

Abstract
This study was performed to determine whether augmented intrarenal angiotensinogen may contribute to the enhanced renal angiotensin II (Ang II) and associated tissue injury in spontaneously hypertensive rats (SHR). SHR and Wistar-Kyoto rats (WKY) were maintained on a normal diet and killed at either 7 or 14 wk of age. Two groups of SHR received either an Ang II type 1 receptor blocker (ARB; olmesartan, 5 mg/d) or a triple therapy (hydralazine 7.5 mg/d, reserpine 0.15 mg/d, and hydrochlorothiazide 3 mg/d [HRH]) during weeks 7 through 14. Systolic BP and renal Ang II were significantly increased in SHR-14 (n = 8) compared with WKY-7, WKY-14, and SHR-7 (n = 8 each), and ARB treatment prevented these increases (n = 8). However, whereas HRH treatment prevented the development of hypertension in SHR, this combination therapy failed to decrease renal Ang II (n = 8). With the use of urine samples or fixed renal sections, renal injuries in rats were quantified in a semiautomated manner by the following six parameters: (1) urinary excretion rate of total protein, (2) glomerular sclerosis, (3) interstitial expansion, (4) and (5) numbers of monocytes/macrophages in interstitium or glomeruli, and (6) arterial proliferation. Angiotensinogen mRNA and protein levels in kidney cortex, measured by real-time reverse transcriptase—PCR and Western blot analysis, respectively, and all six parameters of renal damage were changed in parallel, and ARB treatment also prevented these increases. However, HRH treatment failed to prevent these increases. These results indicate that SHR have enhanced intrarenal angiotensinogen production that contributes to increased Ang II levels leading to the development of hypertension and renal injury in this strain.
Spontaneously hypertensive rats (SHR) have been used as a model of hypertension (1). Mature SHR are reported to have low plasma renin activity (2), which has been interpreted as being indicative of an overall suppression of the renin-angiotensin system (RAS) (3). However, few studies of angiotensinogen have been carried out in these rats. Although generally considered to be characterized by a low activity of circulating RAS, recent studies indicate that treatment with angiotensin I (Ang I)-converting enzyme (ACE) inhibitors and/or Ang II type I (AT1) receptor blocker (ARB) reduces cardiac and/or renal dysfunction in SHR (4-6). These findings suggest that the intrarenal RAS may be inappropriately activated and in turn contribute to the development of hypertension and hypertension-induced renal damages in this animal model.
We recently reported that Dahl-salt sensitive rats on a high-salt diet have an inappropriate augmentation of intrarenal angiotensinogen, which may contribute to impaired sodium excretion and the development of hypertension in this strain (7,8). These results prompted us to perform further experiments to evaluate the intrarenal level of angiotensinogen in other animal models of hypertension. This study was performed to determine whether an augmented intrarenal angiotensinogen contributes to the enhanced renal Ang II and associated early tissue injury in SHR as well as whether the elevated level of intrarenal angiotensinogen in SHR is dependent on the activation of AT1 receptors. Therefore, SHR and age-matched Wistar-Kyoto rats (WKY) were used in this study, and renal parameters of RAS activity and tissue damage were evaluated with or without a chronic ARB treatment.
Materials and Methods
Preparation of Animals
The experimental protocol was approved by the Animal Care and Use Committee of Tulane University and Kagawa University. Male SHR and WKY (7 wk of age; Charles River Laboratories, Wilmington, MA; n = 48) were housed in metabolic cages and maintained in a temperature-controlled room regulated on a 12-h light/dark cycle with free access to water. Rats were maintained on a diet that consisted of commercially available rat chow that contained normal salt (0.3% sodium chloride; #170950 Harlan Teklad, Madison, WI). Two groups of SHR received doses of either an ARB, olmesartan (Sankyo, Tokyo, Japan; 5 mg/d in food), or a triple therapy (hydralazine 7.5 mg/d, reserpine 0.15 mg/d, and hydrochlorothiazide 3 mg/d in food [HRH]; Sigma, St. Louis, MO) during weeks 7 through 14. This amount of olmesartan was shown effectively to block development of hypertension in our previous study performed on Ang II-infused Sprague-Dawley rats (9). Likewise, similar results were previously found by others using the triple therapy in a study performed in SHR (10). Systolic BP was measured in conscious rats using tail-cuff plethysmography once a week as described previously (9,11-14).
Sample Collection
Twenty-four-hour urine samples were collected on the day before the harvesting day in centrifuge tubes that contained a protease inhibitor, 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride (1 mmol/L in final concentration; Sigma). Urine samples were centrifuged, and supernatant was separated and stored at -20°C until assayed for total protein. Urinary concentration of total protein was measured by colorimetric assays using commercially available kits (Bio-Rad, Hercules, CA).
Blood and kidney samples were harvested at either 7 or 14 wk of age. After decapitation, trunk blood was collected into chilled tubes that contained EDTA (5 mmol/L), enalaprilat (20 μmol/L), pepstatin A (10 μmol/L), and 1,10-phenanthrolene (1.25 mmol/L). Plasma was separated and stored at -20°C until assayed for plasma Ang II as described previously (9,11-14). Immediately after removal, one kidney was homogenized in cold methanol and renal Ang II was measured as described previously (9,11-14). The contralateral kidneys were separated into three pieces and immersed in RNAlater (Ambion, Austin, TX) for total RNA extraction, immersed in zinc-saturated formalin (Anatech, Battle Creek, MI) for tissue fixation, and snap-frozen in liquid nitrogen for protein extraction, respectively.
Quantitative Real-Time Reverse Transcriptase-PCR
Total RNA extraction of renal cortex and quantitative real-time reverse transcriptase-PCR for angiotensinogen and renin were performed as described previously (15). Data of quantitative real-time reverse transcriptase-PCR were normalized by glyceraldehyde-3-phosphate dehydrogenase mRNA expression. The information of sequences were as follows: angiotensinogen, forward primer 5′-AGG CAA GAG GTG TAG CCA GT-3′, reverse primer 5′-AGG ACC TTA TGT CCG TCC AG-3′, probe 5′-/56-FAM/TCT TTC TAC CTT GGA TCG TTG GAT CCC/3BHQ-1/-3′; renin, forward primer 5′-TGC TAA AGG AGG AAG TGT TT-3′, reverse primer 5′-TGA TGC TCA CGT AGT GAA AG-3′, probe 5′-/56-FAM/TCT GTC TAC TAC AGC AGG GAG TCC C/3BHQ-1/-3′; glyceraldehyde-3-phosphate dehydrogenase, forward primer 5′-CAG AAC ATC ATC CCT GCA TC-3′, reverse primer 5′-CTG CTT CAC CAC CTT CTT GA-3′, probe 5′-/5-HEX/CCT GGA GAA ACC TGC CAA GTA TGA TGA/3BHQ-2/-3′.
Western Blot Analysis
Protein extraction of renal cortex and Western blot analysis for angiotensinogen and β-actin were performed as described previously (7-9,11-14). Data of Western blot analysis for angiotensinogen protein levels were normalized by β-actin protein levels.
Evaluation of Renal Injury
With the use of urine samples or fixed renal sections, renal injuries in rats were quantified by the following six parameters: (1) Urinary excretion rate of total protein (overall index of renal injury), (2) glomerular sclerosis, (3) interstitial expansion, (4) and (5) numbers of monocytes/macrophages in interstitium or glomeruli, and (6) arterial proliferation.
(1) Urinary concentration of total protein was measured using a commercially available kit as described in the Sample Collection section.
(2) The extent of glomerular sclerosis was evaluated quantitatively by an automatic image analysis to each glomerulus using periodic acid-Schiff-stained sections (Mass Histology Service, Worcester, MA) as described previously (16). The ratio of the affected lesions to each glomerulus was calculated using the Image-Pro plus software (Media Cybernetics, Silver Spring, MD). For each glomerulus, the affected lesion where the intensity was beyond a threshold calculated by the background signal (stronger pink in background pink) was measured automatically by the software, and this affected area in turn was divided by the total area of the glomerulus. Twenty glomeruli were examined for each rat, and the averaged percentages of the affected lesions were obtained for each rat.
(3) The extent of interstitial expansion was evaluated quantitatively by an automatic image analysis to renal cortex occupied by interstitial tissue staining positively for collagen in Masson’s trichrome-stained sections (Mass Histology) as described previously (17). The fraction of renal cortex occupied by interstitial tissue was performed using the Image-Pro plus software. For each microscopic field, the collagen-positive area (blue) was calculated automatically by the software, and this affected area was in turn divided by the total area of the microscopic field. Twenty consecutive microscopic fields were examined for each rat, and the averaged percentages of the collagen-positive lesions were obtained for each rat.
(4) and (5) The numbers of monocytes/macrophages were examined by immunohistochemistry using a commercially available antibody against CD68 (Serotec, Oxford, UK) as described previously (16). Immunohistochemistry was performed by a robotic system (Dako autostainer) as described previously (9,15) and counterstained with hematoxylin-eosin. Twenty consecutive microscopic fields were examined for each rat, and CD68-positive cells (brown) were counted in interstitium or glomeruli in each of the rats. The averaged numbers of monocytes/macrophages in interstitium or glomeruli then were obtained for each rat.
(6) The magnitude of arterial proliferation of afferent arteriolar walls was evaluated by immunohistochemistry using a commercially available antibody against α-smooth muscle isoform of actin (Sigma) as described previously (18). Immunohistochemistry was performed by a robotic system (Dako autostainer) as described previously (9,15). As α-smooth muscle actin was expressed on both afferent and efferent arterioles, elastin was used, which stains only preglomerular vessels, to identify afferent arterioles. The thickness of the wall of afferent arterioles then was measured with microscopy (brown vessels besides a glomerulus in the center of the microscopic field). Twenty afferent arteriolar walls were examined for each rat, and the averaged thickness of the wall of afferent arterioles was obtained for each rat.
The above histologic analyses were performed by an outsourcing company or a robotic system with an automatic image analysis software in a blind manner to avoid any biases.
Statistical Analyses
Statistical analysis was performed using a one-way factorial ANOVA with post hoc Scheffe F test. All data are presented as mean ± SEM. P < 0.05 was considered significant.
Results
Systolic BP
Systolic BP (Figure 1A) values were similar among SHR and WKY at 7 wk of age (126 ± 5 and 127 ± 3 mmHg, respectively). SHR showed a progressive increase in systolic BP up to an average of 206 ± 5 mmHg at 14 wk of age, whereas WKY maintained their systolic BP at the basal levels (123 ± 7 mmHg at 14 wk of age). ARB treatment with olmesartan and HRH from 7 to 14 wk of age equally prevented the development of hypertension in SHR (130 ± 3 and 131 ± 3 mmHg, respectively, at 14 wk of age).
Figure 1.
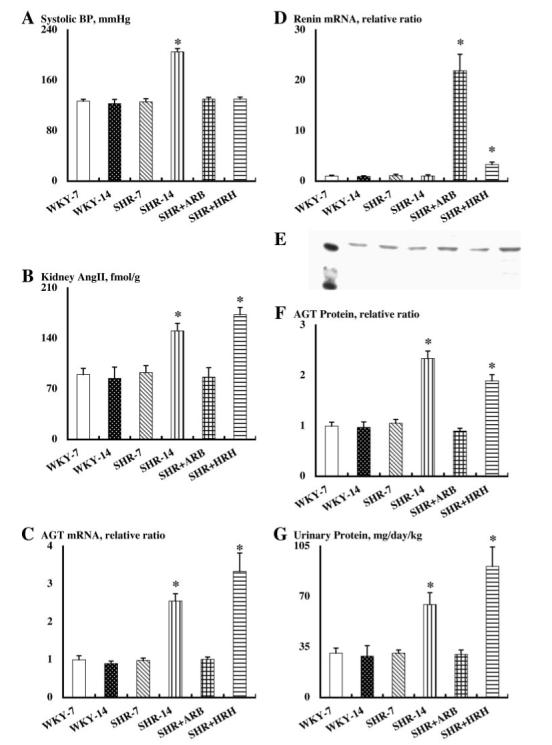
Systolic BP (A), kidney angiotensin II (Ang II) contents (B), angiotensinogen (AGT) mRNA levels in renal cortex (C), renin mRNA levels in renal cortex (D), AGT protein levels in renal cortex (F), and urinary exertion rate of total protein (G) in each group. (E) A representative Western blot analysis of AGT protein levels in renal cortex of each animal group. Each lane represents the following: Molecular size maker at 40 and 50 kD, WKY-7, WKY-14, SHR-7, SHR-14, SHR+ARB, and SHR+HRH. *P < 0.05 versus SHR-14 and WKY-14 groups. WKY, Wistar-Kyoto rats; SHR, spontaneously hypertensive rats; ARB, Ang II type 1 receptor blocker; HRH, triple therapy with hydralazine, reserpine, and hydrochlorothiazide.
Plasma and Kidney Ang II
Plasma Ang II concentrations were unaltered among the WKY groups (84 ± 12 fmol/ml for WKY at 7 wk of age and 86 ± 18 fmol/ml for WKY at 14 wk of age). ARB treatment and HRH treatment showed a tendency to increase plasma Ang II concentrations in the SHR groups, but these changes were not statistically significant (76 ± 10 fmol/ml for SHR at 7 wk of age, 69 ± 17 fmol/ml for SHR at 14 wk of age, 87 ± 9 fmol/ml for SHR at 14 wk of age with ARB treatment, 83 ± 2 fmol/ml for SHR at 14 wk of age with HRH treatment).
Similar to plasma concentrations, intrarenal Ang II levels (Figure 1B) were unchanged in WKY (90 ± 8 fmol/g kidney wt for WKY at 7 wk of age, 85 ± 16 fmol/g kidney wt for WKY at 14 wk of age). However, intrarenal Ang II levels were significantly increased in SHR at 14 wk of age (151 ± 11 fmol/g kidney wt) compared with prehypertensive SHR (93 ± 10 fmol/g kidney wt) and the age-matched WKY. ARB treatment completely blocked this increase in intrarenal Ang II (87 ± 13 fmol/g kidney wt). However, HRH treatment failed to prevent this increase in intrarenal Ang II (173 ± 10 fmol/g kidney wt) even although HRH treatment effectively blocked the development of hypertension in SHR.
Renal Cortical mRNA Expression of Angiotensinogen and Renin
Angiotensinogen mRNA expression in the kidney cortex (Figure 1C) changed in a similar manner to the intrarenal Ang II levels. Angiotensinogen mRNA levels were not changed in WKY (1.0 ± 0.1 relative ratio for WKY at 7 wk of age, 0.9 ± 0.1 relative ratio for WKY at 14 wk of age). However, angiotensinogen mRNA levels were significantly increased in SHR at 14 wk of age (2.6 ± 0.2 relative ratio) compared with SHR at 7 wk of age (1.0 ± 0.1 relative ratio) and the age-matched WKY. ARB treatment prevented the augmentation of angiotensinogen mRNA in the cortex (1.0 ± 0.1 relative ratio). However, HRH treatment failed to prevent this augmentation of angiotensinogen mRNA in cortex (3.3 ± 0.5 relative ratio).
Meanwhile, renin mRNA levels in the kidney cortex (Figure 1D) were not different between SHR and WKY at 7 and 14 wk of age (1.0 ± 0.2 relative ratio for WKY at 7 wk of age, 0.9 ± 0.1 relative ratio for WKY at 14 wk of age, 1.1 ± 0.3 relative ratio for SHR at 7 wk of age, 1.0 ± 0.2 relative ratio for SHR at 14 wk of age). However, ARB treatment to SHR markedly enhanced renin mRNA expression in cortex (21.9 ± 3.3 relative ratio). HRH treatment to SHR also stimulated renin mRNA expression in the cortex (3.3 ± 0.4 relative ratio).
Renal Cortical Protein Expression of Angiotensinogen
Angiotensinogen protein expression in the kidney cortex (Figure 1, E and F) changed in a similar manner to the intrarenal Ang II levels. Angiotensinogen protein levels were not changed in WKY (1.0 ± 0.1 relative ratio for WKY at 7 wk of age, 1.0 ± 0.1 relative ratio for WKY at 14 wk of age). However, angiotensinogen protein levels were significantly increased in SHR at 14 wk of age (2.3 ± 0.1 relative ratio) compared with SHR at 7 wk of age (1.1 ± 0.1 relative ratio) and the age-matched WKY. ARB treatment prevented the augmentation of angiotensinogen protein in the cortex (0.9 ± 0.1 relative ratio). However, HRH treatment failed to prevent this augmentation of angiotensinogen protein in the cortex (1.9 ± 0.1 relative ratio). β-Actin protein levels in the kidney cortex were not altered in any group (data not shown).
Urinary Excretion Rate of Total Protein
Overall renal injury in rats was evaluated by urinary excretion rate of total protein (Figure 1G). Urinary total protein excretion rate changed in a similar manner to intrarenal Ang II levels. Urinary total protein levels were not changed in WKY (31 ± 4 mg/d per kg body wt for WKY at 7 wk of age, 29 ± 7 mg/d per kg body wt for WKY at 14 wk of age). However, urinary total protein excretion rates were significantly increased in SHR at 14 wk of age (65 ± 8 mg/d per kg body wt) compared with SHR at 7 wk of age (31 ± 2 mg/d per kg body wt) and the age-matched WKY. ARB treatment prevented the increase in urinary protein excretion (30 ± 3 mg/d per kg body wt). In contrast, HRH treatment further enhanced the increase in urinary protein excretion (91 ± 13 mg/d per kg body wt).
Glomerular Sclerosis
The extent of glomerular sclerosis was quantified using an automatic image analysis of glomeruli in periodic acid-Schiff-stained sections (Figure 2). The glomerular sclerotic index changed in parallel with urinary protein excretion. The glomerular sclerotic index was found to be similar in WKY at 7 and 14 wk of age (12.7 ± 0.8 and 13.4 ± 0.3%, respectively; Figure 2, A and B). However, SHR at 14 wk of age (17.1 ± 0.2%; Figure 2D) showed a significantly higher glomerular sclerotic index compared with SHR at 7 wk of age (12.7 ± 0.5%; Figure 2C) and the age-matched WKY. ARB treatment normalized the increase in the glomerular sclerotic index (13.2 ± 0.5%; Figure 2E). However, HRH treatment failed to prevent the increase in the glomerular sclerotic index (16.1 ± 0.4%; Figure 2F).
Figure 2.
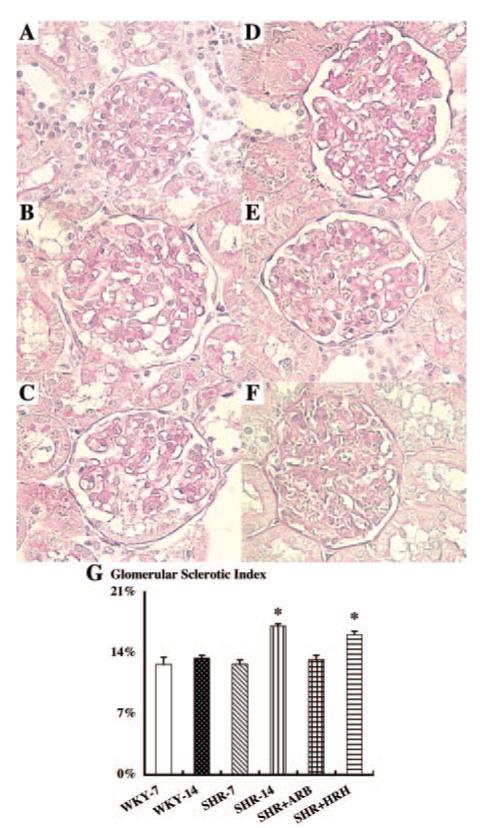
Glomerular sclerotic index using periodic acid-Schiff-stained sections. As evident by A through F, obvious morphologic changes were not observed in each group. However, an automatic image analysis in this study readily detected an increase in glomerular sclerotic index in SHR at 14 wk of age (D) compared with other groups (WKY at 7 wk of age [A]; WKY at 14 wk of age [B]; SHR at 7 wk of age [C]). ARB treatment blocked this increase in SHR at 14 wk of age (E). HRH treatment failed to block this increase in SHR at 14 wk of age (F). *P < 0.05 versus SHR-14 and WKY-14 groups.
Interstitial Expansion
The extent of interstitial expansion was quantified by an automatic image analysis of each microscopic field in the renal cortex using Masson’s trichrome-stained sections (data not shown). The interstitial expansive index changed in parallel with the glomerular sclerotic index. The extent of interstitial expansion was not different in WKY at 7 and 14 wk of age (0.21 ± 0.06 and 0.23 ± 0.03%, respectively). However, SHR at 14 wk of age (0.48 ± 0.03%) showed a significantly higher interstitial expansive index compared with SHR at 7 wk of age (0.21 ± 0.01%) and the age-matched WKY. ARB treatment normalized the increase in the interstitial expansive index (0.22 ± 0.02%). However, HRH treatment failed to prevent the increase in the interstitial expansive index (0.47 ± 0.05%).
Interstitial Inflammation
The extent of the interstitial inflammation was quantified by the cell numbers of monocytes/macrophages stained positively with CD68 antibody in interstitial space using the immunohistochemistry robot (Figure 3). The interstitial inflammation changed in a similar manner to the interstitial expansive index. The extent of the interstitial inflammation was similar between WKY at 7 and 14 wk of age (25 ± 1 and 27 ± 2 counts/mm2, respectively; Figure 3, A and B). Only the SHR at 14 wk of age (41 ± 1 counts/mm2; Figure 3D) showed a significantly higher interstitial inflammation compared with SHR at 7 wk of age (26 ± 1 counts/mm2; Figure 3C) and the age-matched WKY. ARB treatment blocked the increase in interstitial inflammation (22 ± 2 counts/mm2; Figure 3E). However, HRH treatment failed to prevent the increase in interstitial inflammation (38 ± 3 counts/mm2; Figure 3F).
Figure 3.
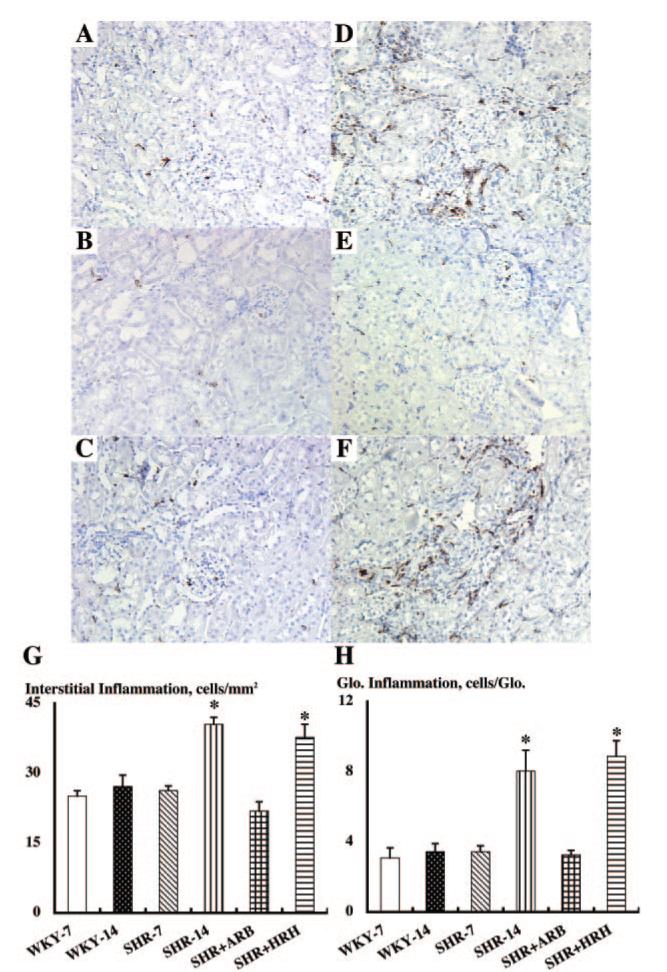
Interstitial (A through G) and glomerular (A through F and H) inflammation by the cell numbers of monocytes/macrophages stained positively with CD68 antibody in interstitial space or glomeruli using immunohistochemistry robot. Again, as evident by A through F, obvious morphologic changes were not observed in each group. However, an automatic image analysis in this study readily detected an increase in interstitial and glomerular inflammation in SHR at 14 wk of age (D) compared with other groups (WKY at 7 wk of age [A]; WKY at 14 wk of age [B]; SHR at 7 wk of age [C]). ARB treatment blocked these increases in SHR at 14 wk of age (E). HRH treatment failed to block this increase in SHR at 14 wk of age (F). *P < 0.05 versus SHR-14 and WKY-14 groups.
Glomerular Inflammation
The extent of the glomerular inflammation was quantified by the cell numbers of monocytes/macrophages stained positively with CD68 antibody in glomeruli using immunohistochemistry robot (Figure 3, A through F and H). The glomerular inflammation changed in parallel with the glomerular sclerotic index. The extent of glomerular inflammation was similar between WKY at 7 and 14 wk of age (3.1 ± 0.6 and 3.4 ± 0.5 counts/glomerulus, respectively; Figure 3, A and B). However, SHR at 14 wk of age (8.1 ± 1.2 counts/glomerulus; Figure 3D) showed significantly higher glomerular inflammation compared with SHR at 7 wk of age (3.4 ± 0.3 counts/glomerulus; Figure 3C) and the age-matched WKY. ARB treatment blocked the increase in glomerular inflammation (3.3 ± 0.3 counts/glomerulus; Figure 3E). However, HRH treatment failed to prevent the increase in glomerular inflammation (8.9 ± 0.9 counts/glomerulus; Figure 3F).
Arterial Proliferation of Afferent Arteriolar Walls
The magnitude of the arterial proliferation was evaluated by the thickness of afferent arteriolar walls, stained positively with α-smooth muscle isoform of actin antibody and elastin, using an immunohistochemistry robot (data not shown). The afferent arteriolar walls’ thickness changed in a similar manner to other parameters. The afferent arteriolar wall thickness was similar between WKY at 7 and 14 wk of age (4.0 ± 0.2 and 4.2 ± 0.1 μm, respectively). However, SHR at 14 wk of age (5.1 ± 0.4 μm) showed significant arterial proliferative change on afferent arteriolar walls compared with SHR at 7 wk of age (4.0 ± 0.3 μm) and the age-matched WKY. ARB treatment normalized the increase in afferent arteriolar walls thickness (4.0 ± 0.1 μm). In contrast, HRH treatment further enhanced the increase in the afferent arteriolar walls thickness (6.7 ± 0.1 μm).
Discussion
In this study, SHR at 14 wk of age exhibited augmentation of systolic BP (Figure 1A) and kidney Ang II contents (Figure 1B) in association with angiotensinogen mRNA (Figure 1C) and protein (Figure 1, E and F) increases in the renal cortex compared with SHR at 7 wk of age. As shown in Figures 1G through 3, all six parameters of renal injury (urinary protein excretion rate, glomerular sclerosis, interstitial expansion, interstitial inflammation, glomerular inflammation, and arterial proliferation of afferent arteriolar wall) were also increased in SHR in the hypertensive stage compared with SHR in the prehypertensive stage. These phenomena were not observed in the age-matched WKY groups. The enhanced renal Ang II levels in SHR compared with WKY were consistent with a previous study (19). Moreover, ARB treatment completely abolished increases in kidney Ang II contents, angiotensinogen mRNA, protein in the renal cortex, and systolic BP, which is consistent with our recent finding in Ang II-infused hypertensive rats (9). ARB treatment also entirely normalized all six parameters of renal injury. It is interesting that HRH treatment failed to prevent these increases even though this combination therapy of HRH effectively blocked the development of hypertension in SHR. These data suggest that angiotensinogen mRNA and protein enhancement in renal cortex participates in the development and the progression of hypertension and/or renal damage in SHR and that angiotensinogen mRNA and protein augmentation in renal cortex is involved in AT1 receptor-mediated mechanisms.
We used urinary protein excretion rate as a marker of overall renal damages. As shown in Figure 1G, SHR at the hypertensive stage showed almost twice the urinary protein excretion compared with SHR in the prehypertensive stage and the age-matched WKY. This increase indicated, somehow, mild renal injury for the severe hypertension in SHR. For instance, we previously reported that deoxycorticosterone acetate salt-induced hypertensive Sprague-Dawley rats, which is a common model of volume-dependent hypertension, showed much higher amounts of urinary protein (166 ± 41 mg/d per kg body wt) even for a mild increase of systolic BP (155 ± 9 mmHg) (14). These data may indicate that other mechanisms than hypertension participate in renal injury in SHR. This interpretation is also supported by the dissociation of systolic BP and urinary protein excretion of SHR that were treated with the triple therapy in this study. Consistent with the mild increase in urinary protein in hypertensive SHR, Figures 2 and 3 did not exhibit severe morphologic changes in kidney tissues of SHR in the hypertensive stage even though these animals showed severe hypertension. Therefore, classical tissue examinations will not work well to evaluate such mild renal damages in this study. It is important to emphasize that our methods to quantify renal injury in multiple aspects would avoid any bias entirely by using an outsourcing company for tissue staining or a robotic system for immunohistochemical analysis equipped with automatic image analysis software, in a blind manner. Successfully, our methods readily detected renal injury in SHR in the hypertensive stage.
We previously demonstrated that intrarenal renin protein levels showed an opposite pattern of intrarenal Ang II levels in Sprague-Dawley rats with chronic Ang II infusion with or without ARB treatment (9). In this study, we showed that intrarenal renin mRNA levels have no correlation with intrarenal Ang II levels. Angiotensinogen is the only known substrate for renin, which is the rate-limiting enzyme of the RAS. Because the level of angiotensinogen in humans is close to the Michaelis-Menten constant value for renin (20,21), angiotensinogen levels can control the activity of the RAS as well as renin levels, and intrarenal angiotensinogen upregulation may lead to elevated Ang II levels in the kidney even though intrarenal renin levels are not particularly elevated.
Within the cortex, there is distribution of Ang II in the interstitial fluid, tubular fluid, and the intracellular compartments. The interstitial as well as the intratubular compartments contribute to the disproportionately high total Ang II levels. Studies using microdialysis probes implanted in the renal cortex demonstrated that Ang II concentrations in the interstitial fluid are much higher than the plasma concentrations found recently by us (22,23) and others (24,25). Therefore, it is speculated that interstitial Ang II and intracellular Ang II play a role in the increased intrarenal Ang II levels in this study. However, another study will be required to clarify this issue.
Ang II-infused rats have increases in renal angiotensinogen mRNA (11,26) and protein (12). Several in vitro studies have demonstrated Ang II-induced augmentation of angiotensinogen mRNA expression. Klett et al. (27) presented evidence that Ang II enhances hepatic angiotensinogen synthesis by inhibiting degradation of angiotensinogen mRNA in hepatocytes. Li and Brasier (28) suggested that activation of the angiotensinogen gene by Ang II is mediated by the NF-κB p65 transcription factor in hepatocytes. Tamura et al. (29) showed that Ang II activates transcription of the angiotensinogen gene exclusively via the AT1 receptor pathway in cardiac myocytes. Mascareno et al. (30) showed that activation of the angiotensinogen promoter by Ang II depends on the signal transducer and activator of transcription protein signal pathway in cardiac myocytes. Although less is known about the amplification mechanisms in renal tissues, Ingelfinger et al. (31) demonstrated an enhanced angiotensinogen mRNA expression by Ang II in an immortalized proximal tubular cell line. These findings support the concept that the elevated Ang II stimulates its precursor, angiotensinogen, thus leading to enhanced intrarenal Ang II formation. When this positive feedback effect of intrarenal Ang II on intrarenal angiotensinogen is taken into consideration, it is not clear whether the enhanced intrarenal angiotensinogen in SHR is a consequence or a cause of the increased intrarenal Ang II. Apparently, it is hard to address in vivo. We therefore propose in vitro studies to address this issue.
In conclusion, this study demonstrates that SHR at 14 wk of age exhibits hypertension and increases in kidney Ang II contents, angiotensinogen mRNA, and protein levels in the renal cortex, which are associated with enhanced parameters of renal injury (urinary protein excretion rate, glomerular sclerosis, interstitial expansion, interstitial inflammation, glomerular inflammation, and arterial proliferation of afferent arteriolar wall). These augmentations are not observed in the age-matched WKY groups and are completely blocked by ARB treatment but not blocked by HRH treatment even though both treatments equally prevent the development of hypertension in SHR. These data indicate that the AT1 receptor-dependent enhancement of intrarenal angiotensinogen contributes to the development and the progression of hypertension and/or renal injury in SHR.
Acknowledgments
This study was supported by grants from the National Center for Research Resources (P20RR017659); the National Heart, Lung, and Blood Institute (R01HL026371); the Health Excellence Fund from Louisiana Board of Regents; and Sankyo Co. Ltd. (Tokyo, Japan).
Portions of this study were presented in abstract form at the 35th Annual Meeting of the American Society of Nephrology (J Am Soc Nephrol 13: 145A, 2002), the 17th Annual Meeting of the Experimental Biology (FASEB J 17: A96, 2003), the 27th Annual Meeting of the Japanese Society of Hypertension (Hypertens Res 27: 138, 2004), the 58th Annual Fall Conference and Scientific Sessions of the Council for High Blood Pressure Research in association with the Council on the Kidney in Cardiovascular Disease (Hypertension 44: 527, 2004), and the 37th Annual Meeting of the American Society of Nephrology (J Am Soc Nephrol 15: 643A, 2004).
We acknowledge critical reviews and valuable comments from L. Gabriel Navar, Ph.D. (Tulane University). We also acknowledge excellent technical assistance from My-Linh Rauv, B.S., Duy V. Tran, B.S., Dale M. Seth, M.S., and Mark A. Cabrera, M.S. (Tulane University).
References
- 1.Okamoto K, Tabei R, Fukushima M, Nosaka S, Yamori Y. Further observations of the development of a strain of spontaneously hypertensive rats. Jpn Circ J. 1966;30:703–716. doi: 10.1253/jcj.30.703. [DOI] [PubMed] [Google Scholar]
- 2.Vincent M, Dupont J, Sassard J. Plasma renin activity as a function of age in two new strains of spontaneously hypertensive and normotensive rats. Clin Sci Mol Med. 1976;50:103–107. doi: 10.1042/cs0500103. [DOI] [PubMed] [Google Scholar]
- 3.Kuriyama S, Kawashima K, Sokabe H. Plasma renin activity determined by two different methods in spontaneously hypertensive rats. Jpn Heart J. 1982;23:587–592. doi: 10.1536/ihj.23.587. [DOI] [PubMed] [Google Scholar]
- 4.Nakamura Y, Ono H, Zhou X, Frohlich ED. Angiotensin type 1 receptor antagonism and ACE inhibition produce similar renoprotection in N(omega)-nitro-L>-arginine methyl ester/spontaneously hypertensive rats. Hypertension. 2001;37:1262–1267. doi: 10.1161/01.hyp.37.5.1262. [DOI] [PubMed] [Google Scholar]
- 5.Teng J, Fukuda N, Suzuki R, Takagi H, Ikeda Y, Tahira Y, Kanmatsuse K. Inhibitory effect of a novel angiotensin II type 1 receptor antagonist RNH-6270 on growth of vascular smooth muscle cells from spontaneously hypertensive rats: Different anti-proliferative effect to angiotensin-converting enzyme inhibitor. J Cardiovasc Pharmacol. 2002;39:161–171. doi: 10.1097/00005344-200202000-00002. [DOI] [PubMed] [Google Scholar]
- 6.Pu Q, Larouche I, Schiffrin EL. Effect of dual angiotensin converting enzyme/neutral endopeptidase inhibition, angiotensin converting enzyme inhibition, or AT1 antagonism on coronary microvasculature in spontaneously hypertensive rats. Am J Hypertens. 2003;16:931–937. doi: 10.1016/s0895-7061(03)01029-x. [DOI] [PubMed] [Google Scholar]
- 7.Kobori H, Nishiyama A, Abe Y, Navar LG. Enhancement of intrarenal angiotensinogen in Dahl salt-sensitive rats on high salt diet. Hypertension. 2003;41:592–597. doi: 10.1161/01.HYP.0000056768.03657.B4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kobori H, Nishiyama A. Effects of tempol on renal angiotensinogen production in Dahl salt-sensitive rats. Biochem Biophys Res Commun. 2004;315:746–750. doi: 10.1016/j.bbrc.2004.01.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kobori H, Prieto-Carrasquero MC, Ozawa Y, Navar LG. AT1 receptor mediated augmentation of intrarenal angiotensinogen in angiotensin II-dependent hypertension. Hypertension. 2004;43:1126–1132. doi: 10.1161/01.HYP.0000122875.91100.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fabris B, Candido R, Armini L, Fischetti F, Calci M, Bardelli M, Fazio M, Campanacci L, Carretta R. Control of glomerular hyperfiltration and renal hypertrophy by an angiotensin converting enzyme inhibitor prevents the progression of renal damage in hypertensive diabetic rats. J Hypertens. 1999;17:1925–1931. doi: 10.1097/00004872-199917121-00023. [DOI] [PubMed] [Google Scholar]
- 11.Kobori H, Harrison-Bernard LM, Navar LG. Expression of angiotensinogen mRNA and protein in angiotensin II-dependent hypertension. J Am Soc Nephrol. 2001;12:431–439. doi: 10.1681/asn.v123431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kobori H, Harrison-Bernard LM, Navar LG. Enhancement of angiotensinogen expression in angiotensin II-dependent hypertension. Hypertension. 2001;37:1329–1335. doi: 10.1161/01.hyp.37.5.1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kobori H, Harrison-Bernard LM, Navar LG. Urinary excretion of angiotensinogen reflects intrarenal angiotensinogen production. Kidney Int. 2002;61:579–585. doi: 10.1046/j.1523-1755.2002.00155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kobori H, Nishiyama A, Harrison-Bernard LM, Navar LG. Urinary angiotensinogen as an indicator of intrarenal angiotensin status in hypertension. Hypertension. 2003;41:42–49. doi: 10.1161/01.hyp.0000050102.90932.cf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prieto-Carrasquero MC, Harrison-Bernard LM, Kobori H, Ozawa Y, Hering-Smith KS, Hamm LL, Navar LG. Enhancement of collecting duct renin in angiotensin II-dependent hypertensive rats. Hypertension. 2004;44:223–229. doi: 10.1161/01.HYP.0000135678.20725.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kanda T, Wakino S, Hayashi K, Homma K, Ozawa Y, Saruta T. Effect of fasudil on Rho-kinase and nephropathy in subtotally nephrectomized spontaneously hypertensive rats. Kidney Int. 2003;64:2009–2019. doi: 10.1046/j.1523-1755.2003.00300.x. [DOI] [PubMed] [Google Scholar]
- 17.Graciano ML, Cavaglieri RDC, Delle H, Dominguez WV, Casarini DE, Malheiros DMAC, Noronha IL. Intrarenal renin-angiotensin system is upregulated in experimental model of progressive renal disease induced by chronic inhibition of nitric oxide synthesis. J Am Soc Nephrol. 2004;15:1805–1815. doi: 10.1097/01.asn.0000131528.00773.a9. [DOI] [PubMed] [Google Scholar]
- 18.Carey AV, Carey RM, Gomez RA. Expression of alpha-smooth muscle actin in the developing kidney vasculature. Hypertension. 1992;19:II168–II175. doi: 10.1161/01.hyp.19.2_suppl.ii168. [DOI] [PubMed] [Google Scholar]
- 19.Meng QC, Durand J, Chen YF, Oparil S. Effects of dietary salt on angiotensin peptides in kidney. J Am Soc Nephrol. 1995;6:1209–1215. doi: 10.1681/ASN.V641209. [DOI] [PubMed] [Google Scholar]
- 20.Gould AB, Green D. Kinetics of the human renin and human substrate reaction. Cardiovasc Res. 1971;5:86–89. doi: 10.1093/cvr/5.1.86. [DOI] [PubMed] [Google Scholar]
- 21.Brasier AR, Li J. Mechanisms for inducible control of angiotensinogen gene transcription. Hypertension. 1996;27:465–475. doi: 10.1161/01.hyp.27.3.465. [DOI] [PubMed] [Google Scholar]
- 22.Nishiyama A, Seth DM, Navar LG. Renal interstitial fluid concentrations of angiotensins I and II in anesthetized rats. Hypertension. 2002;39:129–134. doi: 10.1161/hy0102.100536. [DOI] [PubMed] [Google Scholar]
- 23.Nishiyama A, Seth DM, Navar LG. Renal interstitial fluid angiotensin I and angiotensin II concentrations during local angiotensin-converting enzyme inhibition. J Am Soc Nephrol. 2002;13:2207–2212. doi: 10.1097/01.asn.0000026610.48842.cb. [DOI] [PubMed] [Google Scholar]
- 24.Siragy HM, Howell NL, Ragsdale NV, Carey RM. Renal interstitial fluid angiotensin. Modulation by anesthesia, epinephrine, sodium depletion, and renin inhibition. Hypertension. 1995;25:1021–1024. doi: 10.1161/01.hyp.25.5.1021. [DOI] [PubMed] [Google Scholar]
- 25.Siragy HM, Carey RM. Protective role of the angiotensin AT2 receptor in a renal wrap hypertension model. Hypertension. 1999;33:1237–1242. doi: 10.1161/01.hyp.33.5.1237. [DOI] [PubMed] [Google Scholar]
- 26.Schunkert H, Ingelfinger JR, Jacob H, Jackson B, Bouyounes B, Dzau VJ. Reciprocal feedback regulation of kidney angiotensinogen and renin mRNA expressions by angiotensin II. Am J Physiol. 1992;263:E863–E869. doi: 10.1152/ajpendo.1992.263.5.E863. [DOI] [PubMed] [Google Scholar]
- 27.Klett C, Nobiling R, Gierschik P, Hackenthal E. Angiotensin II stimulates the synthesis of angiotensinogen in hepatocytes by inhibiting adenylylcyclase activity and stabilizing angiotensinogen mRNA. J Biol Chem. 1993;268:25095–25107. [PubMed] [Google Scholar]
- 28.Li J, Brasier AR. Angiotensinogen gene activation by angiotensin II is mediated by the rel A (nuclear factor-kappaB p65) transcription factor: One mechanism for the renin angiotensin system positive feedback loop in hepatocytes. Mol Endocrinol. 1996;10:252–264. doi: 10.1210/mend.10.3.8833654. [DOI] [PubMed] [Google Scholar]
- 29.Tamura K, Umemura S, Nyui N, Hibi K, Ishigami T, Kihara M, Toya Y, Ishii M. Activation of angiotensinogen gene in cardiac myocytes by angiotensin II and mechanical stretch. Am J Physiol. 1998;275:R1–R9. doi: 10.1152/ajpregu.1998.275.1.R1. [DOI] [PubMed] [Google Scholar]
- 30.Mascareno E, Dhar M, Siddiqui MA. Signal transduction and activator of transcription (STAT) protein-dependent activation of angiotensinogen promoter: A cellular signal for hypertrophy in cardiac muscle. Proc Natl Acad Sci U S A. 1998;95:5590–5594. doi: 10.1073/pnas.95.10.5590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ingelfinger JR, Jung F, Diamant D, Haveran L, Lee E, Brem A, Tang SS. Rat proximal tubule cell line transformed with origin-defective SV40 DNA: Autocrine ANG II feedback. Am J Physiol. 1999;276:F218–F227. doi: 10.1152/ajprenal.1999.276.2.F218. [DOI] [PubMed] [Google Scholar]