency. J Thromb Haemost. 2005;3(7):1432–1436. doi: 10.1111/j.1538-7836.2005.01436.x. [DOI] [PubMed] [Google Scholar]
- 45.Peyvandi F, Ferrari S, Lavoretano S, Canciani MT, Mannucci PM. von Willebrand factor cleaving protease (ADAMTS-13) and ADAMTS-13 neutralizing autoantibodies in 100 patients with thrombotic thrombocytopenic purpura. Br J Haematol. 2004;127(4):433–439. doi: 10.1111/j.1365-2141.2004.05217.x. [DOI] [PubMed] [Google Scholar]
- 46.Kokame K, Nobe Y, Kokubo Y, Okayama A, Miyata T. FRETS-VWF73, a first fluorogenic substrate for ADAMTS13 assay. Br J Haematol. 2005;129(1):93–100. doi: 10.1111/j.1365-2141.2005.05420.x. [DOI] [PubMed] [Google Scholar]
- 47.Zheng XL, Kaufman RM, Goodnough LT, Sadler JE. Effect of plasma exchange on plasma ADAMTS13 metalloprotease activity, inhibitor level, and clinical outcome in patients with idiopathic and nonidiopathic thrombotic thrombocytopenic purpura. Blood. 2004;103(11):4043–4049. doi: 10.1182/blood-2003-11-4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Uemura M, Matsuyama T, Ishikawa M, Fujimoto M, Kojima H, Sakurai S, Ishii S, Toyohara M, Yamazaki M, Yoshiji H, Yamao J, Matsumoto M, Ishizashi H, Fujimura Y, Fukui H. Decreased activity of plasma ADAMTS13 may contribute to the development of liver disturbance and multiorgan failure in patients with alcoholic hepatitis. Alcohol Clin Exp Res. 2005;29 12:264S–271S. doi: 10.1097/01.alc.0000192326.08931.cb. [DOI] [PubMed] [Google Scholar]
- 49.Ono T, Mimuro J, Madoiwa S, Soejima K, Kashiwakura Y, Ishiwata A, Takano K, Ohmori T, Sakata Y. Severe secondary deficiency of von Willebrand factor-cleaving protease (ADAMTS13) in patients with sepsis-induced disseminated intravascular coagulation: its correlation with development of renal failure. Blood. 2006;107(2):528–534. doi: 10.1182/blood-2005-03-1087. [DOI] [PubMed] [Google Scholar]
- 50.Nguyen TC, Liu A, Liu L, Ball C, Choi H, May WS, Aboulfatova K, Bergeron AL, Dong JF. Acquired ADAMTS-13 deficiency in pediatric patients with severe sepsis. Haematologica. 2007;92(1):121–124. doi: 10.3324/haematol.10262. [DOI] [PubMed] [Google Scholar]
- 51.Bianchi V, Robles R, Alberio L, Furlan M, Lammle B. Von Willebrand factor-cleaving protease (ADAMTS13) in thrombocytopenic disorders: a severely deficient activity is specific for thrombotic thrombocytopenic purpura. Blood. 2002;100(2):710–713. doi: 10.1182/blood-2002-02-0344. [DOI] [PubMed] [Google Scholar]
- 52.Lisman T, Bongers TN, Adelmeijer J, Janssen HL, De Maat MP, de Groot PG, Leebeek FW. Elevated levels of von Willebrand Factor in cirrhosis support platelet adhesion despite reduced functional capacity. Hepatology. 2006;44(1):53–61. doi: 10.1002/hep.21231. [DOI] [PubMed] [Google Scholar]
- 53.Banno F, Kokame K, Okuda T, Honda S, Miyata S, Kato H, Tomiyama Y, Miyata T. Complete deficiency in ADAMTS13 is prothrombotic, but it alone is not sufficient to cause thrombotic thrombocytopenic purpura. Blood. 2006;107(8):3161–3166. doi: 10.1182/blood-2005-07-2765. [DOI] [PubMed] [Google Scholar]
- 54.Torok TJ, Holman RC, Chorba TL. Increasing mortality from thrombotic thrombocytopenic purpura in the United States--analysis of national mortality data, 1968-1991. Am J Hematol. 1995;50(2):84–90. doi: 10.1002/ajh.2830500203. [DOI] [PubMed] [Google Scholar]
- 55.Miller DP, Kaye JA, Shea K, Ziyadeh N, Cali C, Black C, Walker AM. Incidence of thrombotic thrombocytopenic purpura/hemolytic uremic syndrome. Epidemiology. 2004;15(2):208–215. doi: 10.1097/01.ede.0000113273.14807.53. [DOI] [PubMed] [Google Scholar]
- 56.Schech SD, Brinker A, Shatin D, Burgess M. New-onset and idiopathic thrombotic thrombocytopenic purpura: incidence, diagnostic validity, and potential risk factors. Am J Hematol. 2006;81(9):657–663. doi: 10.1002/ajh.20669. [DOI] [PubMed] [Google Scholar]
- 57.Hymes KB, Karpatkin S. Human immunodeficiency virus infection and thrombotic microangiopathy. Semin Hematol. 1997;34(2):117–125. [PubMed] [Google Scholar]
- 58.Bennett CL, Weinberg PD, Rozenberg-Ben-Dror K, Yarnold PR, Kwaan HC, Green D. Thrombotic thrombocytopenic purpura associated with ticlopidine. A review of 60 cases. Ann Intern Med. 1998;128(7):541–544. doi: 10.7326/0003-4819-128-7-199804010-00004. [DOI] [PubMed] [Google Scholar]
- 59.Tsai HM, Rice L, Sarode R, Chow TW, Moake JL. Antibody inhibitors to von Willebrand factor metalloproteinase and increased binding of von Willebrand factor to platelets in ticlopidine-associated thrombotic thrombocytopenic purpura. Ann Intern Med. 2000;132(10):794–799. doi: 10.7326/0003-4819-132-10-200005160-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bennett CL, Connors JM, Carwile JM, Moake JL, Bell WR, Tarantolo SR, McCarthy LJ, Sarode R, Hatfield AJ, Feldman MD, Davidson CJ, Tsai HM. Thrombotic thrombocytopenic purpura associated with clopidogrel. N Engl J Med. 2000;342(24):1773–1777. doi: 10.1056/NEJM200006153422402. [DOI] [PubMed] [Google Scholar]
- 61.Zakarija A, Bandarenko N, Pandey DK, Auerbach A, Raisch DW, Kim B, Kwaan HC, McKoy JM, Schmitt BP, Davidson CJ, Yarnold PR, Gorelick PB, Bennett CL. Clopidogrel-associated TTP: an update of pharmacovigilance efforts conducted by independent researchers, pharmaceutical suppliers, and the Food and Drug Administration. Stroke. 2004;35(2):533–537. doi: 10.1161/01.STR.0000109253.66918.5E. [DOI] [PubMed] [Google Scholar]
- 62.Ridolfi RL, Hutchins GM, Bell WR. The heart and cardiac conduction system in thrombotic thrombocytopenic purpura. A clinicopathologic study of 17 autopsied patients. Ann Intern Med. 1979;91(3):357–363. doi: 10.7326/0003-4819-91-3-357. [DOI] [PubMed] [Google Scholar]
- 63.Podolsky SH, Zembowicz A, Schoen FJ, Benjamin RJ, Sonna LA. Massive myocardial necrosis in thrombotic thrombocytopenic purpura: a case report and review of the literature. Arch Pathol Lab Med. 1999;123(10):937–940. doi: 10.5858/1999-123-0937-MMNITT. [DOI] [PubMed] [Google Scholar]
- 64.Downes KA, Yomtovian R, Tsai HM, Silver B, Rutherford C, Sarode R. Relapsed thrombotic thrombocytopenic purpura presenting as an acute cerebrovascular accident. J Clin Apheresis. 2004;19(2):86–89. doi: 10.1002/jca.20007. [DOI] [PubMed] [Google Scholar]
- 65.Kennedy SS, Zacharski LR, Beck JR. Thrombotic thrombocytopenic purpura: analysis of 48 unselected cases. Semin Thromb Hemost. 1980;6(4):341–349. doi: 10.1055/s-2007-1005107. [DOI] [PubMed] [Google Scholar]
- 66.Bukowski RM. Thrombotic thrombocytopenic purpura. A review. Prog Hemost Thromb. 1982;6:287–337. [PubMed] [Google Scholar]
- 67.Mannucci PM, Vanoli M, Forza I, Canciani MT, Scorza R. Von Willebrand factor cleaving protease (ADAMTS-13) in 123 patients with connective tissue diseases (systemic lupus erythematosus and systemic sclerosis) Haematologica. 2003;88(8):914–918. [PubMed] [Google Scholar]
- 68.Sill H, Hofler G, Kaufmann P, Horina J, Spuller E, Kleinert R, Beham-Schmid C. Angiotropic large cell lymphoma presenting as thrombotic microangiopathy (thrombotic thrombocytopenic purpura) Cancer. 1995;75(5):1167–1170. doi: 10.1002/1097-0142(19950301)75:5<1167::aid-cncr2820750517>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 69.Lin YC, Chang HK, Sun CF, Shih LY. Microangiopathic hemolytic anemia as an initial presentation of metastatic cancer of unknown primary origin. South Med J. 1995;88(6):683–687. doi: 10.1097/00007611-199506000-00021. [DOI] [PubMed] [Google Scholar]
- 70.Fontana S, Gerritsen HE, Kremer HJ, Furlan M, Lammle B. Microangiopathic haemolytic anaemia in metastasizing malignant tumours is not associated with a severe deficiency of the von Willebrand factor-cleaving protease. Br J Haematol. 2001;113(1):100–102. doi: 10.1046/j.1365-2141.2001.02704.x. [DOI] [PubMed] [Google Scholar]
- 71.Forman RB, Benkel SA, Novik Y, Tsai HM. Presence of ADAMTS13 activity in a patient with metastatic cancer and thrombotic microangiopathy. Acta Haematol. 2003;109(3):150–152. doi: 10.1159/000069291. [DOI] [PubMed] [Google Scholar]
- 72.Sinha A, Rai R. Haemolytic uraemic syndrome following acute pancreatitis. JOP. 2005;6(4):365–368. [PubMed] [Google Scholar]
- 73.Moake JL, McPherson PD. Abnormalities of von Willebrand factor multimers in thrombotic thrombocytopenic purpura and the hemolytic-uremic syndrome. Am J Med. 1989;87(3N):9N–15N. [PubMed] [Google Scholar]
- 74.Tsai HM, Chandler WL, Sarode R, Hoffman R, Jelacic S, Habeeb RL, Watkins SL, Wong CS, Williams GD, Tarr PI. von Willebrand factor and von Willebrand factor-cleaving metalloprotease activity in Escherichia coli O157:H7-associated hemolytic uremic syndrome. Pediatr Res. 2001;49(5):653–659. doi: 10.1203/00006450-200105000-00008. [DOI] [PubMed] [Google Scholar]
- 75.Veyradier A, Nishikubo T, Humbert M, Wolf M, Sitbon O, Simonneau G, Girma JP, Meyer D. Improvement of von Willebrand factor proteolysis after prostacyclin infusion in severe pulmonary arterial hypertension. Circulation. 2000;102(20):2460–2462. doi: 10.1161/01.cir.102.20.2460. [DOI] [PubMed] [Google Scholar]
- 76.Vincentelli A, Susen S, Le TT, Six I, Fabre O, Juthier F, Bauters A, Decoene C, Goudemand J, Prat A, Jude B. Acquired von Willebrand syndrome in aortic stenosis. N Engl J Med. 2003;349(4):343–349. doi: 10.1056/NEJMoa022831. [DOI] [PubMed] [Google Scholar]
- 77.Tripodi A, Chantarangkul V, Bohm M, Budde U, Dong JF, Friedman KD, Galbusera M, Girma JP, Moake J, Rick ME, Studt JD, Turecek PL, Mannucci PM. Measurement of von Willebrand factor cleaving protease (ADAMTS -13): results of an international collaborative study involving 11 methods testing the same set of coded plasmas. J Thromb Haemost. 2004;2(9):1601–1609. doi: 10.1111/j.1538-7836.2004.00879.x. [DOI] [PubMed] [Google Scholar]
- 78.Rieger M, Ferrari S, Kremer Hovinga JA, Konetschny C, Herzog A, Koller L, Weber A, Remuzzi G, Dockal M, Plaimauer B, Scheiflinger F. Relation between ADAMTS13 activity and ADAMTS13 antigen levels in healthy donors and patients with thrombotic microangiopathies (TMA) Thromb Haemost. 2006;95(2):212–220. doi: 10.1160/TH05-08-0550. [DOI] [PubMed] [Google Scholar]
- 79.Shelat SG, Smith P, Ai J, Zheng XL. Inhibitory autoantibodies against ADAMTS-13 in patients with thrombotic thrombocytopenic purpura bind ADAMTS-13 protease and may accelerate its clearance in vivo. J Thromb Haemost. 2006;4(8):1707–1717. doi: 10.1111/j.1538-7836.2006.02025.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tsai HM, Li A, Rock G. Inhibitors of von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura. Clin Lab. 2001;47(78):387–392. [PubMed] [Google Scholar]
- 81.Rieger M, Mannucci PM, Hovinga JA, Herzog A, Gerstenbauer G, Konetschny C, Zimmermann K, Scharrer I, Peyvandi F, Galbusera M, Remuzzi G, Bohm M, Plaimauer B, Lammle B, Scheiflinger F. ADAMTS13 autoantibodies in patients with thrombotic microangiopathies and other immunomediated diseases. Blood. 2005;106(4):1262–1267. doi: 10.1182/blood-2004-11-4490. [DOI] [PubMed] [Google Scholar]
- 82.Byrnes JJ, Moake JL, Klug P, Periman P. Effectiveness of the cryosupernatant fraction of plasma in the treatment of refractory thrombotic thrombocytopenic purpura. Am J Hematol. 1990;34(3):169–174. doi: 10.1002/ajh.2830340303. [DOI] [PubMed] [Google Scholar]
- 83.Yarranton H, Lawrie AS, Purdy G, MacKie IJ, Machin SJ. Comparison of von Willebrand factor antigen, von Willebrand factor-cleaving protease and protein S in blood components used for treatment of thrombotic thrombocytopenic purpura. Transfus Med. 2004;14(1):39–44. doi: 10.1111/j.0958-7578.2004.00478.x. [DOI] [PubMed] [Google Scholar]
- 84.Scott EA, Puca KE, Pietz BC, Duchateau BK, Friedman KD. Comparison and stability of ADAMTS13 activity in therapeutic plasma products. Transfusion. 2007;47(1):120–125. doi: 10.1111/j.1537-2995.2007.01074.x. [DOI] [PubMed] [Google Scholar]
- 85.Allford SL, Harrison P, Lawrie AS, Liesner R, MacKie IJ, Machin SJ. Von Willebrand factor--cleaving protease activity in congenital thrombotic thrombocytopenic purpura. Br J Haematol. 2000;111(4):1215–1222. doi: 10.1046/j.1365-2141.2000.02503.x. [DOI] [PubMed] [Google Scholar]
- 86.Zeigler ZR, Shadduck RK, Gryn JF, Rintels PB, George JN, Besa EC, Bodensteiner D, Silver B, Kramer RE. Cryoprecipitate poor plasma does not improve early response in primary adult thrombotic thrombocytopenic purpura (TTP) J Clin Apheresis. 2001;16(1):19–22. doi: 10.1002/jca.1003. [DOI] [PubMed] [Google Scholar]
- 87.Rock G, Anderson D, Clark W, LeBlond P, Palmer D, Sternbach M, Sutton D, Wells G. Does cryosupernatant plasma improve outcome in thrombotic thrombocytopenic purpura? No answer yet. Br J Haematol. 2005;129(1):79–86. doi: 10.1111/j.1365-2141.2005.05418.x. [DOI] [PubMed] [Google Scholar]
- 88.Tsai HM. Physiologic cleavage of von Willebrand factor by a plasma protease is dependent on its conformation and requires calcium ion. Blood. 1996;87(10):4235–4244. [PubMed] [Google Scholar]
- 89.Bobbio-Pallavicini E, Gugliotta L, Centurioni R, Porta C, Vianelli N, Billio A, Tacconi F, Ascari E. Antiplatelet agents in thrombotic thrombocytopenic purpura (TTP). Results of a randomized multicenter trial by the Italian Cooperative Group for TTP. Haematologica. 1997;82(4):429–435. [PubMed] [Google Scholar]
- 90.Allford SL, Hunt BJ, Rose P, Machin SJ. Guidelines on the diagnosis and management of the thrombotic microangiopathic haemolytic anaemias. Br J Haematol. 2003;120(4):556–573. doi: 10.1046/j.1365-2141.2003.04049.x. [DOI] [PubMed] [Google Scholar]
- 91.George JN, Woodson RD, Kiss JE, Kojouri K, Vesely SK. Rituximab therapy for thrombotic thrombocytopenic purpura: a proposed study of the Transfusion Medicine/Hemostasis Clinical Trials Network with a systematic review of rituximab therapy for immune-mediated disorders. J Clin Apher. 2006;21(1):49–56. doi: 10.1002/jca.20091. [DOI] [PubMed] [Google Scholar]
- 92.Cataland SR, Wu HM. Immunotherapy for thrombotic thrombocytopenic purpura. Curr Opin Hematol. 2005;12(5):359–363. doi: 10.1097/01.moh.0000170534.33517.99. [DOI] [PubMed] [Google Scholar]
- 93.Wyllie BF, Garg AX, Macnab J, Rock GA, Clark WF. Thrombotic thrombocytopenic purpura/haemolytic uraemic syndrome: a new index predicting response to plasma exchange. Br J Haematol. 2006;132(2):204–209. doi: 10.1111/j.1365-2141.2005.05857.x. [DOI] [PubMed] [Google Scholar]
- 94.Vesely SK, Li X, McMinn JR, Terrell DR, George JN. Pregnancy outcomes after recovery from thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Transfusion. 2004;44(8):1149–1158. doi: 10.1111/j.1537-2995.2004.03422.x. [DOI] [PubMed] [Google Scholar]
- 95.Lattuada A, Rossi E, Calzarossa C, Candolfi R, Mannucci PM. Mild to moderate reduction of a von Willebrand factor cleaving protease (ADAMTS -13) in pregnant women with HELLP microangiopathic syndrome. Haematologica. 2003;88(9):1029–1034. [PubMed] [Google Scholar]
- 96.Sanchez-Luceros A, Farias CE, Amaral MM, Kempfer AC, Votta R, Marchese C, Salviu MJ, Woods AI, Meschengieser SS, Lazzari MA. von Willebrand factor-cleaving protease (ADAMTS13) activity in normal non-pregnant women, pregnant and post-delivery women. 2004;92(6):1320–1326. doi: 10.1160/TH03-11-0683. [DOI] [PubMed] [Google Scholar]
- 97.Schulman I, Pierce M, Lukens A, Currimbhoy Z. Studies on thrombopoiesis. I. A factor in normal human plasma required for platelet production; Chronic thrombocytopenia due to its deficiency. Blood. 1960;16(7):943–957. [PubMed] [Google Scholar]
- 98.Upshaw JD., Jr Congenital deficiency of a factor in normal plasma that reverses microangiopathic hemolysis and thrombocytopenia. N Engl J Med. 1978;298(24):1350–1352. doi: 10.1056/NEJM197806152982407. [DOI] [PubMed] [Google Scholar]
- 99.Tsai HM. Current concepts in thrombotic thrombocytopenic purpura. Annu Rev Med. 2006;57:419–436. doi: 10.1146/annurev.med.57.061804.084505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Plaimauer B, Fuhrmann J, Mohr G, Wernhart W, Bruno K, Ferrari S, Konetschny C, Antoine G, Rieger M, Scheiflinger F. Modulation of ADAMTS13 secretion and specific activity by a combination of common amino acid polymorphisms and a missense mutation. Blood. 2006;107(1):118–125. doi: 10.1182/blood-2005-06-2482. [DOI] [PubMed] [Google Scholar]
- 101.Shibagaki Y, Matsumoto M, Kokame K, Ohba S, Miyata T, Fujimura Y, Fujita T. Novel compound heterozygote mutations (H234Q/R1206X) of the ADAMTS13 gene in an adult patient with Upshaw-Schulman syndrome showing predominant episodes of repeated acute renal failure. Nephrol Dial Transplant. 2006;21(5):1289–1292. doi: 10.1093/ndt/gfk072. [DOI] [PubMed] [Google Scholar]
- 102.Tao Z, Anthony K, Peng Y, Choi H, Nolasco L, Rice L, Moake JL, Dong JF. Novel ADAMTS-13 mutations in an adult with delayed onset thrombotic thrombocytopenic purpura. J Thromb Haemost. 2006;4(9):1931–1935. doi: 10.1111/j.1538-7836.2006.02098.x. [DOI] [PubMed] [Google Scholar]
- 103.Donadelli R, Banterla F, Galbusera M, Capoferri C, Bucchioni S, Gastoldi S, Nosari S, Monteferrante G, Ruggeri ZM, Bresin E, Scheiflinger F, Rossi E, Martinez C, Coppo R, Remuzzi G, Noris M. In-vitro and in-vivo consequences of mutations in the von Willebrand factor cleaving protease ADAMTS13 in thrombotic thrombocytopenic purpura. Thromb Haemost. 2006;96(4):454–464. [PubMed] [Google Scholar]
- 104.Schneppenheim R, Kremer Hovinga JA, Becker T, Budde U, Karpman D, Brockhaus W, Hrachovinova I, Korczowski B, Oyen F, Rittich S, von RJ, Tjonnfjord GE, Pimanda JE, Wienker TF, Lammle B. A common origin of the 4143insA ADAMTS13 mutation. Thromb Haemost. 2006;96(1):3–6. doi: 10.1160/TH05-12-0817. [DOI] [PubMed] [Google Scholar]
- 105.Noris M, Bucchioni S, Galbusera M, Donadelli R, Bresin E, Castelletti F, Caprioli J, Brioschi S, Scheiflinger F, Remuzzi G. Complement factor H mutation in familial thrombotic thrombocytopenic purpura with ADAMTS13 deficiency and renal involvement. J Am Soc Nephrol. 2005;16(5):1177–1183. doi: 10.1681/ASN.2005010086. [DOI] [PubMed] [Google Scholar]