

Abstract
Protein tyrosine phosphatases (PTPs) are signaling enzymes that control a diverse array of cellular processes. Further insight into the specific functional roles of PTPs in cellular signaling requires detailed understanding of the molecular basis for substrate recognition by the PTPs. A central question is how PTPs discriminate between multiple structurally diverse substrates that they encounter in the cell. Although x-ray crystallography is capable of revealing the intimate structural details for molecular interaction, structures of higher order PTP•substrate complexes are often difficult to obtain. Hydrogen/deuterium exchange mass spectrometry (H/DX-MS) is a powerful tool for mapping protein-protein interfaces, as well as identifying conformational and dynamic perturbations in proteins. In addition, H/DX-MS enables analysis of large protein complexes at physiological concentrations and provides insight into the solution behavior of these complexes that can not be gleaned from crystal structures. We have utilized H/DX-MS to probe PTP dynamics, ligand binding, and the structural basis of substrate recognition. In this article, we review general principles of H/DX-MS technology as applied to study protein-protein interactions and dynamics. We also provide protocols for H/DX-MS successfully used in our laboratory to define the molecular basis of ERK2 substrate recognition by MKP3. Many of the aspects that we cover in detail should be applicable to the study of other PTPs with their specific targets.
Keywords: H/D exchange, mass spectrometry, protein tyrosine phosphatases (PTPs), dynamics, ligand binding, substrate specificity, MKP3, ERK2, protein-protein interaction
1. Introduction
Protein tyrosine phosphatases (PTPs) are a large and structurally diverse family of signaling enzymes that, together with protein tyrosine kinase, modulate the cellular level of protein tyrosine phosphorylation. An appropriate level of tyrosine phosphorylation is essential for regulating cell growth, differentiation, metabolism, progression through the cell cycle, cell-cell communication, cell migration, gene transcription, ion channel activity, the immune response, and apoptosis/survival decisions [1, 2]. Defective or inappropriate regulation of PTP activity leads to aberrant tyrosine phosporylation, which contributes to the development of many human diseases including cancers and diabetes [3]. Biochemical and structural studies have shown that all PTPs share a common mechanism of action to catalyze the hydrolysis of pTyr [4]. However, to fully appreciate the role of PTPs in signal transduction, it is necessary to have a detailed understanding of how they recognize their physiological substrates. This is because that although the PTPs have conserved catalytic domains and share a common mechanism of action (hydrolysis of pTyr), the cellular processes in which they are involved can be both highly specialized and fundamentally important, due in large part to the distinct substrate specificity of individual PTPs. In addition, in contrast to our understanding of the structure and chemical mechanism, very little is known about the dynamics associated with PTP substrate binding and catalysis. Many PTP enzymes are characterized by fluctuating conformations and dynamic motions, which are coupled to the binding and release of substrate or product. Such conformational changes and motions may function to exclude solvent, recruit essential amino acids for catalysis or to stabilize and/or prevent loss of reactive intermediates.
The detection and characterization of noncovalent protein-protein complexes and protein dynamics has been a challenging problem in traditional structural biology. Indeed, very few structural studies involve the analysis of PTPs with their physiological substrates due to the obvious difficulty in obtaining sufficient quantities of stoichiometrically phosphorylated proteins and the uncertainty of crystallizing the enzyme•substrate complex. Hydrogen/deuterium exchange mass spectrometry (H/DX-MS) has emerged in recent years as a powerful tool for mapping protein-ligand and protein-protein interfaces, as well as identifying conformational and dynamic perturbations in proteins [5-11]. Using this approach, we uncovered conformational and dynamic changes of Yesinia PTP induced by ligand binding and active site mutations [5]. We also established the binding mode of a high affinity small molecule inhibitor for PTP1B using H/DX-MS, which provides a structural framework for further development of potent and selective PTP inhibitory agents [6]. More recently, we employed H/DX-MS to map the interaction surfaces between MAP kinase phosphatase 3 (MKP3) and it physiological substrate ERK2 [12]. By combining the information gained from the H/DX-MS experiments and direct biochemical analyses, coupled with molecular modeling, we were able to define the structural elements in both MKP3 and ERK2 that are important for specific recognition. The results also yielded a structural model for understanding how efficient and precise docking interactions between ERK2 and its cognate substrates and regulators can be achieved. In this review, we describe the general principles of H/DX-MS technology as applied to study protein-protein interactions and dynamics. We then present protocols for H/DX-MS tailored to define the molecular basis of ERK2 substrate recognition by MKP3.
2. Principles of H/DX-MS
The exchangeable hydrogens in proteins include polar side-chain hydrogens bound to heteroatoms (N, O, and S), the N- and C-terminal hydrogens, and the backbone amide hydrogens [13, 14]. The H/D exchange rates for polar side-chain hydrogens are 103-106 times faster than those of the backbone amide hydrogens at neutral pH and so are usually too fast to be readily determined. Thus, of all exchangeable hydrogens present in a protein, only backbone amide hydrogens are measurable for H/D exchange studies. The backbone amide H/D exchange rate is dependent on a variety of factors including protein structure, pH and temperature. In general, solvent exposed amide hydrogens can exchange with solvent relatively rapidly, while amide protons that are inaccessible to solvent or are participating in stable hydrogen bonds must undergo transient distortion of local structure to allow exchange with significantly slower kinetics. In addition, ligand-binding or protein-protein interaction can also perturb protein structure and thus solvent accessibility of contact regions and exchange kinetics of amide protons in the proteins. This dependence of amide proton exchange rate on protein structure and protein-protein interaction makes H/D exchange an excellent and sensitive probe for monitoring protein conformational changes and mapping protein binding interfaces.
The use of mass spectrometry to analyze protein H/D exchange was initially developed by Smith and coworkers [15]. Over the past decade, this methodology has been optimized and successfully applied to study various proteins and has greatly contributed to our understanding of protein dynamics and recognition [10]. In a typical H/DX-MS experiment (Figure 1), the PTP of choice, either alone or in complex with its substrate, is incubated in D2O to allow exchange of protons with solvent deuterium, and mass spectrometry can be used to monitor in-exchange rates. To identify the segments of the PTP that display altered solvent accessibility, deuterium uptake in the PTP, either alone or in complex with its substrate, is locked in place at various times by rapidly lowering the pH and temperature. Global exchange measures the total number of amide protons that are protected form deuterium exchange upon complex formation. The overall deuterium level in protein or peptide can be obtained from the increase in molecular mass after solvent exchange in D2O. To identify regions in the PTP that display altered H/D exchange rate, the PTP is digested with pepsin, and the resulting peptides shall be separated by HPLC and analyzed by electrospray ionization-mass spectrometry. This measurement reports time-dependent changes in weighted average peptide masses, which yields rates of H/D exchange within different regions of the protein [10]. The binding interfaces and/or conformational changes can be localized by comparing the rates of H/D exchange on the PTP in bound and unbound states. In the following section, we provide detailed experimental procedures for H/DX-MS analysis of interactions between MKP3 and ERK2.
Figure 1.

A schematic for H/DX-MS experiment.
3. Reagents and Instrument
3.1 Reagents
Pepsin was obtained from Worthington, Co. (Lakewood, NJ) and pepsin solution was made freshly by dissolving pepsin in cold 0.5 M phosphate buffer, pH 2.5.
D2O (99.9 atom %D) from Aldrich Chemical Co. (Milwaukee, WI).
All other chemicals and reagents were of the highest-grade commercially available.
3.2 HPLC-MS System
A Shimazu HPLC equipped with two LC-10AD pumps was used to generate acetonitrile gradients. A solvent pre-cooling coil, a static mixing tee, and a Rheodyne injector were also equipped with the HPLC system. A Vydac1.0 × 50 mm C4 column was used for protein analysis, and a C18 or C8 Vydac 1.0 × 50 mm column was used for peptic peptides separation. During the HPLC run, the solvent pre-cooling coil, static mixing tee, Rheodyne injector, and column were immersed in an ice bath (0°C) to minimize back change with HPLC solvent. A Thermo Finnigan (Riviera Beach, FL) LCQ Deca XP plus ion trap mass spectrometer was used for mass analysis.
4. H/DX-MS Analysis of the MKP3-ERK2 Complex
ERK2 plays a central role in regulating cell growth and differentiation. MKP3, an ERK2 specific phosphatase, terminates ERK2 signaling. The structural basis for the exquisite specificity of ERK2 dephosphorylation by MKP3 has not been elucidated. In fact, it has been notoriously difficult to obtain co-crystals of ERK2 in complex with any of its interacting proteins. We sought to define the MKP3-ERK2/pTpY (ERK2 phosphorylated on both Thr183 and Tyr185) binding sites by determining the changes in solvent accessibility of the protein backbone amides as a result of complex formation between the full-length proteins in solution. However, given the transient nature of the enzyme•substrate complex, it is difficult to study the binding interaction between the wild-type MKP3 and the doubly phosphorylated ERK2/pTpY. Fortunately, the catalytic inactive Cys to Ser mutant PTP (e.g., Cys293 in MKP3) retains the ability to bind substrates but is unable to carry out substrate dephosphorylation. In fact, the Cys to Ser mutant binds substrates/ligands with the same affinity as the wild-type enzyme [5, 16, 17]. Consequently, we employed the catalytically inactive MKP3/C293S to capture the enzyme•substrate complex and to prevent the hydrolysis of ERK2/pTpY during H/DX-MS analysis.
4.1 Expression and Purification of ERK2/pTpY
ERK2/pTpY was expressed by transforming the plasmid pET-His6-ERK2-MEK1(R4F) (a generous gift of Dr. Melanie Cobb) in E. coli BL21(DE3). The expression and purification of ERK2/pTpY were carried out as described [18]. After the final FPLC step, approximately 3 mg of ERK2/pTpY was obtained from 6 liters of culture. The purified ERK2/pTpY was phosphorylated stoichiometrically to a ratio of 2 mol of phosphate per mole of ERK2, which was confirmed by mass spectrometry.
4.2 Expression and Purification of MKP3/C293S
The catalytically inactive MKP3/C293S with a C-terminal His6-tag was expressed in E. coli and purified as described previously [19]:
The plasmid pET21a-MKP3/C293S-His6 was used to transform E. coli BL21(DE3). An overnight culture (10 ml) of the transformed cells was diluted 1:100 into 1 liter of LB medium (supplemented with 100 μg/ml ampicillin) and allowed to grow at 37 °C until the absorbance at 600 nm was 0.6. Following the addition of isopropyl-thio-β-D-galactopyranoside to a final concentration of 1 mM, the culture was incubated at room temperature with shaking for an additional 6 hr.
The cells were harvested by centrifugation at 5,000 rpm (Sorvall Centrifuge, SS-34 rotor) for 5 min at 4°C. The cell pellets were resuspended in 30 ml of lysate buffer (20 mM Tris, pH 7.9, 500 mM NaCl, 5 mM imidazole), and lysed by passage through a French pressure cell press at 1,200 psi twice. After centrifuging the lysates at 16,000 rpm for 30 min at 4°C, the supernatant was collected.
A 4-ml 50% slurry of Ni2+-nitrilotriacetic acid agarose (Qiagen; preequilibrated by lysate buffer) was added to the supernatant. After incubating with gentle agitation at 4°C for 1 hr, the resin was transferred to a column and washed by 20 bed volumes of lysate buffer and 20 bed volumes of 20 mM Tris, pH 7.9, 500 mM NaCl, 20 mM imidazole. By eluting with 10 bed volumes of 20 mM Tris, pH 7.9, 500 mM NaCl, 100 mM imidazole, the His6-taged protein was collected and concentrated with a Centriprep-30 filtration unit (Amicon), and the buffer was changed to 25 mM Tris, pH 7.5, 1 mM EDTA, and 2 mM DTT. Protein concentration of MKP3/C293S was determined using the Bradford dye binding assay (Bio-Rad) diluted according to the manufacturer's recommendations with bovine serum albumin as standard.
4.3 Preparation of Controls
A “zero-deuteration” control was prepared by adding 1 μl of a 200 μM protein solution into a 19:20 (v/v) mixture of deuterated buffer and quenching buffer (0.5 M phosphate, pH 2.5). A “full-deuteration” control was prepared by incubating the protein sample for 16 hr at room temperature in 3M deuterated guanidine HCl followed by addition of an equal volume of cold quenching buffer. Peptic peptide controls were prepared by digesting the protein controls.
4.4 HPLC Conditions
Acetonitrile gradients 50 μl/min were generated with a Shimazu HPLC equipped with two LC-10AD pumps. Solvent A was 94.4% H2O containing 5% acetonitrile and 0.1% formic acid, and solvent B was 95% acetontirle containing 4.9% H2O and 0.1% formic acid. A low volume static mixing tee and a solvent pre-cooling coil were used to minimize the delay period for HPLC. The solvent pre-cooling coil, static mixing tee, Rheodyne injector, and the column were immersed in an ice bath (0°C) to minimize back exchange with HPLC solvents.
Protein sample: desalted at 5% solvent B for 5 min, the protein was then eluted with a 2 min 30-50% B gradient. The column effluent was delivered directly to the mass spectrometry.
Pepsin digests: the peptic peptides were eluted with a 0.5 min 5-15% B gradient followed by an 8 min 15-45% B gradient. The column effluent was delivered directly to the mass spectrometry.
4.5 MS Data Analysis
The column effluent (50 μl/min) was delivered directly to a Thermo Finnigan (Riviera Beach, FL) LCQ Deca XP plus ion trap mass spectrometer for mass analyses of deuterated protein and its peptic fragments. The deuterium content of a protein or a peptide was determined from the centriod of the molecular ion isotope peaks. MagTran 1.0 software (written by Dr. Zhongqi Zhang) was used to determine the centriod value for each peak in the list processed from the mass spectrum. The extent of H/D exchange was calculated using equation 1.
| (1) |
where m is the measured mass of deutered protein or peptide; m0% is the measured mass of a “zero-deuteration” control; m100% is the measured mass of a “full-deuteration” control; and H is the total exchangeable amide hydrogens excluding the N-terminal residue and prolines present in the protein or peptide [5].
The sequence of the peptide fragments obtained after pepsin digestion was identified using tandem mass spectrometry (MS/MS). Briefly, the total ion intensity of the peptides was detected in the m/z range of 400-2000. Mass scanning was followed by collision-induced dissociation to acquire a MS/MS spectrum within a peak during the HPLC run. MS/MS spectra were interpreted by searching peptide databases using SEQUEST algorithm.
4.6 Global H/D Exchange in Intact MKP3/C293S and ERK2/pTpY
H/D exchange in MKP3/C293S and ERK2/pTpY, either alone or in the complex form, was initiated by diluting 1 μl of the stock solution (200 μM) to 19 μl of the D2O buffer (20 mM Tris, 100 mM NaCl, pD 7.5). The final concentration for both proteins was 10 μM. Given the Kd of 31 nM for complex formation, 95% of MKP3/C293S and ERK2/pTpY should be in the bound state under the experimental conditions [12]. At appropriate time intervals, the H/D exchange reaction was quenched by addition of an equal volume of cold 0.5 M phosphate buffer, pH 2.5. One μl of the deuterated protein solution was loaded immediately onto a Vydac 1.0 × 50-mm C4 column and analyzed by mass spectrometry for global exchange. The C-terminal His6-tagged MKP3/C293S contains 387 amino acids (MW=43,117.5 Da) with 365 exchangeable amide hydrogens, while the N-terminal His6-tagged ERK2/pYpT has 364 residues (MW= 42,328.0 Da) with 343 exchangeable amide hydrogens. Figure 2 shows the total deuterium incorporation into MKP3/C293S and ERK2/pTpY either in the free or the bound form. Within 40 min, a total of 190 amide hydrogens in MKP3/C293S were replaced with deuterium in the absence of ERK2/pTpY while 136 of the MKP3/C293S amide protons were exchanged with deuterium in the presence of ERK2/pTpY, indicating that 54 amide protons in MKP3/C293S were protected form deuterium exchange upon complex formation. Compared to ERK2/pTpY alone, 30 of ERK2 amide protons were protected from exchange in the MKP3/C293S•ERK2/pTpY complex. These results indicate that an overall decrease in solvent accessibility occurred in MKP3/C293S and ERK2/pTpY as a result of protein-protein interaction. By determining the changes in solvent accessibility of MKP3/C293S and ERK2/pTpY resulting from complex formation, one can identify the MKP3-ERK2 binding interfaces and the structural changes that accompany the complex formation.
Figure 2.
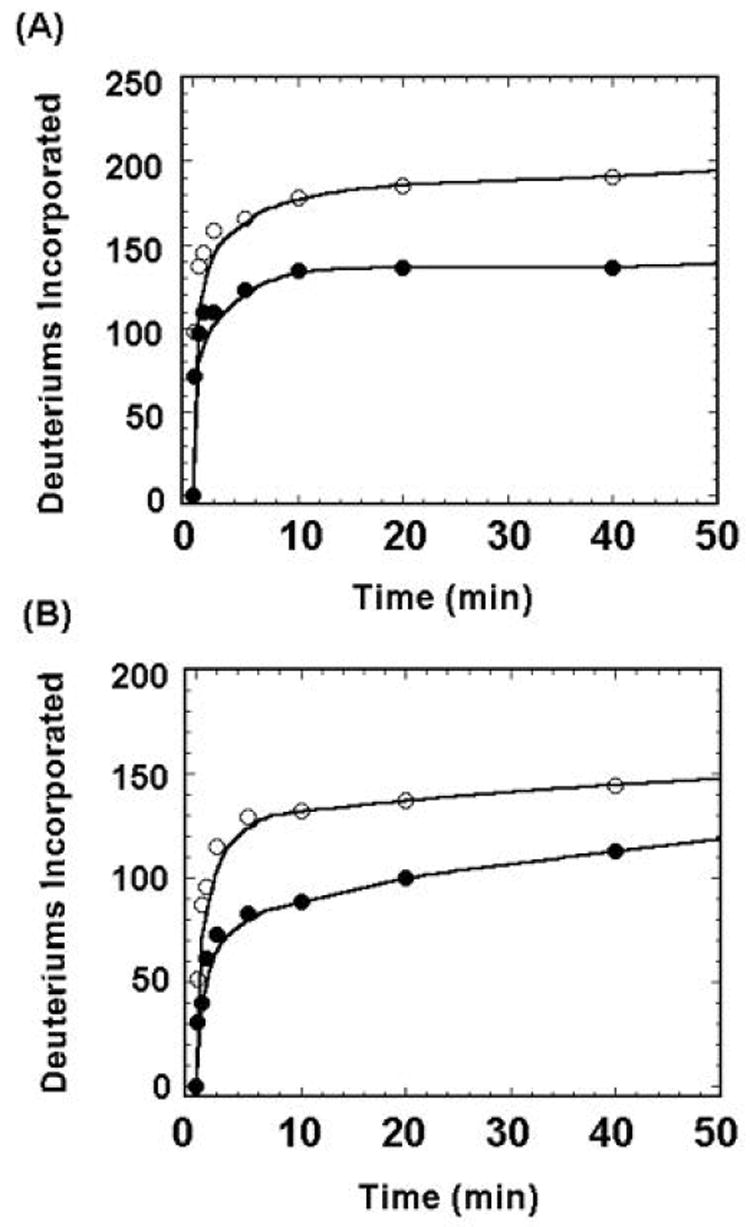
Global H/D exchange for MKP3/C293S (A) and ERK2/pTpY (B) alone (◯) or in complex (●).
4.7 Determination of Changes in Solvent Accessibility in Specific Segments of MKP3/C293S and ERK2/pTpY upon Complex Formation
H/DX-MS combined with peptidic mapping permits segment-specific identification of solvent-accessible exchange sites in proteins. The principle for this approach is that ligand-binding or protein-protein interaction perturbs protein structure and thus solvent accessibility of the contact regions. The binding interfaces can be localized by comparing the rates of H/D exchange on proteins in bound and unbound states. To identify the segments of MKP3/C293S and ERK2/pTpY that displayed altered solvent accessibility, deuterium uptake in MKP3/C293S and ERK2/pTpY was locked in place at various times by rapidly lowering the pH and temperature, the proteins were digested with pepsin, and the resulting peptides were separated by HPLC and analyzed by electrospray ionization-mass spectrometry. To analyze H/D exchange in segments of MKP3/C293S and ERK2/pTpY, the H/D exchange reaction was quenched by the addition of an equal volume of cold 0.5 M phosphate buffer, pH 2.5 in the presence of 20 μM pepsin. Immediately, 5 μl of the sample was injected into the HPLC sample loop where pepsin digestion proceeded at 0°C for 4 min. The pepsin digests were subsequently separated on a Vydac 1.0×50 mm C18 column.
Overall, 49 peptides (including those with overlapping sequences), covering 92% of MKP3 amino acid sequence, were identified by tandem mass spectrometry (MS/MS), as shown by the coverage map (Figure 3). Out of the 49 peptides, 30 displayed reduced exchange rates, 2 exhibited an increase in the exchange rates, and 17 had no change in deuterium incorporation upon binding to ERK2/pTpY. For ERK2/pTpY, 47 peptides were identified, effectively covering 92.5% of the primary structure [12]. Upon complex formation with MKP3/C293S, there were 22 peptides displaying a decrease in deuterium incorporation, 22 showing no change, and 3 peptides exhibiting an increase in deuterium incorporation.
Figure 3.
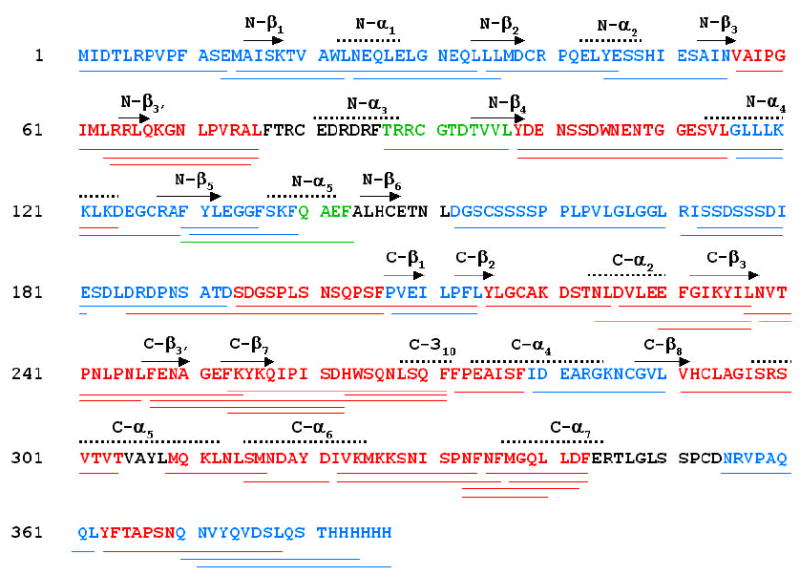
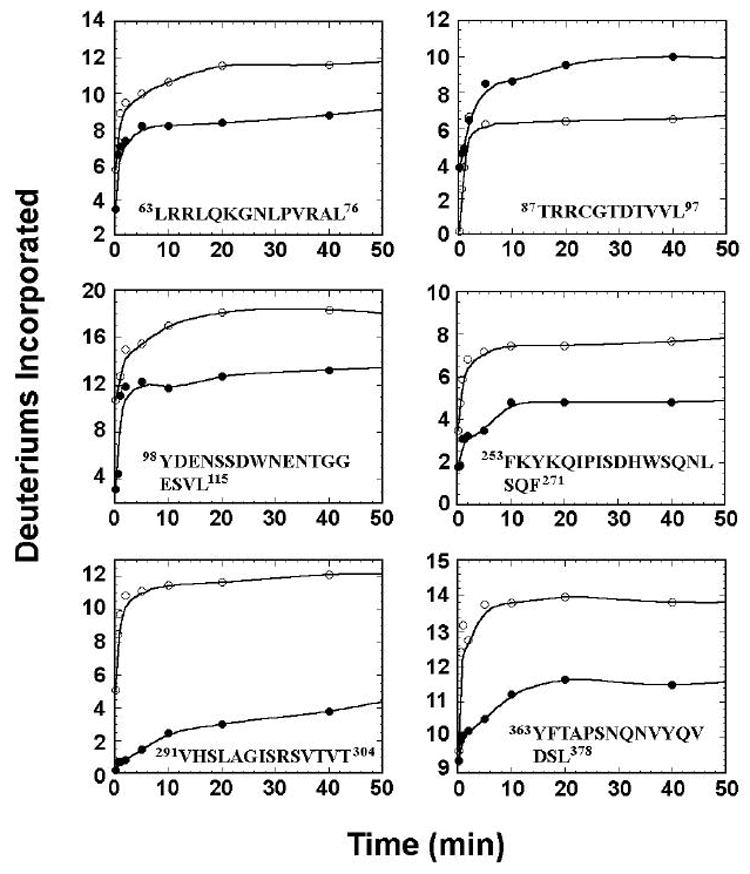
H/DX-MS analysis of MKP/C293S. (A) sequence coverage map of MKP3, showing 49 identified peptides in the MS/MS experiments. Indicated are peptides observed by H/DX-MS that exhibited a decrease (colored in red), an increase (colored in green), or no change (color in blue) in H/D exchange rates upon complex formation. Peptides in black were not identified in the MS/MS experiments. (B) time courses of deuterium uptake are shown for a number of peptides in MKP3/C293S that underwent significant changes in H/D exchange rates between the free (○) and ERK2/pTpY bound state (•).
4.8 Identification of the Binding Interfaces between ERK2/pTpY and MKP3
It should be noted that the H/DX-MS technique can not differentiate changes in solvent accessibility as a result of direct binding from those due to conformational or dynamic perturbations. Thus, peptide segments that show altered H/D exchange after complex formation can either be directly involved in ERK2/MKP3 binding or conformational/dynamic changes. Additional biochemical experiments in combination with the H/DX-MS data are required to identify the ERK2 binding sites in MKP3 [12]. Using a combination of H/DX-MS, mutagenesis and direct assessment of binding, a structural model was constructed for the MKP3-ERK2/pTpY complex (Figure 4). It appears that the exquisite specificity of MKP3 for phospho-ERK2 is governed by two distinctive protein-protein interactions. To increase the “effective concentration” of the interacting molecules, the kinase interaction motif (KIM) in MKP3 (64RRLQKGNLPVR74) and an MKP3-specific segment (101NSSDWNE107) bind the common docking (CD) site in ERK2 defined by residues in L16, L5, β7-β8, and αd-L8-αe, located opposite of the kinase active site. In addition to this “tethering” effect, additional interactions between the 364FTAP367 sequence in MKP3 and the ERK2 substrate-binding site, formed by residues in the activation lip and the P+1 site (β9-αf loop), L13 (αf-αg loop), and the MAP kinase insert (L14-α1L14-α2L14), are essential for allosteric activation of MKP3 and formation of a productive complex whereby the MKP3 catalytic site is correctly juxtaposed to carry out ERK2 dephosphorylation.
Figure 4.
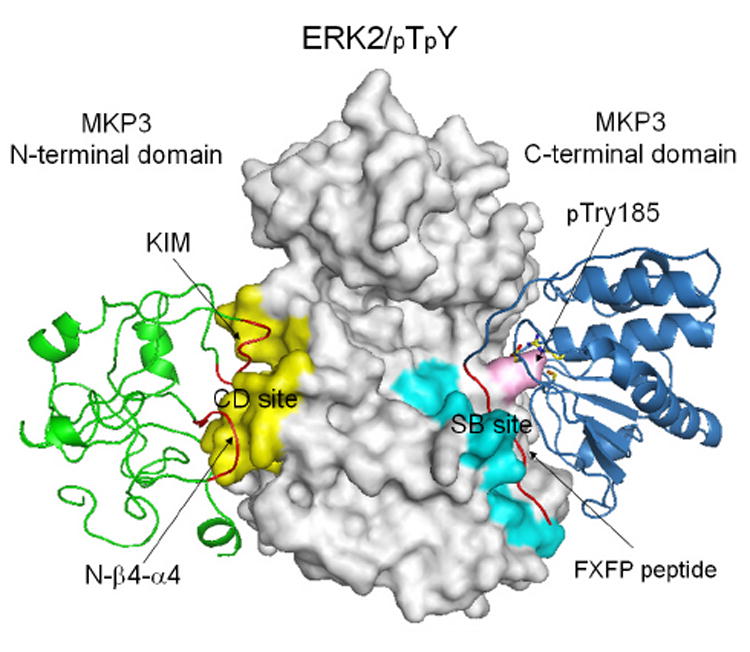
A structural model for the ERK2/pTpY-MKP3 complex. ERK2/pTpY was shown in gray, MKP3 N-terminal domain in green, and MKP3 C-terminal domain in blue. The common docking (CD) and substrate-binding (SB) site were colored yellow and cyan, respectively, and pTyr185 was colored pink. The kinase interaction motif (KIM) peptide, the N-β4-α4 loop, and the FXFP peptide were highlighted in red. MKP3 active site residues Asp262, Cys293, and Arg299 were also depicted as stick model in atomic colors.
5.Concluding Remarks
Given the importance of PTPs in cellular signaling, the interaction of PTPs with their physiological substrates must be rather specific to ensure proper integration of diverse biological stimuli. Further insight into the specific functional roles of PTPs in cellular signaling requires detailed understanding of the molecular basis for substrate recognition by the PTPs. A central question is how PTPs discriminate between multiple structurally diverse substrates that they encounter in the cell. Although x-ray crystallography is capable of revealing the intimate structural details for protein interaction, structures of higher order PTP•substrate complexes are often difficult to obtain. H/DX-MS enables analysis of large protein complexes at physiological concentrations and provides insight into the solution behavior of these complexes that can not be gleaned from crystal structures. In this article, we have reviewed general principles of H/DX-MS technology as applied to study protein-protein interactions and dynamics. We have also provided protocols for H/DX-MS successfully used in our laboratory to define the molecular basis of ERK2 substrate recognition by MKP3. Many of the aspects that we discussed in detail should be applicable to the study of recognition and interaction of PTPs with their specific targets.
Acknowledgments
Work in the author's laboratory was supported by NIH grants CA69202 and DK68447.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hunter T. Cell. 2000;100:113–127. doi: 10.1016/s0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- 2.Tonks NK, Benjamin GN. Cell Biology. 2001;13:182–195. doi: 10.1016/s0955-0674(00)00196-4. [DOI] [PubMed] [Google Scholar]
- 3.Zhang ZY. Curr Opin Chem Biol. 2001;5:416–423. doi: 10.1016/s1367-5931(00)00223-4. [DOI] [PubMed] [Google Scholar]
- 4.Zhang ZY. Annu Rev Pharmacol Toxicol. 2002;42:209–234. doi: 10.1146/annurev.pharmtox.42.083001.144616. [DOI] [PubMed] [Google Scholar]
- 5.Wang F, Li W, Emmett MR, Hendrickson CL, Marshall AG, Zhang YL, Wu L, Zhang ZY. Biochemistry. 1998;37:15289–15299. doi: 10.1021/bi981481q. [DOI] [PubMed] [Google Scholar]
- 6.Guo XL, Shen K, Wang F, Lawrence DS, Zhang ZY. J Biol Chem. 2002;277:41014–41022. doi: 10.1074/jbc.M207347200. [DOI] [PubMed] [Google Scholar]
- 7.Engen JR, Smith DL. Anal Chem. 2001;73:256A–265A. doi: 10.1021/ac012452f. [DOI] [PubMed] [Google Scholar]
- 8.Anand GS, Law D, Mandell JG, Snead AN, Tsigelny I, Taylor SS, Ten Eyck LF, Komives EA. Proc Natl Acad Sci USA. 2003;100:13264–12369. doi: 10.1073/pnas.2232255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoofnagle AN, Resing KA, Goldsmith EJ, Ahn NG. Proc Natl Acad Sci USA. 2001;98:956–961. doi: 10.1073/pnas.98.3.956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoofnagle AN, Resing KA, Ahn NG. Annu Rev Biophys Biomol Struct. 2003;32:1–25. doi: 10.1146/annurev.biophys.32.110601.142417. [DOI] [PubMed] [Google Scholar]
- 11.Lee T, Hoofnagle AN, Kabuyama Y, Stroud J, Min X, Goldsmith EJ, Chen L, Resing KA, Ahn NG. Mol Cell. 2004;14:43–55. doi: 10.1016/s1097-2765(04)00161-3. [DOI] [PubMed] [Google Scholar]
- 12.Zhou B, Zhang J, Liu S, Reddy S, Wang F, Zhang ZY. J Biol Chem. 2006;281:38834–38844. doi: 10.1074/jbc.M608916200. [DOI] [PubMed] [Google Scholar]
- 13.Englander SW, Kallenbach N. Quart Rev Biophys. 1984;16:521–655. doi: 10.1017/s0033583500005217. [DOI] [PubMed] [Google Scholar]
- 14.Woodward C, Simon I, Tuechsen E. Mol Cell Biochem. 1982;48:135–160. doi: 10.1007/BF00421225. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Z, Smith DL. Protein Sci. 1993;2:522–531. doi: 10.1002/pro.5560020404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang YL, Yao ZJ, Sarmiento M, Wu L, Burke TR, Jr, Zhang ZY. J Biol Chem. 2000;275:34205–34212. doi: 10.1074/jbc.M004490200. [DOI] [PubMed] [Google Scholar]
- 17.Xie L, Zhang YL, Zhang ZY. Biochem. 2002;41:4032–4039. doi: 10.1021/bi015904r. [DOI] [PubMed] [Google Scholar]
- 18.Wilsbacher JL, Cobb MH. Methods Enzymol. 2001;302:387–400. doi: 10.1016/s0076-6879(01)32217-6. [DOI] [PubMed] [Google Scholar]
- 19.Liu S, Sun JP, Zhou B, Zhang ZY. Proc Natl Acad Sci USA. 2006;103:5326–5331. doi: 10.1073/pnas.0510506103. [DOI] [PMC free article] [PubMed] [Google Scholar]