

SUMMARY
Endocannabinoids (eCB) have emerged as key activity-dependent signals that, by activating presynaptic cannabinoid receptors (CB1) coupled to Gi/o protein, can mediate short-term and long-term synaptic depression (LTD). While the presynaptic mechanisms underlying eCB-dependent short-term depression have been identified, the molecular events linking CB1 receptors to LTD are unknown. Here we show in the hippocampus that long-term, but not short-term, eCB-dependent depression of inhibitory transmission requires presynaptic cAMP/PKA signaling. We further identify the active zone protein RIM1α as a key mediator of both CB1 receptor effects on the release machinery, as well as eCB-dependent LTD in the hippocampus. Moreover, we show that eCB-dependent LTD in the amygdala and hippocampus shares major mechanistic features. These findings reveal for the first time the signaling pathway by which CB1 receptors mediate long-term effects of eCBs in two crucial brain structures. Further, our results highlight a conserved mechanism of presynaptic plasticity in the brain.
INTRODUCTION
The endogenous ligands of cannabinoid receptors (endocannabinoids or eCBs) are important regulators of synaptic function (Alger, 2002; Freund et al., 2003) mediating short- and long-term plasticity at both excitatory and inhibitory synapses in a number of brain structures (for a recent review, see Chevaleyre et al., 2006). These lipid signaling molecules are generally synthesized “on demand” from membrane precursors in the postsynaptic cell, and diffuse retrogradely to the presynaptic terminal (Piomelli, 2003). Once bound to the presynaptic cannabinoid receptor CB1, eCBs suppress transmitter release either transiently (<1 min) (Diana and Marty, 2004; Kreitzer and Regehr, 2002; Wilson and Nicoll, 2002), or in a consolidated long-term form (>1 hour) (Chevaleyre et al., 2006; Gerdeman and Lovinger, 2003) that persists long after CB1 receptor activation has ceased (Chevaleyre and Castillo, 2003; Robbe et al., 2002; Ronesi et al., 2004; Sjostrom et al., 2003). How activation of the same presynaptic receptor leads to transient versus long-lasting depression remains unknown.
At present, eCB-mediated long-term depression (eCB-LTD) probably constitutes the best documented and most widely distributed form of activity-dependent, long-term presynaptic plasticity in the brain (Chevaleyre et al., 2006; Gerdeman and Lovinger, 2003). Examples of presynaptic eCB-LTD have been reported at excitatory synapses in dorsal striatum (Gerdeman et al., 2002; Kreitzer and Malenka, 2005), nucleus accumbens (Robbe et al., 2002), neocortex (Bender et al., 2006; Sjostrom et al., 2003) and cerebellum (Soler-Llavina and Sabatini, 2006), as well as at inhibitory synapses in hippocampus (Chevaleyre and Castillo, 2003; Chevaleyre and Castillo, 2004) and amygdala (Azad et al., 2004; Marsicano et al., 2002). This prevalence suggests that eCB-LTD may be a fundamental mechanism for making long-term modifications to neural circuits and behavior. On the other hand, transient, eCB-mediated depression of synaptic transmission is exemplified by the phenomenon known as Depolarization-induced Suppression of Inhibition or Excitation (DSI or DSE), a phenomenon well-characterized in the hippocampus and cerebellum, but also observed in other brain areas (reviewed in: Alger, 2002; Diana and Marty, 2004; Kreitzer and Regehr, 2002; Wilson and Nicoll, 2002). It is worth noting that by regulating GABA release, eCBs can also modify the induciblity of non-eCB-mediated forms of synaptic plasticity such as long-term potentiation (LTP) (Azad et al., 2004; Carlson et al., 2002; Chevaleyre and Castillo, 2004). Therefore, whether eCBs engage transient or long-lasting depression may profoundly impact neural circuit processing.
A mechanism for the transient suppression of release in DSI/DSE has been suggested by studies in hippocampus (Lenz et al., 1998; Varma et al., 2002; Wilson et al., 2001) and in cerebellum (Kreitzer and Regehr, 2001). Namely, CB1 receptor activation transiently inhibits presynaptic voltage-gated calcium channels (VGCC) for a period of seconds to minutes, a process likely mediated by the βγ G protein subunits (Wilson et al., 2001). One of the most striking requirements distinguishing I-LTD induction from that of DSI is the extended duration of CB1 receptor activity (Chevaleyre and Castillo, 2003). This increased time requirement for I-LTD induction could reflect the recruitment of a distinct signaling pathway by CB1 receptors. A likely candidate pathway for I-LTD is the αi/o effector limb of the CB1 receptor G protein signaling cascade, i.e. the cAMP/Protein Kinase A (PKA) pathway (Childers and Deadwyler, 1996; Howlett et al., 1986). Indeed, modulation of the cAMP/PKA pathway has been implicated in other presynaptic forms of plasticity in both hippocampus (Huang et al., 1994; Tzounopoulos et al., 1998; Villacres et al., 1998; Weisskopf et al., 1994) and cerebellum (Linden and Ahn, 1999; Salin et al., 1996; Storm et al., 1998). A common theme of these long-term forms of plasticity is that their expression mechanism relies on changes in the transmitter release machinery (Castillo et al., 1997; Castillo et al., 2002; Lonart et al., 2003; Tzounopoulos et al., 1998). In particular, the active zone protein RIM1α (Wang et al., 1997) has been shown to be necessary for PKA-dependent LTP at several excitatory synapses including the mossy fiber to CA3 pyramidal cell synapse (Castillo et al., 2002), the parallel fiber to Purkinje cell synapse (Castillo et al., 2002; Lonart et al., 2003), and the Schaffer collateral to CA1 pyramidal synapse (Huang et al., 2005). These previous studies prompted us to ask whether eCB-LTD also involves downregulation of the cAMP/PKA pathway and/or a RIM1α-dependent modification of the release machinery.
In this study, we compared the dependence of DSI and I-LTD on cAMP/PKA signaling, and on the active zone protein RIM1α. We found that hippocampal I-LTD, but not DSI, requires cAMP/PKA signaling and RIM1α. In addition, we report that CB1 receptors, via PKA, regulate GABA release downstream of VGCC-mediated calcium influx, and that both CB1 receptors and PKA can target the release machinery in a RIM1α-dependent manner. Furthermore, we tested the generality of these mechanisms in the basolateral amygdala (BLA) where eCBs mediate I-LTD (Azad et al., 2004; Marsicano et al., 2002). At inhibitory synapses in the BLA, we also found that CB1 receptors target the release machinery via PKA and that RIM1α is necessary for I-LTD. Taken together, these findings raise the intriguing prospect of a core mechanism of presynaptic plasticity spanning multiple brain areas and synapse types (i.e. excitatory and inhibitory), and also provide a potential substrate for the hippocampus- and amygdala-dependent memory deficits reported in RIM1α KO mice (Powell et al., 2004).
RESULTS
During hippocampal DSI, CB1 receptor-mediated block of the N-type VGCC is believed to depress GABA release (Varma et al., 2002; Wilson et al., 2001). On the other hand, the CB1 receptors’ effectors during I-LTD are completely unknown. Because CB1 receptors are Gi/o coupled (Howlett et al., 2002) and have a well documented inhibitory effect on adenylyl cyclase and PKA activity (Childers and Deadwyler, 1996; Howlett et al., 1986), we tested whether the cAMP/PKA pathway might be involved in eCB-mediated long-term plasticity in slices of mouse hippocampus. First, cAMP levels were raised by continuous activation of adenylyl cyclase with forskolin (FSK), a manipulation that also increased the amplitude of evoked inhibitory postsynaptic currents (IPSCs) by 25.0 ± 5.4 % (n = 6). Theta-burst stimulation (TBS) of presynaptic inputs, which triggered I-LTD under control conditions (Fig. 1A; 23.8 ± 2.0 %, n = 4), failed in the presence of FSK (Fig. 1A; 3.8 ± 3.1 %, n = 5, p = 0.0014). Changes in cAMP level can affect synaptic transmission through both PKA-dependent and -independent actions (Seino and Shibasaki, 2005), so we next tested the involvement of PKA with two different blockers, the cell-permeable myristoylated form of Protein Kinase A inhibitor 14-22 amide (PKI), and H89. I-LTD was markedly reduced after incubating slices >30 min with either of these compounds (Fig. 1B, PKA blocker 5.2 ± 1.8 %, n = 11, 7 cells in PKI and 4 cells in H89, data were combined since no differential effect was observed by these blockers, vs. 20.8 ± 2.0 %, n = 6 in interleaved control slice, p<0.0001). This result likely represents an occlusion of I-LTD, given that CB1 receptors can downregulate PKA activity and PKA inhibition acutely depresses synaptic transmission (see below, Fig. 5B). In order to rule out a postsynaptic role for PKA in I-LTD induction, I-LTD was also tested with the non-myristoylated, membrane impermeant form of PKI, delivered to the postsynaptic cell via the recording pipette. Blocking postsynaptic PKA did not affect I-LTD (Fig. 1B), indicating that the PKA requirement is presynaptic. In marked contrast to these effects on long term plasticity, neither FSK nor PKA blockers altered DSI in the same CA1 pyramidal cells (Fig. 1C), in accordance with previous findings (Morishita et al., 1998; Wilson et al., 2001). We obtained results similar to these with FSK and PKA inhibitors for I-LTD and DSI in brain slices from rats (data not shown). We considered the possibility that interfering with cAMP/PKA signaling blocked I-LTD by reducing glutamate release, which is the initial trigger for I-LTD (Chevaleyre and Castillo, 2003). However, neither PKA blockers nor FSK depressed excitatory synaptic transmission (i.e. CA1 field potentials) after 50 min bath application (PKI: 1.3 ± 2.1 %, n = 6; H89: -0.5 ± 3.2 %, n = 5; FSK: 24.4 ± 5.3 %, n = 4, data not shown). Taken together, these results show that the presynaptic cAMP/PKA pathway is required downstream of CB1 receptor activation for long-term, but not short-term eCB-mediated plasticity.
Figure 1.
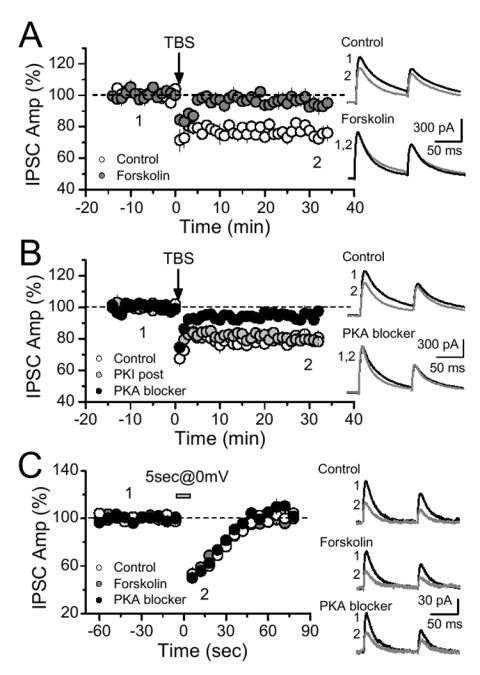
Endocannabinoid-mediated long-term, but not short-term, plasticity requires cAMP/PKA signaling. (A) A persistent increase in cAMP level by incubation (>30 min) and continuous bath application of the adenylyl cyclase activator forskolin (10 μM) blocked I-LTD. (B) Incubation and continous application of either of two different PKA inhibitors (10 μM H-89, n = 4; 1 μm PKI 14-22 amide, n = 7) also impaired I-LTD induction. Direct application of a membrane impermeant PKA blocker (1 μM PKI 6-22 amide, n = 7 cells, 5 mice) to the postsynaptic pyramidal cell, via recording pipette, did not affect I-LTD. (C) None of these manipulations interfered with DSI. Averaged sample traces taken at times indicated by numbers are shown on the right.
Figure 5.
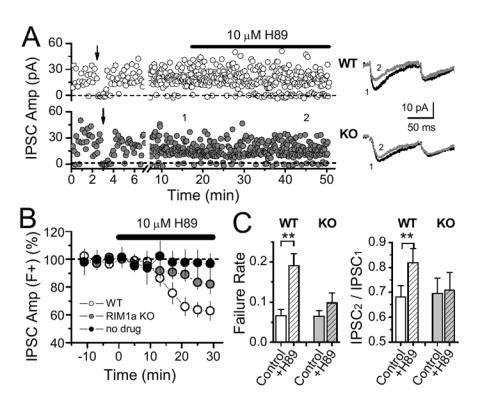
PKA regulation of GABA release at CB1+ fibers requires RIM1α. (A) Representative experiments showing the effect of H-89 in WT and RIM1α KO mice using focal stimulation of CB1R+ fibers. Averaged sample traces at times indicated by numbers are shown on the right. (B) Summary graph showing significantly greater depression of synaptic transmission (IPSC amplitude, including failures) by H-89 in WT vs. RIM1α KO mice (8 cells, 8 mice; 9 cells, 8 mice respectively). Without drug application, no significant depression was observed over time. (C) H-89 significantly increased failure rate and aired-pulse ratio (IPSC2/IPSC1) in WT, but not RIM1α KO mice (same cells as B).
If CB1 receptors can regulate GABA release, as shown above, by two divergent signalling cascades, it stands to reason that this receptor may also independently target distinct steps of neurotransmitter release. In the hippocampus, previous studies have suggested that CB1 receptors suppress GABA release mainly by targeting VGCCs rather than the vesicle release machinery (Hoffman and Lupica, 2000; Varma et al., 2002; Wilson et al., 2001; Wilson and Nicoll, 2001). In the cerebellum however, CB1 receptors can target the release machinery in a manner that presumably depends on basal calcium levels in the presynaptic terminal (Yamasaki et al., 2006). For this reason, we chose to examine miniature IPSCs (mIPSCs) in CA1 in the presence of 5 mM extracellular Ca2+, blocking action potentials and VGCCs with 500 nM TTX and 100 μM CdCl2, respectively. Application of 5 μM WIN 55,212-2 (WIN), a CB1 receptor agonist, significantly decreased mIPSC frequency in the hippocampus (Fig. 2A, 17.6 ± 1.2 %, n = 5, p = 0.0002). This effect was blocked by a CB1 receptor antagonist, AM 251 or SR 141716 (Fig. 2A). In addition, WIN had no effect on mIPSC amplitude (Fig. 2B), indicating a presynaptic action of CB1 receptors on GABA release. As the cAMP/PKA pathway is required for I-LTD induction, we tested whether it is also involved in CB1 receptor-mediated depression of mIPSC frequency. In presence of FSK, mIPSC frequency was increased (by 58.1 %, n = 6, p = 0.003) and WIN was unable to depress mIPSC frequency (Fig. 2C). Furthermore, blocking PKA with H89 reduced mIPSC frequency to the same extent as WIN alone, and the effects of WIN and H89 did not summate, again suggesting occlusion (Fig. 2C). These results show that in addition to presynaptic VGCCs, CB1 receptors can also target the release machinery via the cAMP/PKA pathway.
Figure 2.
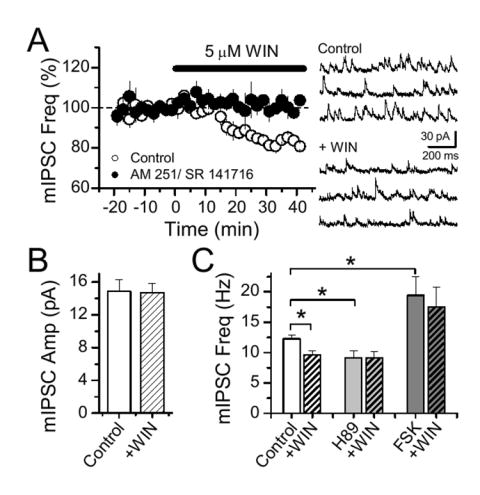
CB1 receptors target the release machinery via the cAMP/PKA pathway. (A) WIN application depressed mIPSC frequency, an effect blocked by CB1 receptor antagonists (2 μM AM 251, n = 2; 10 μM SR141716, n = 3). Sample traces before and during WIN application are shown at right. (B) WIN had no effect on mIPSC amplitude. (C) The WIN-mediated depression of mIPSC frequency is blocked in the continuous presence of forskolin (n = 6) or H89 (n = 5).
What lies downstream PKA in I-LTD? To address this question, we tested whether RIM1α, a presynaptic active zone protein (Schoch et al., 2002; Wang et al., 1997) which is phosphorylated by PKA (Lonart et al., 2003) and is required for presynaptic forms of LTP at excitatory synapses (Castillo et al., 2002; Lonart et al., 2003), could also be involved in I-LTD. Notably, I-LTD induced by a TBS was abolished in RIM1α KO mice (Fig. 3A, 3.9 ± 1.3 %, n = 18 cells, 10 KO mice, vs. 19.1 ± 1.8 %, n = 14 cells, 9 WT mice, p<0.001). This effect does not seem to result from deficient CB1 receptor function, as DSI remained unaffected in RIM1α KO mice (Fig. 3B). The differential sensitivity of I-LTD and DSI to RIM1α deletion parallels the differential involvement of the cAMP/PKA pathway in these processes and supports the idea that different targets downstream of the CB1 receptor mediate short- and long-term eCB-plasticity.
Figure 3.
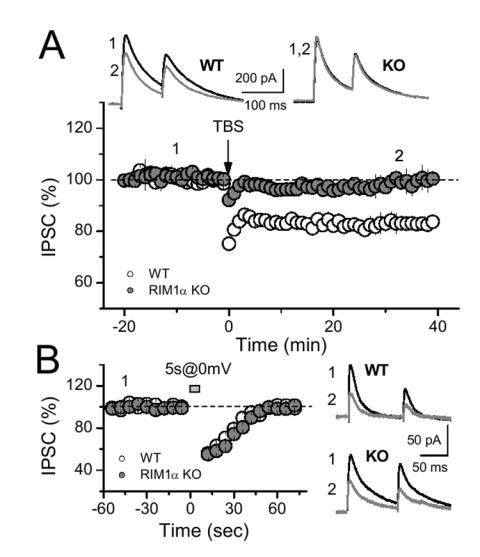
Long-term, but not short-term, eCB-mediated plasticity requires the presynaptic active zone protein RIM1α. (A) TBS delivered in slices from RIM1α KO mice triggered no depression compared to the I-LTD obtained in WT littermates (n = 18, 10 mice and n = 14, 9 mice respectively). (B) In contrast, DSI was identical in both WT and KO animals. Averaged sample traces are shown above (A) or on the right (B).
I-LTD is a heterosynaptic form of plasticity triggered by glutamate release from Schaffer collaterals, and postsynaptic activation of group I metabotropic glutamate receptors (mGluR-I) which stimulates eCB production (Chevaleyre and Castillo, 2003). Because the probability of glutamate release in area CA1 of the hippocampus is reduced in RIM1α KO mice (Schoch et al., 2002), the loss of I-LTD could simply be due to a subthreshold activation of mGluRs. We addressed this possibility in several ways. First, we found that I-LTD was normal using 10 μM Cd2+, a non-selective VGCC blocker, that depressed excitatory transmission in WT mice to a level similar to that of KO mice (~50%, data not shown) (Schoch et al., 2002). Second, a 10 min bath application of the mGluR-I agonist DHPG (50 μM), which bypasses glutamate release and also triggers I-LTD upon DHPG washout (Chevaleyre and Castillo, 2003; Edwards et al., 2006), failed to elicit I-LTD in KO mice (Fig. 4A, 4.9 ± 3.3 %, n = 5 cells, 4 KO mice, vs. 21.7 ± 1.5 %, n = 5 cells, 4 WT mice, p=0.0002). Finally, we directly tested the effect of CB1 receptor activation with the CB1 receptor agonist WIN. Because CB1 receptor agonists are lipophilic compounds with slow diffusion kinetics in acute brain slices, for these experiments we monitored extracellular field IPSPs (fIPSPs) (Arai et al., 1995; Lambert et al., 1991) that allow for stable long-term recordings (see methods). In WT mice, bath application of WIN, subsequently chased with the CB1 receptor antagonist SR 141716, led to a lasting depression of fIPSPs (Fig. 4B, 23.9 ± 2.8 %, n = 4, 3 mice, p = 0.0006). In RIM1α KO mice, WIN acutely depressed the fIPSP to the same extent as observed in WT mice, but fIPSP amplitude subsequently recovered to baseline (5.5 ± 1.5 %, n = 3, 2 mice, p = 0.0001 compared to WT). The similar transient depression, but differential long-term effect of WIN at inhibitory synapses strongly echoes the dissociation of DSI and I-LTD in these mice. Together, these results show that impaired glutamate or eCB release cannot account for the lack of I-LTD in RIM1α KO animals.
Figure 4.
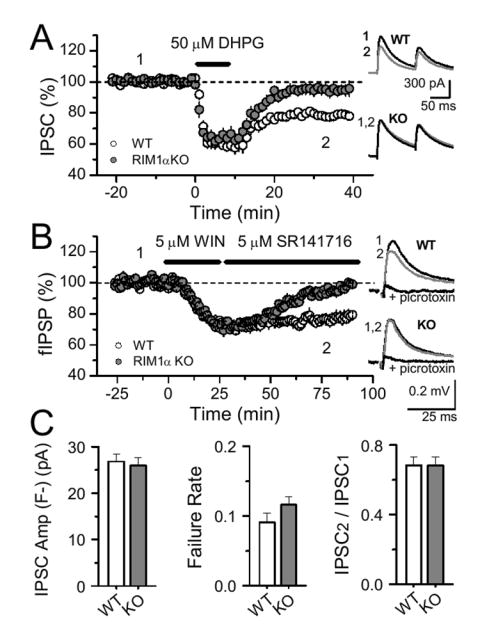
Altered basal transmission at excitatory or inhibitory synapses cannot explain the lack of I-LTD in RIM1α KO mice. (A) Pharmacological induction of I-LTD by DHPG (50 μM, 10 min) was normal in WT mice (n = 5 cells, 4 mice) but impaired in RIM1α KO mice (n = 5 cells, 4 mice).(B) Similarly, application of the CB1 receptor agonist WIN (5 µM, 25 min) subsequently chased with the CB1 receptor antagonist SR 141716 (5 µM) induced a lasting depression of field IPSPs in WT (n = 4, 3 mice) but not in RIM1α KO mice (n = 5, 3 mice). Sample traces before, following WIN application, and in 100 µM picrotoxin are shown at right. (C) The average IPSC amplitude (excluding transmission failures), the failure rate and the paired-pulse ratio (IPSC2/IPSC1) at CB1R+ fibers isolated by focal stimulation were identical in WT (10 cells, 10 mice) and RIM1α KO mice (13 cells, 11 mice).
Perhaps in slices from RIM1α KO mice, CB1 receptor-expressing fibers (CB1R+) are already depressed, thereby occluding attempts to induce I-LTD. Because not all inhibitory inputs express CB1 receptors, the bulk stimulation used in previous experiments (see Methods), by activating a mixture of CB1R+ and CB1R− fibers, may have led us to underestimate potential changes specific to CB1R+ fibers. To isolate CB1R+ synaptic inputs, we lowered the stimulation intensity and selected those inputs that exhibited complete block of transmission during DSI (Chevaleyre and Castillo, 2004) (Fig. 4C). We found that the IPSC amplitude (excluding failures), failure rate, and paired-pulse ratio of two successive IPSCs were identical in WT and KO mice (Fig. 4D). Taken together, these results show that the lack of I-LTD in RIM1α KO is not due to an initial change in release probability at CB1R+ inhibitory synapses.
Our results so far show that PKA and RIM1α are required for CB1 receptor-mediated LTD. To confirm that RIM1α is indeed required for both CB1 receptor and PKA action on GABA release, we performed two additional sets of experiments. First, we tested the acute effect of a PKA blocker on GABA release in both WT and KO mice. Because PKA may differentially control GABA release at synapses that do not express the CB1 receptor, we performed this experiment after isolating CB1R+ fibers with focal stimulation. As shown in figure 5, blockade of PKA by H89 depressed GABAergic synaptic transmission at CB1R+ inputs, a depression (including failures) that was significantly attenuated in RIM1α KO mice (Fig. 5A,B; 35.3 ± 5.8 %, n = 8 cells, 8 WT mice vs. 15.2 ± 4.8 %, n = 9 cells, 8 KO mice, p<0.01). In the absence of PKA blocker, transmission remained stable throughout the experiment (Fig. 5B, 95.8 ± 4.7 %, n = 7), suggesting that the remaining depression in KO mice probably reflects an action of PKA on targets other than RIM1α. A major component of the H89-mediated depression of GABA release is likely RIM1α-dependent, as indicated by the increase in failure rate and paired-pulse ratio in WT, but not KO mice (Fig. 5C,D).
To investigate whether the CB1 receptor-mediated effect on GABAergic release machinery (Fig. 2A) similarly requires RIM1α, in addition to cAMP/PKA signaling, we tested the effect of CB1 receptor activation on mIPSC activity in WT and KO mice. Again, in order to rule out a contribution of VGCC inhibition to a CB1 receptor-mediated effect, these experiments were performed in presence of 100 μM Cd2+. As shown in figure 6, WIN-induced depression of mIPSC activity was absent in RIM1α KO mice (Fig. 6). Together, these results indicate that the PKA/RIM1α pathway is required for CB1 receptor-mediated effects on the release machinery and long-term, but not short-term, synaptic plasticity.
Figure 6.
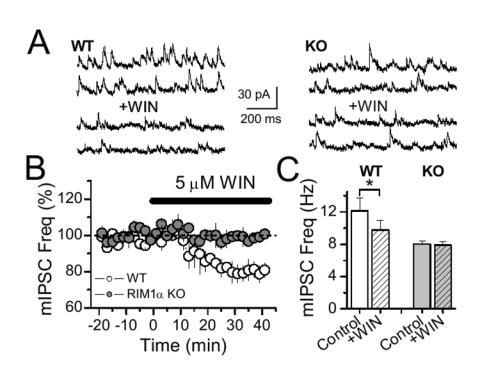
RIM1α mediates CB1 receptor action on the release machinery. (A) Sample trace before and after WIN application in WT and RIM1α KO mice. (B) Averaged mIPSC frequency plot before and after WIN application in WT (4 cells, 3 animals) and RIM1α KO mice (5 cells, 3 animals). (C) Histogram of mIPSC frequency showing the lack of effect of WIN in RIM1α KO mice.
Given the widespread prevalence of eCB-dependent LTD, we tested whether the RIM1α-dependent mechanism of long-term plasticity is conserved in other brain structures. In the basolateral amygdala (BLA), where eCBs also mediate a form of I-LTD (Azad et al., 2004; Marsicano et al., 2002), we first investigated whether CB1 receptors could suppress GABA release by targeting the release machinery. Indeed, activation of CB1 receptors by WIN decreased mIPSC frequency in BLA neurons in 5 mM Ca2+, an effect blocked by CB1 receptor antagonists (Fig. 7A). WIN induced no significant effect on mIPSC amplitude (data not shown), indicating a presynaptic action of CB1 receptors. Importantly, H89 blocked the WIN-mediated effect on mIPSC frequency (Fig. 7B). These results strongly suggest that at BLA inhibitory synapses, CB1 receptors target the release machinery via PKA to depress GABA release.
Figure 7.
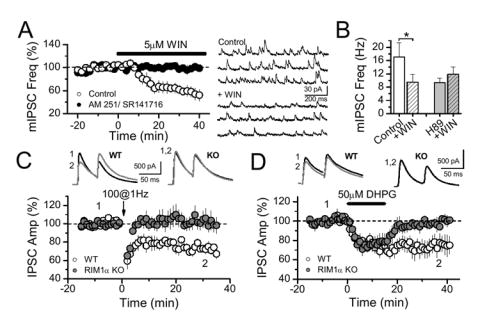
RIM1α and PKA control GABA release and inhibitory synaptic plasticity in basolateral amygdala. (A) WIN depressed mIPSC frequency in BLA (n = 5), an effect blocked by CB1 receptor antagonists (5 μM AM 251, n = 2; 5 μM SR131716, n = 2). Sample traces are shown on the right. (B) WIN-mediated depression was blocked by H-89 (n =5). (C) LTD induction was blocked in RIM1α KO mice (11 cells, 5 mice), compared to their WT littermates (9 cells, 6 mice). (D) LTD induced pharmacologically by DHPG (50 µM, 15 min) was also abolished in RIM1α KO mice compared to WT mice (n = 7, 3 WT mice and n = 6, 4 KO mice).
The coupling of CB1 receptors to GABA release in BLA parallels our findings in hippocampus, and suggests that CB1 receptor mediated plasticity in these two structures may share important features. We first verified that low frequency stimulation (LFS, 100 stimuli, 1 Hz) within the amygdala, close to the external capsule, triggers a CB1 receptor-dependent LTD of IPSCs in the BLA (data not shown) (Azad et al., 2004; Marsicano et al., 2002). As in the hippocampus (Fig. 3A), BLA I-LTD was abolished in RIM1α KO mice (Fig. 7C, -1.7 ± 6.3 %, n = 11 cells, 5 KO mice, vs. 25.3 ± 2.4 %, n = 9 cells, 6 WT mice, p<0.0016). In addition, to rule out a potential decrease in glutamate release during I-LTD induction in KO mice, we tested whether direct activation of group I mGluRs with an agonist could trigger I-LTD in these mice. Similar to the results observed with LFS, we found that DHPG-evoked LTD was also abolished in RIM1α KO mice (Fig. 7D, 27.6 ± 8.0 %, n = 7, 3 WT mice, vs. 3.4 ± 4.0 %, n = 6, 4 KO mice, p = 0.02). Interestingly, as observed in the hippocampus (Fig. 4A), the IPSC depression during DHPG application was identical in both WT and RIM1α KO mice, indicating that the acute effect of CB1 receptor activation on GABA release (Azad et al., 2004) is also independent of RIM1α in the amygdala. In conclusion, our results show that RIM1α is necessary for the long-term reduction of GABA release induced by CB1 receptors not only in the hippocampus, but also in the amygdala, and that in both of these structures, the CB1 receptor-mediated effect on release machinery requires PKA.
DISCUSSION
In this study we investigated the mechanism by which activation of CB1 receptors triggers a long-lasting reduction of transmitter release. First, we report in the hippocampus that eCB-mediated long-term, but not short-term depression, of inhibitory transmission requires cAMP/PKA signaling. Second, we identify the active zone protein RIM1α as a critical player in CB1 receptor-mediated suppression of GABA release. Finally, we show that RIM1α is necessary for eCB-dependent LTD in both the hippocampus and amygdala. Our findings support the notion that enduring changes in the release machinery via cAMP/PKA signaling and RIM1α represent a general mechanism underlying presynaptic forms of long-term plasticity.
CB1 receptors engage different pathways during short and long term eCB-plasticity
One of the main conclusions of our study is that the signaling pathways downstream of the CB1 receptor that trigger short- and long-term plasticity differ (Fig. 8). The mechanism underlying transient suppression of transmitter release by eCBs, i.e. DSI/DSE, has been extensively studied in several brain areas (reviewed in Alger, 2002; Chevaleyre et al., 2006; Diana and Marty, 2004; Freund et al., 2003; Kreitzer and Regehr, 2002; Wilson and Nicoll, 2002). Most experimental evidence indicates that eCB-mediated short-term plasticity, at least in the hippocampus and cerebellum, is mainly due to a CB1 receptor-mediated reduction of presynaptic Ca2+ influx through VGCCs. Indeed, at climbing fiber inputs to Purkinje cells, action potential–induced Ca2+ influx into presynaptic terminals was directly visualized and decreased during DSE to an extent that fully accounted for the decrease in EPSC amplitude (Kreitzer and Regehr, 2001). In the hippocampus, inhibition of presynaptic N-type VGCCs reportedly mediates DSI, and this inhibition likely occurs as a result of the direct action of the Gi/o βγ subunits (Wilson et al., 2001). In support of this mechanism, early studies in expression systems showed that Gβγ dimers, but not the Gα subunit are responsible for the modulation of N-type VGCCs (Herlitze et al., 1996; Ikeda, 1996), and that cannabinoids inhibit N-type VGCC (Mackie and Hille, 1992). In addition, the fast onset of DSI (~1 s) (Heinbockel et al., 2005; Wilson et al., 2001), is more consistent with a fast, membrane-delimited pathway of presynaptic inhibition than a phosphorylation-induced change in channel activity which typically requires many seconds and may involve soluble second messengers (Dolphin, 2003; Hescheler and Schultz, 1993; Hille, 1992). Consistent with this idea, DSI was found to be unaffected by broad spectrum blockers of protein kinases and phosphatases (Wilson et al., 2001) and by manipulation of the cAMP pathway (Morishita et al., 1998; Wilson et al., 2001). Our study confirms these observations, thereby supporting the notion that DSI is unlikely to be mediated by adenylyl cyclase inhibition. Furthermore, we demonstrate that both cAMP/PKA signaling and RIM1α are necessary for I-LTD but not for DSI.
Figure 8.
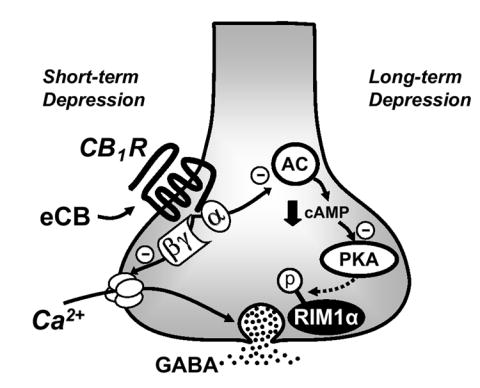
Model of the divergent signaling pathways downstream of the CB1 receptor that triggers eCB-mediated short- or long-term depression. During eCB-mediated short-term depression, brief activation of CB1 receptors reversibly depresses neurotransmitter release mainly by blocking presynaptic VGCC, presumably via Gβ/γ subunits. In contrast, during the induction of eCB-mediated LTD, a longer-lasting (i.e., few minutes) (Chevaleyre and Castillo, 2003; Ronesi et al., 2004) activation of CB1 receptors inhibits adenylyl cyclase (AC) activity via Gα subunits, thereby reducing presynaptic cAMP levels and PKA activity. This modification “gates” an enduring change of the release machinery that requires the active zone protein RIM1α. PKA may directly phosphorylate RIM1α and/or other associated proteins.
Previous studies at both inhibitory (Chevaleyre and Castillo, 2003) and excitatory (Ronesi et al., 2004) synapses have shown that the induction of eCB-mediated LTD requires CB1 receptor activation for several minutes, and that once this form of plasticity is induced, it becomes independent of CB1 receptor activation. The relatively long induction period suggests that CB1 receptors may engage a different signaling process than the one mediating transient suppression of transmitter release. Indeed, the fast time course of DSI (<1 min) likely reflects the transient profile of eCB production and CB1 receptor engagement since CB1 receptor desensitization does not occur within this time window. Therefore, it is tempting to speculate that DSI requires neither cAMP/PKA signaling nor RIM1α, because CB1 receptor activation is too brief to significantly engage the αi/o effector limb of the CB1 receptor G protein signaling cascade. Our findings are consistent with a model (Fig. 8) whereby brief activation of CB1 receptors, presumably by blocking presynaptic VGCCs via Gβγ subunits (Wilson et al., 2001), leads to short-term depression of transmitter release. In contrast, a more sustained, but still transient activation of CB1 receptors (Chevaleyre and Castillo, 2003; Ronesi et al., 2004), downregulates the cAMP/PKA signaling pathway (Childers and Deadwyler, 1996; Howlett et al., 1986) perhaps via the Gα subunit, thereby triggering a RIM1α-dependent modification in the release machinery to suppress transmitter release in a long-lasting manner.
CB1 receptors can control synaptic transmission by targeting the release machinery in a PKA- and RIM1α-dependent manner
Most studies have shown that CB1 receptor-mediated suppression of GABA release mainly occurs as a result of presynaptic VGCC modulation (Diana and Marty, 2003; Hajos et al., 2000; Hoffman and Lupica, 2000; Varma et al., 2002; Wilson et al., 2001). Consistent with this mechanism, activation of CB1 receptors depressed the frequency of spontaneous IPSCs (action potential-driven events), but not mIPSC frequency monitored under normal extracellular Ca2+ concentration not only in the hippocampus (Alger et al., 1996; Hajos et al., 2000; Hoffman and Lupica, 2000; Pitler and Alger, 1994; Varma et al., 2002; Wilson et al., 2001) but also in the amygdala (Azad et al., 2003; Katona et al., 2001). Taken together, these observations could be interpreted to mean that CB1 receptors do not target the release machinery. In the cerebellum however, it has been reported that CB1 receptors can act downstream of Ca2+ influx (Diana and Marty, 2003; Takahashi and Linden, 2000). A recent report demonstrated that the CB1 receptor-mediated effect on miniature synaptic events depends on basal Ca2+ in the presynaptic terminal (Yamasaki et al., 2006). A similar observation was also reported in the amygdala (Azad et al., 2003). Thus, in the absence of action potentials -i.e. in presence of TTX- and in blockers of presynaptic Ca2+ influx, quantal release may still depend on basal Ca2+ in the terminal (Yamasaki et al., 2006), and it is likely that Ca2+ released from intracellular stores play an important role in this process (Collin et al., 2005). It is therefore conceivable that decreasing presynaptic Ca2+, a likely consequence of continuous action potential blockade with TTX, may mask any CB1 receptor-mediated (Ca2+-dependent) effect on the release machinery in normal extracellular Ca2+.
We now show in the CA1 area of the hippocampus that after blocking action potentials and Ca2+ influx through VGCCs, but in 5 mM extracellular Ca2+, CB1 receptors suppress mIPSC frequency. Early studies have demonstrated that presynaptic Gi/o protein-coupled receptors can suppress transmitter release downstream Ca2+-influx, presumably by targeting the release machinery (Cohen et al., 1992; Dittman and Regehr, 1996; Gereau and Conn, 1995; Jarolimek and Misgeld, 1997; Poncer et al., 1995; Scanziani et al., 1992; Scanziani et al., 1995; Scholz and Miller, 1992; Umemiya and Berger, 1995). While a similar mechanism may contribute to the CB1 receptor-mediated suppression of transmitter release at some synapses (Szabo and Schlicker, 2005), the precise signaling pathway and identity of CB1 receptor targets have been largely unexplored. Our results demonstrate that CB1 receptors can target the release machinery via the cAMP/PKA pathway. Moreover, we provide evidence that both PKA and CB1 receptor action on GABA release depends on the presence of the active zone protein RIM1α.
Previous studies have suggested that CB1 receptor agonists suppress action-potential driven synaptic responses independently of PKA (Azad et al., 2003; Daniel et al., 2004; Huang et al., 2002; Robbe et al., 2001). While these data seem to conflict with our model (Fig. 8), agonist application likely engages multiple pathways downstream of CB1 receptors. Any one of these mechanisms could suppress transmitter release to such an extent that a concurrent PKA-dependent change at the level of the release machinery could go undetected. Finally, our findings are consistent with previous reports showing that cAMP/PKA signaling enhances inhibitory synaptic transmission by increasing release probability independently of presynaptic Ca2+ influx not only in the hippocampus (Capogna et al., 1995; Sciancalepore and Cherubini, 1995; Trudeau et al., 1996) but also in cerebellum (Kondo and Marty, 1997; Llano and Gerschenfeld, 1993; Saitow et al., 2005).
It is likely that PKA-dependent modulation of transmitter release occurs as a result of the phosphorylation of presynaptic proteins involved in exocytosis (for a recent review see Seino and Shibasaki, 2005). RIM1α emerges as a unique candidate as this presynaptic protein is phosphorylated by PKA and its phosphorylation reportedly enhances glutamate release in cerebellum (Lonart et al., 2003). RIM1α has been exclusively localized to presynaptic nerve terminals in rodents (Wang et al., 1997) and in C. elegans (Koushika et al., 2001; Weimer et al., 2006). In situ hybridization studies suggest that RIM1α is expressed in all neurons (Schoch et al., 2006). Furthermore, immunohistochemistry evidence in the hippocampus shows that RIM1α is co-expressed with GAD67 (S. Schoch, personal communication), indicating that RIM1α is present in GABAergic interneurons. In this context, it is tempting to speculate that activation of CB1 receptors, presumably by reducing PKA activity, may lead to a decrease in RIM1α phosphorylation. However, it is formally possible that some other RIM1α-partner, also phosphorylated by PKA, may mediate the CB1 receptor-induced effects on the release machinery.
PKA and RIM1α as a general mechanism for presynaptic plasticity
RIM1α is a key protein in the active zone (Wang et al., 1997) that regulates neurotransmitter release (Calakos et al., 2004; Koushika et al., 2001; Schoch et al., 2002) and is thought to scaffold and organize the synaptic release machinery (Sudhof, 2004). Its selective localization to active zones and its interactions with multiple presynaptic molecules places RIM1α in a strategic position to modulate presynaptic release and plasticity (Kaeser and Sudhof, 2005). In addition, RIM1α knockout mice show significant cognitive deficits (Powell et al., 2004). Previous studies reported that RIM1α is critical for the long-term enhancement of glutamate release associated with PKA-dependent, NMDA receptor-independent presynaptic forms of LTP at excitatory synapses in both the hippocampus (Castillo et al., 2002) and cerebellum (Castillo et al., 2002; Lonart et al., 2003). More recently, Huang et al (2005) have described a role for RIM1α in the PKA-dependent late phase of LTP at Schaffer collateral to CA1 pyramidal cell synapses. It is worth noting that all these studies identified a role for RIM1α exclusively in excitatory LTP. We now demonstrate that CB1 receptor-dependent LTD at inhibitory synapses requires RIM1α . In addition, we show that cAMP/PKA signaling is necessary for hippocampal I-LTD. Enduring changes in the release machinery mediated by PKA signaling and RIM1α represent a widespread mechanism underlying presynaptic regulation of long-term plasticity (LTP and LTD) at both excitatory and inhibitory synapses.
I-LTD as a substrate for learning and memory
Disrupting eCB-CB1 receptor signaling has a variety of effects on both hippocampus- and amygdala-dependent memory tasks (Chhatwal et al., 2005; Kamprath et al., 2006; Marsicano et al., 2002; Mikics et al., 2006; Suzuki et al., 2004; Varvel et al., 2005; Varvel and Lichtman, 2002; Varvel et al., 2006). However, the precise role of this pathway, let alone I-LTD, in learning and memory still remains to be fully understood. Unlike manipulations involving the CB1 receptor, the RIM1α KO maintains short term eCB mediated plasticity (DSI) without the long term form (I-LTD). These mice have striking deficits in acquiring both hippocampus- and amygdala-dependent memories (Powell et al., 2004). I-LTD strongly modulates excitability and induction of LTP in both brain areas (Azad et al., 2004; Chevaleyre and Castillo, 2004), so it is possible that this process, together with other deficits in synaptic function (Huang et al., 2005; Kaeser and Sudhof, 2005) could contribute to the RIM1α KO phenotype. Selective disruption of the RIM1α pathway in a subset of synaptic inputs (e.g. those expressing CB1 receptors) may help to establish the contribution of eCB-mediated long-term plasticity to learning and memory.
METHODS
Experiments were performed either in C57Bl/6 mice (3-6 weeks; Charles River), paired RIM1α KO and WT littermates (3-6 weeks), or in Wistar rats (3-5 weeks; Charles River). RIM1α KO mice were generated as described previously (Schoch et al., 2002). Transverse hippocampal slices were prepared as described elsewhere (Chevaleyre and Castillo, 2003). Animals were killed by decapitation in accordance with institutional regulations. Slices (400 μm thickness) were cut on a vibrating slicer (Dosaka, Kyoto, Japan) in ice-cold extracellular solution containing (in mM): 215 Sucrose, 2.5 KCl, 20 glucose, 26 NaHCO3, 1.6 NaH2PO4, 1 CaCl2, 4 MgSO4 and 4 MgCl2. The cutting medium was gradually switched to the recording solution (ACSF) that contained (in mM): 124 NaCl, 2.5 KCl, 10 glucose, 26 NaHCO3, 1 NaH2PO4, 2.5 CaCl2 and 1.3 MgSO4. Slices were kept at room temperature for at least 1.5 hr before recording. Cutting and recording solutions were saturated with 95% O2 and 5% CO2, pH 7.4. Experiments were performed at 25.0 ± 0.1 °C except for mIPSC recordings (28.0 °C).
CA1 pyramidal cells were blind-patched and recorded in whole-cell voltage clamp mode. For recording IPSCs, hippocampal slices were completely submerged and continuously superfused at a flow rate of 2 ml/min with ACSF containing NMDA and AMPA/Kainate receptor antagonists (25 μM d-APV and 10 μM NBQX). Constant-voltage, synaptic stimulation (200 μs pulse width), was delivered in pairs (100 ms inter-stimulus interval) via a patch-type pipette filled with ACSF and placed in the middle third of s. radiatum. For experiments with “bulk” stimulation, stimulation pipette tips were broken to ~20 μm and IPSCs were recorded at -60 mV for DSI or +10 mV for I-LTD with an internal solution containing (in mM): 123 cesium gluconate, 1 CaCl2, 10 EGTA, 10 Hepes, 10 glucose, 5 ATP, 0.4 GTP (pH 7.2; 280-290 mOsm). We adjusted stimulus intensity to evoke IPSCs of comparable amplitudes across experiments (0.5-1.4 nA). All mIPSCs were recorded with the same internal solution at +20 mV, and ACSF-containing 500 nM TTX, 100 μM CdCl2, 5 mM CaCl2, 25 μM d-APV and 10 μM NBQX. For focal stimulation experiments, IPSCs were recorded at -60 mV with an internal solution containing CsCl instead of CsGluconate. The stimulus strength for these experiments was reduced to elicit small amplitude IPSCs (~30 pA) and a ~10 % failure rate over >10 min baseline. Series resistance (Rs, typically 8-15 MΩ) was continuously monitored throughout each experiment with a -5 mV, 80 ms command pulse delivered prior to each stimulus pair; cells with more than 10 % change in Rs were excluded from analysis. I-LTD was induced after a stable baseline by theta-burst stimulation (TBS) consisting of a series of 10 bursts of 5 stimuli (100 Hz within each burst, 200 ms inter-burst interval, repeated 4 times, 5 s apart). I-LTD magnitude was quantified by averaging IPSC amplitudes, for 10 min periods right before and 20 min after TBS. DSI was evoked by a 5 second voltage step from -60 to 0 mV. Typically 2-3 DSIs per cell were averaged prior to group statistical analysis. The peak inhibition during DSI was quantified by averaging IPSC amplitudes 6 and 12 seconds after depolarization. IPSCs were monitored every 6 seconds for DSI and every 20 seconds for I-LTD. 5-10 s epochs were obtained every 15 s for mIPSC analysis. Extracellular field recordings were performed using recording pipettes filled with 1M NaCl. For field EPSPs (fEPSPs) stimulating and recording pipettes were placed in the middle third of CA1 s. radiatum. Field IPSP (fIPSP) experiments in CA1 were performed under the same recording conditions as whole-cell IPSC experiments, except that stimulation and recording pipette tips were both placed in s. pyramidale ~150 um apart. Stimulating and recording pipettes were placed at a similar depth within the tissue for each experiment (80-100 μm). Stimulation intensity was adjusted to produce 0.2-0.4 mV fIPSP. In each experiment, we confirmed that these potentials were completely blocked by the GABA-A receptor antagonist picrotoxin (100 μM), in accordance with previously published reports (Lambert et al, 1991; Arai et al, 1995).
For experiments performed in basolateral amygdala (BLA), coronal slices were prepared using similar methods as described above from 4-8 week old male C57Bl/6 mice. Principal cells in the BLA were identified by their morphological and electrophysiological characteristics (Washburn and Moises, 1992). IPSCs were recorded from principal cells in the basal nucleus using the same internal solution as for focal stimulation in hippocampus, voltage clamped between -60 and -70 mV. Synaptic responses were evoked as paired pulse stimuli (200 μs pulse width, 50 ms apart) via monopolar ACSF-filled patch-type pipettes with broken tips (~20 μm) placed just medial to the external capsule. I-LTD was triggered with low frequency stimulation (100 stimuli at 1 Hz, intensity twice that of test pulses). Recording conditions for mIPSCs were identical to those for hippocampus.
Whole-cell and extracellular recordings were performed with a MultiClamp 700A amplifier (Axon Instruments Inc., Union City, CA) whose output signals were filtered at 3 kHz, acquired at 5 kHz and analyzed on-line using customized software for IgorPro (Wavemetrics Inc., Lake Oswego, OR). Results are reported as mean ± SEM. Statistical analyses were performed using Student’s t-test. Unless otherwise stated, drugs were bath applied following dilution into the ACSF from concentrated stock solutions. SR 141716 (NIMH), H-89 (Sigma-Aldrich), forskolin (Calbiochem), NBQX, AM 251, WIN 55,212-2 and CP 55,940 (Tocris) were dissolved in DMSO. d-APV, DHPG (Tocris), Protein Kinase A Inhibitor 14-22 amide (cell permeable, myristoylated) and Protein Kinase A Inhibitor 6-22 Amide (Calbiochem) were dissolved in water. For experiments involving drug treatment of slices, interleaved controls were performed in the same concentration of the respective solvent. Stock solutions of drugs were stored at -20 °C prior to use. RIM1α KO and WT littermates were shipped to Albert Einstein College of Medicine (Bronx, NY) unidentified, and data were acquired and analyzed in a blind fashion; the KO mice exhibit no obvious phenotype (Schoch et al., 2002). Genotype was confirmed afterward by PCR using tail DNA.
Acknowledgments
We thank Donald S. Faber, Michael V.L. Bennett and Kanji Takahashi for their constructive comments on the manuscript. We are also grateful to Pankaj Sah for his advice on BLA recordings. This work was supported by the US National Institutes of Health/NIDA (P.E.C.) and the Howard Hughes Medical Institutes (T.C.S.). V.C. was partially supported by the Epilepsy Foundation. P.E.C. is a Pew Biomedical Scholar.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alger BE. Retrograde signaling in the regulation of synaptic transmission: focus on endocannabinoids. Prog Neurobiol. 2002;68:247–286. doi: 10.1016/s0301-0082(02)00080-1. [DOI] [PubMed] [Google Scholar]
- Alger BE, Pitler TA, Wagner JJ, Martin LA, Morishita W, Kirov SA, Lenz RA. Retrograde signalling in depolarization-induced suppression of inhibition in rat hippocampal CA1 cells. J Physiol. 1996;496(Pt 1):197–209. doi: 10.1113/jphysiol.1996.sp021677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai A, Silberg J, Lynch G. Differences in the refractory properties of two distinct inhibitory circuitries in field CA1 of the hippocampus. Brain Res. 1995;704:298–306. doi: 10.1016/0006-8993(95)01137-4. [DOI] [PubMed] [Google Scholar]
- Azad SC, Eder M, Marsicano G, Lutz B, Zieglgansberger W, Rammes G. Activation of the cannabinoid receptor type 1 decreases glutamatergic and GABAergic synaptic transmission in the lateral amygdala of the mouse. Learn Mem. 2003;10:116–128. doi: 10.1101/lm.53303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azad SC, Monory K, Marsicano G, Cravatt BF, Lutz B, Zieglgansberger W, Rammes G. Circuitry for associative plasticity in the amygdala involves endocannabinoid signaling. J Neurosci. 2004;24:9953–9961. doi: 10.1523/JNEUROSCI.2134-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender VA, Bender KJ, Brasier DJ, Feldman DE. Two coincidence detectors for spike timing-dependent plasticity in somatosensory cortex. J Neurosci. 2006;26:4166–4177. doi: 10.1523/JNEUROSCI.0176-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calakos N, Schoch S, Sudhof TC, Malenka RC. Multiple roles for the active zone protein RIM1alpha in late stages of neurotransmitter release. Neuron. 2004;42:889–896. doi: 10.1016/j.neuron.2004.05.014. [DOI] [PubMed] [Google Scholar]
- Capogna M, Gahwiler BH, Thompson SM. Presynaptic enhancement of inhibitory synaptic transmission by protein kinases A and C in the rat hippocampus in vitro. J Neurosci. 1995;15:1249–1260. doi: 10.1523/JNEUROSCI.15-02-01249.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson G, Wang Y, Alger BE. Endocannabinoids facilitate the induction of LTP in the hippocampus. Nat Neurosci. 2002;5:723–724. doi: 10.1038/nn879. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Janz R, Sudhof TC, Tzounopoulos T, Malenka RC, Nicoll RA. Rab3A is essential for mossy fibre long-term potentiation in the hippocampus. Nature. 1997;388:590–593. doi: 10.1038/41574. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Schoch S, Schmitz F, Sudhof TC, Malenka RC. RIM1alpha is required for presynaptic long-term potentiation. Nature. 2002;415:327–330. doi: 10.1038/415327a. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron. 2003;38:461–472. doi: 10.1016/s0896-6273(03)00235-6. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Endocannabinoid-mediated metaplasticity in the hippocampus. Neuron. 2004;43:871–881. doi: 10.1016/j.neuron.2004.08.036. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Takahashi KA, Castillo PE. Endocannabinoid-Mediated Synaptic Plasticity in the CNS. Annu Rev Neurosci. 2006;29:37–75. doi: 10.1146/annurev.neuro.29.051605.112834. [DOI] [PubMed] [Google Scholar]
- Chhatwal JP, Davis M, Maguschak KA, Ressler KJ. Enhancing cannabinoid neurotransmission augments the extinction of conditioned fear. Neuropsychopharmacology. 2005;30:516–524. doi: 10.1038/sj.npp.1300655. [DOI] [PubMed] [Google Scholar]
- Childers SR, Deadwyler SA. Role of cyclic AMP in the actions of cannabinoid receptors. Biochem Pharmacol. 1996;52:819–827. doi: 10.1016/0006-2952(96)00419-4. [DOI] [PubMed] [Google Scholar]
- Cohen GA, Doze VA, Madison DV. Opioid inhibition of GABA release from presynaptic terminals of rat hippocampal interneurons. Neuron. 1992;9:325–335. doi: 10.1016/0896-6273(92)90171-9. [DOI] [PubMed] [Google Scholar]
- Collin T, Marty A, Llano I. Presynaptic calcium stores and synaptic transmission. Curr Opin Neurobiol. 2005;15:275–281. doi: 10.1016/j.conb.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Daniel H, Rancillac A, Crepel F. Mechanisms underlying cannabinoid inhibition of presynaptic Ca2+ influx at parallel fibre synapses of the rat cerebellum. J Physiol. 2004;557:159–174. doi: 10.1113/jphysiol.2004.063263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diana MA, Marty A. Characterization of depolarization-induced suppression of inhibition using paired interneuron--Purkinje cell recordings. J Neurosci. 2003;23:5906–5918. doi: 10.1523/JNEUROSCI.23-13-05906.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diana MA, Marty A. Endocannabinoid-mediated short-term synaptic plasticity: depolarization-induced suppression of inhibition (DSI) and depolarization-induced suppression of excitation (DSE) Br J Pharmacol. 2004;142:9–19. doi: 10.1038/sj.bjp.0705726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittman JS, Regehr WG. Contributions of calcium-dependent and calcium-independent mechanisms to presynaptic inhibition at a cerebellar synapse. J Neurosci. 1996;16:1623–1633. doi: 10.1523/JNEUROSCI.16-05-01623.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin AC. G protein modulation of voltage-gated calcium channels. Pharmacol Rev. 2003;55:607–627. doi: 10.1124/pr.55.4.3. [DOI] [PubMed] [Google Scholar]
- Edwards DA, Kim J, Alger BE. Multiple mechanisms of endocannabinoid response initiation in hippocampus. J Neurophysiol. 2006;95:67–75. doi: 10.1152/jn.00813.2005. [DOI] [PubMed] [Google Scholar]
- Freund TF, Katona I, Piomelli D. Role of endogenous cannabinoids in synaptic signaling. Physiol Rev. 2003;83:1017–1066. doi: 10.1152/physrev.00004.2003. [DOI] [PubMed] [Google Scholar]
- Gerdeman GL, Lovinger DM. Emerging roles for endocannabinoids in long-term synaptic plasticity. Br J Pharmacol. 2003;140:781–789. doi: 10.1038/sj.bjp.0705466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdeman GL, Ronesi J, Lovinger DM. Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci. 2002;5:446–451. doi: 10.1038/nn832. [DOI] [PubMed] [Google Scholar]
- Gereau RWt, Conn PJ. Multiple presynaptic metabotropic glutamate receptors modulate excitatory and inhibitory synaptic transmission in hippocampal area CA1. J Neurosci. 1995;15:6879–6889. doi: 10.1523/JNEUROSCI.15-10-06879.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajos N, Katona I, Naiem SS, MacKie K, Ledent C, Mody I, Freund TF. Cannabinoids inhibit hippocampal GABAergic transmission and network oscillations. Eur J Neurosci. 2000;12:3239–3249. doi: 10.1046/j.1460-9568.2000.00217.x. [DOI] [PubMed] [Google Scholar]
- Heinbockel T, Brager DH, Reich CG, Zhao J, Muralidharan S, Alger BE, Kao JP. Endocannabinoid signaling dynamics probed with optical tools. J Neurosci. 2005;25:9449–9459. doi: 10.1523/JNEUROSCI.2078-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA. Modulation of Ca2+ channels by G-protein beta gamma subunits. Nature. 1996;380:258–262. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- Hescheler J, Schultz G. G-proteins involved in the calcium channel signalling system. Curr Opin Neurobiol. 1993;3:360–367. doi: 10.1016/0959-4388(93)90129-m. [DOI] [PubMed] [Google Scholar]
- Hille B. G protein-coupled mechanisms and nervous signaling. Neuron. 1992;9:187–195. doi: 10.1016/0896-6273(92)90158-a. [DOI] [PubMed] [Google Scholar]
- Hoffman AF, Lupica CR. Mechanisms of cannabinoid inhibition of GABA(A) synaptic transmission in the hippocampus. J Neurosci. 2000;20:2470–2479. doi: 10.1523/JNEUROSCI.20-07-02470.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, Felder CC, Herkenham M, Mackie K, Martin BR, et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev. 2002;54:161–202. doi: 10.1124/pr.54.2.161. [DOI] [PubMed] [Google Scholar]
- Howlett AC, Qualy JM, Khachatrian LL. Involvement of Gi in the inhibition of adenylate cyclase by cannabimimetic drugs. Mol Pharmacol. 1986;29:307–313. [PubMed] [Google Scholar]
- Huang CC, Chen YL, Lo SW, Hsu KS. Activation of cAMP-dependent protein kinase suppresses the presynaptic cannabinoid inhibition of glutamatergic transmission at corticostriatal synapses. Mol Pharmacol. 2002;61:578–585. doi: 10.1124/mol.61.3.578. [DOI] [PubMed] [Google Scholar]
- Huang YY, Li XC, Kandel ER. cAMP contributes to mossy fiber LTP by initiating both a covalently mediated early phase and macromolecular synthesis-dependent late phase. Cell. 1994;79:69–79. doi: 10.1016/0092-8674(94)90401-4. [DOI] [PubMed] [Google Scholar]
- Huang YY, Zakharenko SS, Schoch S, Kaeser PS, Janz R, Sudhof TC, Siegelbaum SA, Kandel ER. Genetic evidence for a protein-kinase-A-mediated presynaptic component in NMDA-receptor-dependent forms of long-term synaptic potentiation. Proc Natl Acad Sci U S A. 2005;102:9365–9370. doi: 10.1073/pnas.0503777102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda SR. Voltage-dependent modulation of N-type calcium channels by G-protein beta gamma subunits. Nature. 1996;380:255–258. doi: 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- Jarolimek W, Misgeld U. GABAB receptor-mediated inhibition of tetrodotoxin-resistant GABA release in rodent hippocampal CA1 pyramidal cells. J Neurosci. 1997;17:1025–1032. doi: 10.1523/JNEUROSCI.17-03-01025.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeser PS, Sudhof TC. RIM function in short- and long-term synaptic plasticity. Biochem Soc Trans. 2005;33:1345–1349. doi: 10.1042/BST0331345. [DOI] [PubMed] [Google Scholar]
- Kamprath K, Marsicano G, Tang J, Monory K, Bisogno T, Di Marzo V, Lutz B, Wotjak CT. Cannabinoid CB1 receptor mediates fear extinction via habituation-like processes. J Neurosci. 2006;26:6677–6686. doi: 10.1523/JNEUROSCI.0153-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona I, Rancz EA, Acsady L, Ledent C, Mackie K, Hajos N, Freund TF. Distribution of CB1 cannabinoid receptors in the amygdala and their role in the control of GABAergic transmission. J Neurosci. 2001;21:9506–9518. doi: 10.1523/JNEUROSCI.21-23-09506.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo S, Marty A. Protein kinase A-mediated enhancement of miniature IPSC frequency by noradrenaline in rat cerebellar stellate cells. J Physiol. 1997;498:165–176. doi: 10.1113/jphysiol.1997.sp021849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koushika SP, Richmond JE, Hadwiger G, Weimer RM, Jorgensen EM, Nonet ML. A post-docking role for active zone protein Rim. Nat Neurosci. 2001;4:997–1005. doi: 10.1038/nn732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Dopamine modulation of state-dependent endocannabinoid release and long-term depression in the striatum. J Neurosci. 2005;25:10537–10545. doi: 10.1523/JNEUROSCI.2959-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG. Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron. 2001;29:717–727. doi: 10.1016/s0896-6273(01)00246-x. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG. Retrograde signaling by endocannabinoids. Curr Opin Neurobiol. 2002;12:324–330. doi: 10.1016/s0959-4388(02)00328-8. [DOI] [PubMed] [Google Scholar]
- Lambert NA, Borroni AM, Grover LM, Teyler TJ. Hyperpolarizing and depolarizing GABAA receptor-mediated dendritic inhibition in area CA1 of the rat hippocampus. J Neurophysiol. 1991;66:1538–1548. doi: 10.1152/jn.1991.66.5.1538. [DOI] [PubMed] [Google Scholar]
- Lenz RA, Wagner JJ, Alger BE. N- and L-type calcium channel involvement in depolarization-induced suppression of inhibition in rat hippocampal CA1 cells. J Physiol. 1998;512(Pt 1):61–73. doi: 10.1111/j.1469-7793.1998.061bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden DJ, Ahn S. Activation of presynaptic cAMP-dependent protein kinase is required for induction of cerebellar long-term potentiation. J Neurosci. 1999;19:10221–10227. doi: 10.1523/JNEUROSCI.19-23-10221.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llano I, Gerschenfeld HM. Beta-adrenergic enhancement of inhibitory synaptic activity in rat cerebellar stellate and Purkinje cells. J Physiol. 1993;468:201–224. doi: 10.1113/jphysiol.1993.sp019767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonart G, Schoch S, Kaeser PS, Larkin CJ, Sudhof TC, Linden DJ. Phosphorylation of RIM1alpha by PKA triggers presynaptic long-term potentiation at cerebellar parallel fiber synapses. Cell. 2003;115:49–60. doi: 10.1016/s0092-8674(03)00727-x. [DOI] [PubMed] [Google Scholar]
- Mackie K, Hille B. Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proc Natl Acad Sci U S A. 1992;89:3825–3829. doi: 10.1073/pnas.89.9.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann C, Zieglgansberger W, et al. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- Mikics E, Dombi T, Barsvari B, Varga B, Ledent C, Freund TF, Haller J. The effects of cannabinoids on contextual conditioned fear in CB1 knockout and CD1 mice. Behav Pharmacol. 2006;17:223–230. doi: 10.1097/00008877-200605000-00003. [DOI] [PubMed] [Google Scholar]
- Morishita W, Kirov SA, Alger BE. Evidence for metabotropic glutamate receptor activation in the induction of depolarization-induced suppression of inhibition in hippocampal CA1. J Neurosci. 1998;18:4870–4882. doi: 10.1523/JNEUROSCI.18-13-04870.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piomelli D. The molecular logic of endocannabinoid signalling. Nat Rev Neurosci. 2003;4:873–884. doi: 10.1038/nrn1247. [DOI] [PubMed] [Google Scholar]
- Pitler TA, Alger BE. Depolarization-induced suppression of GABAergic inhibition in rat hippocampal pyramidal cells: G protein involvement in a presynaptic mechanism. Neuron. 1994;13:1447–1455. doi: 10.1016/0896-6273(94)90430-8. [DOI] [PubMed] [Google Scholar]
- Poncer JC, Shinozaki H, Miles R. Dual modulation of synaptic inhibition by distinct metabotropic glutamate receptors in the rat hippocampus. J Physiol. 1995;485(Pt 1):121–134. doi: 10.1113/jphysiol.1995.sp020717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell CM, Schoch S, Monteggia L, Barrot M, Matos MF, Feldmann N, Sudhof TC, Nestler EJ. The presynaptic active zone protein RIM1alpha is critical for normal learning and memory. Neuron. 2004;42:143–153. doi: 10.1016/s0896-6273(04)00146-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbe D, Alonso G, Duchamp F, Bockaert J, Manzoni OJ. Localization and mechanisms of action of cannabinoid receptors at the glutamatergic synapses of the mouse nucleus accumbens. J Neurosci. 2001;21:109–116. doi: 10.1523/JNEUROSCI.21-01-00109.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbe D, Kopf M, Remaury A, Bockaert J, Manzoni OJ. Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proc Natl Acad Sci U S A. 2002;99:8384–8388. doi: 10.1073/pnas.122149199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronesi J, Gerdeman GL, Lovinger DM. Disruption of endocannabinoid release and striatal long-term depression by postsynaptic blockade of endocannabinoid membrane transport. J Neurosci. 2004;24:1673–1679. doi: 10.1523/JNEUROSCI.5214-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitow F, Suzuki H, Konishi S. beta-Adrenoceptor-mediated long-term up-regulation of the release machinery at rat cerebellar GABAergic synapses. J Physiol. 2005;565:487–502. doi: 10.1113/jphysiol.2005.084384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salin PA, Malenka RC, Nicoll RA. Cyclic AMP mediates a presynaptic form of LTP at cerebellar parallel fiber synapses. Neuron. 1996;16:797–803. doi: 10.1016/s0896-6273(00)80099-9. [DOI] [PubMed] [Google Scholar]
- Scanziani M, Capogna M, Gahwiler BH, Thompson SM. Presynaptic inhibition of miniature excitatory synaptic currents by baclofen and adenosine in the hippocampus. Neuron. 1992;9:919–927. doi: 10.1016/0896-6273(92)90244-8. [DOI] [PubMed] [Google Scholar]
- Scanziani M, Gahwiler BH, Thompson SM. Presynaptic inhibition of excitatory synaptic transmission by muscarinic and metabotropic glutamate receptor activation in the hippocampus: are Ca2+ channels involved? Neuropharmacology. 1995;34:1549–1557. doi: 10.1016/0028-3908(95)00119-q. [DOI] [PubMed] [Google Scholar]
- Schoch S, Castillo PE, Jo T, Mukherjee K, Geppert M, Wang Y, Schmitz F, Malenka RC, Sudhof TC. RIM1alpha forms a protein scaffold for regulating neurotransmitter release at the active zone. Nature. 2002;415:321–326. doi: 10.1038/415321a. [DOI] [PubMed] [Google Scholar]
- Schoch S, Mittelstaedt T, Kaeser PS, Padgett D, Feldmann N, Chevaleyre V, Castillo PE, Hammer RE, Han W, Schmitz F, et al. Redundant functions of RIM1alpha and RIM2alpha in Ca(2+)-triggered neurotransmitter release. Embo J. 2006;25:5852–5863. doi: 10.1038/sj.emboj.7601425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz KP, Miller RJ. Inhibition of quantal transmitter release in the absence of calcium influx by a G protein-linked adenosine receptor at hippocampal synapses. Neuron. 1992;8:1139–1150. doi: 10.1016/0896-6273(92)90134-y. [DOI] [PubMed] [Google Scholar]
- Sciancalepore M, Cherubini E. Protein kinase A-dependent increase in frequency of miniature GABAergic currents in rat CA3 hippocampal neurons. Neurosci Lett. 1995;187:91–94. doi: 10.1016/0304-3940(95)11348-7. [DOI] [PubMed] [Google Scholar]
- Seino S, Shibasaki T. PKA-dependent and PKA-independent pathways for cAMP-regulated exocytosis. Physiol Rev. 2005;85:1303–1342. doi: 10.1152/physrev.00001.2005. [DOI] [PubMed] [Google Scholar]
- Sjostrom PJ, Turrigiano GG, Nelson SB. Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron. 2003;39:641–654. doi: 10.1016/s0896-6273(03)00476-8. [DOI] [PubMed] [Google Scholar]
- Soler-Llavina GJ, Sabatini BL. Synapse-specific plasticity and compartmentalized signaling in cerebellar stellate cells. Nat Neurosci. 2006;9:798–806. doi: 10.1038/nn1698. [DOI] [PubMed] [Google Scholar]
- Storm DR, Hansel C, Hacker B, Parent A, Linden DJ. Impaired cerebellar long-term potentiation in type I adenylyl cyclase mutant mice. Neuron. 1998;20:1199–1210. doi: 10.1016/s0896-6273(00)80500-0. [DOI] [PubMed] [Google Scholar]
- Sudhof TC. The synaptic vesicle cycle. Annu Rev Neurosci. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Josselyn SA, Frankland PW, Masushige S, Silva AJ, Kida S. Memory reconsolidation and extinction have distinct temporal and biochemical signatures. J Neurosci. 2004;24:4787–4795. doi: 10.1523/JNEUROSCI.5491-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo B, Schlicker E. Effects of cannabinoids on neurotransmission. Handb Exp Pharmacol. 2005:327–365. doi: 10.1007/3-540-26573-2_11. [DOI] [PubMed] [Google Scholar]
- Takahashi KA, Linden DJ. Cannabinoid receptor modulation of synapses received by cerebellar Purkinje cells. J Neurophysiol. 2000;83:1167–1180. doi: 10.1152/jn.2000.83.3.1167. [DOI] [PubMed] [Google Scholar]
- Trudeau LE, Emery DG, Haydon PG. Direct modulation of the secretory machinery underlies PKA-dependent synaptic facilitation in hippocampal neurons. Neuron. 1996;17:789–797. doi: 10.1016/s0896-6273(00)80210-x. [DOI] [PubMed] [Google Scholar]
- Tzounopoulos T, Janz R, Sudhof TC, Nicoll RA, Malenka RC. A role for cAMP in long-term depression at hippocampal mossy fiber synapses. Neuron. 1998;21:837–845. doi: 10.1016/s0896-6273(00)80599-1. [DOI] [PubMed] [Google Scholar]
- Umemiya M, Berger AJ. Presynaptic inhibition by serotonin of glycinergic inhibitory synaptic currents in the rat brain stem. J Neurophysiol. 1995;73:1192–1201. doi: 10.1152/jn.1995.73.3.1192. [DOI] [PubMed] [Google Scholar]
- Varma N, Brager D, Morishita W, Lenz RA, London B, Alger B. Presynaptic factors in the regulation of DSI expression in hippocampus. Neuropharmacology. 2002;43:550–562. doi: 10.1016/s0028-3908(02)00168-5. [DOI] [PubMed] [Google Scholar]
- Varvel SA, Anum EA, Lichtman AH. Disruption of CB(1) receptor signaling impairs extinction of spatial memory in mice. Psychopharmacology (Berl) 2005;179:863–872. doi: 10.1007/s00213-004-2121-2. [DOI] [PubMed] [Google Scholar]
- Varvel SA, Lichtman AH. Evaluation of CB1 receptor knockout mice in the Morris water maze. J Pharmacol Exp Ther. 2002;301:915–924. doi: 10.1124/jpet.301.3.915. [DOI] [PubMed] [Google Scholar]
- Varvel SA, Wise LE, Niyuhire F, Cravatt BF, Lichtman AH. Inhibition of Fatty-Acid Amide Hydrolase Accelerates Acquisition and Extinction Rates in a Spatial Memory Task. Neuropsychopharmacology. 2006 doi: 10.1038/sj.npp.1301224. [DOI] [PubMed] [Google Scholar]
- Villacres EC, Wong ST, Chavkin C, Storm DR. Type I adenylyl cyclase mutant mice have impaired mossy fiber long-term potentiation. J Neurosci. 1998;18:3186–3194. doi: 10.1523/JNEUROSCI.18-09-03186.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Okamoto M, Schmitz F, Hofmann K, Sudhof TC. Rim is a putative Rab3 effector in regulating synaptic-vesicle fusion. Nature. 1997;388:593–598. doi: 10.1038/41580. [DOI] [PubMed] [Google Scholar]
- Washburn MS, Moises HC. Electrophysiological and morphological properties of rat basolateral amygdaloid neurons in vitro. J Neurosci. 1992;12:4066–4079. doi: 10.1523/JNEUROSCI.12-10-04066.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weimer RM, Gracheva EO, Meyrignac O, Miller KG, Richmond JE, Bessereau JL. UNC-13 and UNC-10/rim localize synaptic vesicles to specific membrane domains. J Neurosci. 2006;26:8040–8047. doi: 10.1523/JNEUROSCI.2350-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisskopf MG, Castillo PE, Zalutsky RA, Nicoll RA. Mediation of hippocampal mossy fiber long-term potentiation by cyclic AMP. Science. 1994;265:1878–1882. doi: 10.1126/science.7916482. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Kunos G, Nicoll RA. Presynaptic specificity of endocannabinoid signaling in the hippocampus. Neuron. 2001;31:453–462. doi: 10.1016/s0896-6273(01)00372-5. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA. Endocannabinoid signaling in the brain. Science. 2002;296:678–682. doi: 10.1126/science.1063545. [DOI] [PubMed] [Google Scholar]
- Yamasaki M, Hashimoto K, Kano M. Miniature synaptic events elicited by presynaptic Ca2+ rise are selectively suppressed by cannabinoid receptor activation in cerebellar Purkinje cells. J Neurosci. 2006;26:86–95. doi: 10.1523/JNEUROSCI.2258-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]