

Abstract
The current study aimed to understand the anti-apoptotic effect of overexpressed gap junction forming protein connexin (Cx) 43 in C6 glioma cells. C6 cells exposed to hydrogen peroxide (H2O2) or staurosporine demonstrated morphological and biochemical changes consistent with apoptosis, whereas C6 cells expressing Cx43 demonstrated relative resistance to H2O2, but not to staurosporine. This selective protection against H2O2 was due to inhibition of caspase 3 activation in Cx43 expressing cells. siRNA knockdown experiments in rat primary astrocytes confirmed the presence of endogenous Cx43-mediated anti-apoptotic effect. Cx43 interacts with the upstream apoptosis signal-regulating kinase 1 known to mediate H2O2-induced apoptosis providing a possible mechanism for protection. These findings provided new evidence for regulation of the mitogen activated protein kinase pathway and apoptosis by Cx43 implicating this protein in intracellular signaling beyond its role as a gap junction forming protein on the plasma membrane.
Keywords: connexin 43, apoptosis, C6 glioma, primary astrocytes, siRNA knock down, hydrogen peroxide, apoptosis signal-regulating kinase 1
Introduction
Connexin 43 (Cx43) is a gap junction forming protein found in the plasma membrane in a variety of cell types, including astrocytes. Gap junctions allow the transfer of small hydrophilic molecules less than 1.2 kDa, including ATP and Ca2+ [1], from one cell to adjacent cells. Gap junctions are composed of six connexin (Cx) proteins, which together make a connexon hemichannel. A hemichannel may interact with a hemichannel from a neighboring cell and together they form a gap junction. There have been 20 identified mammalian connexin proteins, and in addition to the prototypical role as the proteins forming the gap junction, they play diverse physiological roles including proliferation, differentiation, and development [2].
Gap junctions have previously been shown to propagate cellular injury in models of ischemia, traumatic brain injury, and chemotherapy-induced apoptosis, suggesting gap junctions may allow the passage of “death” signals directly from one cell to other. This phenomenon has be termed bystander death [3]. In contrast, gap junctions may aid the survival of cells, thought to be mediated through the dilution of the “death signal” through neighboring cells [3] or maintenance of homeostatic milieu [4].
Cx43 also appears to regulate apoptosis outside the traditional context of gap junctions. Expression of Cx43-(enhanced green fluorescent protein) EGFP fusion protein in HeLa cells usually devoid of functional gap junction protein resulted in an acceleration of injured cells exhibiting irreversible damage [5]. However, the degree and timing of the apoptotic process did not correlate with plasma membrane permeability through the hemichannel, assessed by ethidium bromide entry from the cell exterior into the cytoplasm, suggesting an alternative non-gap junction mechanism for Cx43 regulation of apoptosis. In the bone-forming osteoblasts and osteocytes, bisphosphonates, a class of drug clinically used to prevent bone loss, was shown to exert its anti-apoptotic effect through activation of Cx43 hemichannels. Cx43 transduction of this anti-apoptotic action of bisphosphonates required the pore-forming transmembrane domains as well as the cytoplasmic C-terminal of the Cx43 protein [6]. Conversely, camptothecin-induced apoptosis was three-fold higher in primary osteoblasts isolated from Cx43-null mutant mice compared to the wild-type control [7]. A site-specific proteolytic cleavage of Cx45.6 by caspase-3 in the developing lens suggested the presence of a reciprocal regulation of connexin by an apoptotic effector caspase further providing evidence for a link between apoptosis and Cx [8]. An association between Cx43 expression and apoptosis was also suggested by a gene chip study comparing genes differentially expressed between wild-type and Cx43-null mutant astrocytes [9]. Among the 252 mRNAs affected by the loss of Cx43 expression were genes associated with apoptosis and cell cycle regulation. The Cx43 protein itself, independent of its ability to form gap junctions, may serve a protective function conferring resistance to glial cell injury [10] again suggesting a role of Cx43 in cell death most likely independent of gap junctions. These observations suggest an under-explored role of Cx43 in the cell biology of growth and apoptosis and a deeper understanding of the cellular mechanisms by which Cx43 affects cell survival may reveal novel roles of this protein as a bona fide signal transduction molecule. The current study examined the role of Cx43 in apoptosis in C6 glioma cells caused by exposure to H2O2, a well established model for oxidative stress-induced injury.
Materials and methods
Culture
Rat C6 glioma cells stably expressing wild type connexin 43 (designated C6-Cx) or mock transfected cell (designated Cx-null) lines have been described [10]. Cultures were grown in DMEM supplemented with 10% fetal bovine serum and antibiotics. Low passage number cells were used for all experiments. Cells were seeded at a final density of 0.5 × 106 cells/ cm2 24 h before used in experiments. Cells were exposed to H2O2 in DMEM containing no serum for 5 - 15 min, media was aspirated and cells were left in DMEM containing no serum for indicated times. STS exposure was conducted in a serum containing medium for indicated times to prevent excessive cell death.
Primary cultures of rat brain astrocytes were obtained from cerebral cortices of 2 day old rat pups by trypsinization (0.05% for 20 minutes at 37 C°) and trituration before being plated onto 75 cm2 flasks. After the astrocytes became confluent, the cultures were washed 3 times in ice cold PBS to remove any microglia, oligodendroglia, and any remaining neurons and re-seeded onto 6 well plates for further experimentation.
Immunohistochemical staining with anti- glial fibrillary acidic protein antibody (Chemicon International, Temecula, CA, USA) confirmed greater than 95% purity for astrocytes.
siRNA knock down
siRNA targeting rat Cx43 (Accession number X06656) was designed using a web-based program (http://www.dharmacon.com/sidesign) and manufactured by Dharmacon Inc. (Chicago, IL, USA). Three siRNA were tested and after preliminary experiments, the sequence ACAUCAUUGAGCUCUUCUA and its complimentary strand were chosen for further use. For siRNA experiments, the astrocytes were plated onto 6 well dishes no more than 24 h prior to transfection. 200 pmol of double stranded annealed siRNA or its scrambled control was combined with Lipofectamine 2000 (3 μg/ well) in Opti-MEM media. The apoptotic cell count experiments were conducted 24-48 h after transfection when the target knockdown was verified to be maximal by Western blotting.
Caspase Assay
Cells treated with H2O2 or STS and incubated for variable times as dictated by the experiment were washed in PBS, lysed in ice-cold caspase lysis buffer (10mM Tris-HCl, 10mM NaH2PO4, 130mM NaCl, 1% Triton x-100, 10mM NaPPi, pH 7.5), for 15 min, and clarified by centrifugation. Protein concentration was standardized to 1 mg/ ml in caspase lysis buffer and 50 μg of protein was used for the assays. The caspase enzymatic reactions were carried out at 37°C in 20 mM PIPES, 100 mM KCl, 1 mM EDTA, 1% Chaps, 5 mM DTT (pH 7.2) containing either 0.1 mM Ac-DEVD-AMC or 0.1mM Ac-IETD-AMC (Biomol Research). The rate of fluorescence increase was measured kinetically over 1h (ex: 360 nm, em: 460 nm) using a Synergy II microplate reader (Bio-Tek Instruments, Winooski, VT, USA) and converted to AMC released/ min/ mg protein using the calibration standard correlating fluorescence to AMC.
TUNEL Assay
The TUNEL assay was carried out as per manufacturer’s instructions (Dead End Fluorimetric TUNEL Assay, Promega, Madison, WI, USA). Positive staining was visualized under an inverted microscope and random fields of view photographed and positive cells were counted and expressed as a percentage of total cells, determined by DAPI staining. At least 6 random fields with a total cell count >600 from 3 independent experiments were photographed and counted.
Western Blotting
Western blotting was carried out by standard methods. Proteins from cell lysates were separated by electrophoresis, and transferred to a nitrocellulose membrane (BioRad, Hercules, CA, USA). The membrane was blocked in 3% milk-TBST, probed with the anti-Cx43 antibody (1:500, Chemicon International; MAB3067) in 3% milk-PBST, and reacted with the horseradish peroxidase-conjugated secondary antibody (1:2000) in 1% milk-TBST. After reaction with the Western Lightning chemiluminescence reagent (NEN Life Science Products, Boston, MA, USA), the images were captured on the EpiChemi Darkroom System (UVP Inc, Upland, CA).
For immunoprecipitation, 200 μg cell lysate was incubated with 25 μl each of Agarose A and G (Invitrogen) in 500 μl total volume for 1 h and immunoprecipitating antibody (4 μg) added with additional 1 h incubation at 4C° with constant rotation. The reaction was washed 3 times with RIPA buffer and the pellet was resuspended in SDS loading buffer. After boiling, the supernatant was loaded for Western blotting as described above. Parallel experiments but omitting the immunoprecipitating antibody served as a negative control for all experiments. The co-immunoprecipitation experiments (Figure 3) are presented in 3 lanes consisting of In (input lysate), - (no antibody), and + (with antibody) where the total protein in input lysate was approximately 1/10 of the amount used for immunoprecipitation. Antibodies used were: JNK (Cell Signaling Technology, Beverly, MA, USA, #9252); MKK4 (Cell Signaling Technology; #9152), MKK7 (Cell Signaling Technology; #4172), c-Src (Cell Signaling Technology; #2107), EGFP (Roche Molecular, Indianapolis, IN, USA; #1814460), pan-ASK1 (Santa Cruz Biotech, Santa Cruz, CA, USA; sc-7931), pThr845-ASK1 (Cell Signaling Technology; #3765).
Figure 3. Cx43 co-immunoprecipitates with endogenous ASK1 but not with other members of the JNK-signaling cascade.
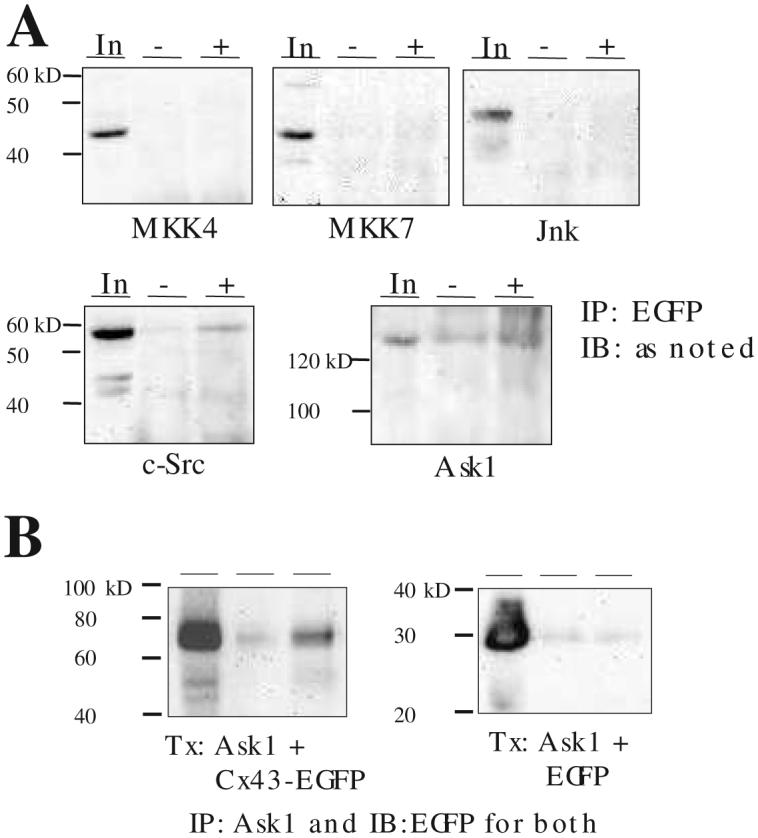
A. HEK293 cells were transfected with a Cx43-EGFP plasmid, cells harvested 48 h after transfection, lysates subjected to immunoprecipitation (IP) with anti-EGFP antibody, and the Western blot probed with the respective anti-MKK4, -MKK7, -JNK, -cSrc, or anti-ASK1 antibodies. B. Same as above but in cells transfected (Tx) with both ASK1 and Cx43-EGFP (left) or EGFP (right). In: input lysate approximately 1:10 of the protein amount used for IP, -: no IP antibody control, +: with IP antibody. Representative of 3 independent experiments.
Data analysis
Data are presented as mean ± SD and the statistical significance determined by the Mann-Whitney nonparametric test at the indicated p-level.
Results
Expression of Cx43 inhibits H2O2-induced caspase 3 activation in C6 cells
Exposure of C6-null cells to 100 μM H2O2 resulted in an increase in morphological changes consistent with apoptosis, including cellular shrinkage and nuclear condensation. As previously described [10], C6-Cx cells demonstrated a marked resistance to H2O2 (Figure 1A). Pretreatment with 10μM DEVD-FMK or LEHD-FMK, to inhibit caspase -3 or -8, respectively, demonstrated a caspase-3-mediated cell death induced by H2O2 in C6-null cells (Figure 1B). Consistent with the observation that DEVD-FMK reduced H2O2-induced apoptosis, C6-null cells demonstrated a marked increase in caspase-3 like activity, in a time and H2O2 concentration-dependent manner, but with little change in caspase-8 like activity (Figure 1C, D). Western blot of lysates from the two cell lines (Figure 1C, inset) demonstrated the presence of similar amounts of caspase-3 protein indicating that the difference in the caspase-3 like activity must be due to a difference in H2O2 activation of this protease. To ensure that the relative-apoptotic resistant C6-Cx cells could indeed induce caspase-3 activity and that protection was not due to the inability of C6-Cx cells to activate this protease, the caspase 3 activity was measured in STS treated lysates. C6-Cx treated with STS demonstrated the expected increase in caspase-3 like and caspase-8 like activity not different from the C6-null cells (Figure 1E, F). Taken together, these results suggested Cx43-mediated cellular protection against H202-induced injury was specific and pre-mitochondrial likely to involve upstream kinases.
Figure 1. Expression of Cx43 confers relative protection against H2O2-induced apoptosis.

A. C6-null and -Cx cells were exposed to 100μM H2O2 for 15 min and left in serum-free media for an additional 24 h. After fixation, cells were TUNEL labeled and co-stained with DAPI. TUNEL positive cells are clearly seen in C6-null cultures treated with H2O2 (arrows), but not in C6-Cx cells. B. TUNEL labeling was quantified by counting cells from random fields. Where used, caspase inhibitors were pre-incubated for 30 min prior to H2O2 insult and left throughout the duration of the experiment. Greater than 400 cells, from n = 6 experiments were counted. Lysates were prepared from H2O2-injured cells harvested at the indicated times, and proteolysis of (C) Ac-DEVD-AMC (caspase 3-like reporter) or (D) Ac-IETD-AMC (caspase 8-like reporter) was kinetically recorded at 37 C° at 1 min intervals for 1h. Data represents mean ± SD obtained from 3 separate experiments and * indicates significant difference at p<0.05. Inset in C is a Western blot probed with anti-caspase 3 antibody demonstrating equivalent amounts of the caspase protein present in both cell lines. Lysates from STS (1 μM for 6 h) treated cells demonstrated a marked increase in both (E) caspase-3 like and (F), caspase 8-like activity for both cell lines. The means were not significantly different between the C6-null and C6-Cx cell lines under control or STS stimulation. In contrast, STS stimulation significantly increased both caspase-like activities over control.
Cx43 knockdown in primary astrocytes enhances H2O2 toxicity
To demonstrate the presence of a similar Cx43 regulation of apoptosis in non-transformed cells, we examined the effect of siRNA knockdown of Cx43 on H2O2-induced apoptosis in cultured primary astrocytes. If Cx43 is protective against H2O2, knockdown of this protective protein should increase the occurrence of apoptotic events. After transient transfection with siRNA, Cx43 protein expression was decreased in a time-dependent manner demonstrating maximum inhibition at times longer than 24 hours (Figure 2A) consistent with the rapid turnover of Cx43 protein [11]. Cells mock transfected demonstrated a marked resistance to H2O2-induced apoptosis, whereas cells treated with siRNA, resulting in a knock down of the Cx43 protein level, showed an increase in sensitivity to H2O2 consistent with a protective role of the endogenously expressed Cx43 in primary astrocytes (Figure 2B). Therefore the cytoprotective effect of Cx43 is not an epiphenomenon of a transformed C6 glioma cell line engineered to over express this protein.
Figure 2. siRNA knock down of endogenous Cx43 sensitizes cultured primary astrocytes to H2O2-induced apoptosis.

A. Cultured primary astrocytes were transfected with Cx43 siRNA (left) or control siRNA (right) (200 pmoles/ well) and the level of Cx43 protein in cell lysates harvested at the indicated time points after transfection determined by a Western blot. The blot shown is representative of 3 replicate experiments. GAPDH serves as a loading control. B. Photographs of control C6 cells with or without H2O2 treatment. Note the clear correlation between cells with nuclear condensation (arrows) and the TUNEL positive cells. Scale bar = 30 μm. C. 48 h after siRNA transfection, cultures were treated with H2O2 (0, 300, or 1000 μM) for 24h and subsequently fixed. Hoechst stained nuclei were counted from random fields and apoptotic nuclei expressed as a percentage of total. Data represent mean ± SD from four independent experiments. * denotes p<0.05 or ** p <0.01 between the control (open) and 200 pmoles siRNA transfected (hatched) conditions.
Cx43 protein interacts with apoptosis signal-regulating kinase (ASK) 1
Cx43 has been shown to directly interact with members of the MAPK family including ERK1/2 [12] and ERK5 [13]. Therefore, we sought for a potential physical interaction between the Cx43 protein and the kinases comprising the ASK1 pathway well documented to mediate H2O2-induced apoptosis [14]. Since the commercially available antibodies for the ASK1 pathway kinases only recognized the human isoform and C6 cells are difficult to transfect, the human embryonic kidney (HEK) 293 cells were transfected with the expression plasmid encoding Cx43-EGFP and probed with antibodies against one of the following: JNK, MKK4, MKK7, or ASK1. The presence of EGFP epitope-tag allowed for monitoring of transfection efficiency while not interfering with the Cx43 function [15] nor its interaction with ERK5 [13]. Lysates harvested 48 hours later were immunoprecipitated with the anti-EGFP antibody and probed for co-immunoprecipitation of the endogenous ASK1 pathway kinases. We chose to use the anti-EGFP antibody for immunoprecipitation and the respective anti-kinase antibody for immunoblotting in an effort to minimize problems due to potential differences in immunoprecipitating efficiency of the different anti-kinase antibodies. Figure 3A shows a co-immunoprecipitation between Cx43-EGFP and endogenous ASK1. Identical results were obtained when anti-Cx43 antibody was used rather than the anti-EGFP targeting the epitope-tag (data not shown). While the Cx43-ASK1 interaction was weak, this protein-protein interaction was comparable to the Cx43-c-Src interaction previously demonstrated [16]. In contrast, Cx43-EGFP did not co-immunoprecipitate with JNK or the upstream MKK 4 or 7. Thus Cx43 protein interacted with the ASK1 protein hinting at a possible mechanism responsible for the relative resistance to apoptosis seen in the C6-Cx cells. When both Ask1 and Cx43-EGFP were overexpressed in transfected HEK293 cells, the Cx43-ASK1 protein-protein interaction was more robust while a negative-control experiment with transfection of ASK1 and EGFP confirmed that the epitope-tag was not responsible for the co-immunoprecipitation (Figure 3B). Further control experiments supplementing the lysis buffer with 10 mM N-methyl-maleamide to maintain a strong reducing environment to minimize cross-linking of oxidized cysteine residues possibly resulting in spurious co-immunoprecipitation of the two proteins had no effect (data not shown).
In an effort to better understand the consequence of Cx43-ASK1 interaction, we studied the effect of Cx43 co-expression on the status of ASK1 protein phosphorylation at Thr845 as a reporter for kinase activation in response to H2O2 stimulation [17]. HEK293 cells transfected with either human ASK1 and Cx43 or empty vector were treated with 1 mM H2O2 for 5 minutes and assayed for pThr845 at the indicated times. Western Blot analysis revealed a Cx43-dependent decrease in pThr845 indicative of inhibition of ASK1 activation in the presence of Cx43 (Figure 4).
Figure 4. Co-expression of Cx43 inhibits H2O2-induced phosphorylation of ASK1 Thr845.

A. HEK293 cells were co-transfected with ASK1 and Cx43 (open bar) plasmids or empty vector (solid bar) and 48 h after transfection, cells were serum starved and exposed to 1 mM H2O2 for 5 min and left in serum-free medium for the indicated times until harvested. Lysates were analyzed by a Western blot probed with the pThr845-specific ASK1 or pan-ASK1 antibodies. The pan-ASK immunoreactivity confirmed equal loading of the lanes and transfection efficiency. B. A histogram of densitometric analyses (mean ± SD) for pThr845 from three independent experiments normalized to the control no H2O2-treatment condition. * denotes statistical significance at p<0.05 for a comparison between the solid and open bars at the indicated times.
Discussion
Our work confirmed the earlier observation that Cx43 expression protected the C6 cells from oxidative challenge with H2O2. We propose that by selectively interfering with a signaling pathway which leads to caspase-3 activation, Cx43 protected against H2O2-induced apoptosis. Furthermore, we demonstrated that by reducing the expression of Cx43 with siRNA in cultured astrocytes, we can sensitize these normally resistant cells to H2O2-mediated apoptosis, indicating that the anti-apoptotic effect of Cx43 expression was present in normal astrocytes.
Cx43-mediated protection was upstream of caspase activation, as there was little activation of caspase-3 after stimulation with H2O2 in C6-Cx cells. There was no difference between the cell types in STS-induced caspase-3-like activity, nor in overall expression of the caspase 3 protein, indicating that the failure of H2O2 to induce caspase-3 activity in C6-Cx was selective. In contrast, caspase-8 was not activated by H2O2 in neither C6-null nor C6-Cx lines, indicating that H2O2-induced apoptosis in this glioma cell line is driven mainly by the intrinsic apoptotic pathway.
It is now known that ASK1, an upstream activator of JNK, plays a central role in H2O2-induced apoptosis (reviewed in Matsukawa et. al., [14]). ASK1 activity is regulated by many kinases, phosphatases, and binding proteins and it is likely that more regulators exist yet to be discovered. Cx43 interacts with various protein kinases, including c-Src [16], PKC [18, 19], ERK1/2 [12] and ERK5 [13]; it is not known whether Cx43 interacts with any of the MAPKs comprising the ASK1 cascade. We demonstrated that Cx43 does co-immunoprecipitate selectively with ASK1 but not other downstream kinases. A direct inhibition of the H2O2-induced apoptotic cascade by a protein-protein interaction between Cx43 and the components of the ASK1 cascade similar to the action of other apoptosis-modulating proteins such as the adaptor protein Daxx [20], thioredoxin [21], TNF-associated factor 2 [21], ASK1-interacting protein 1 [23] and glutaredoxin [24], is a plausible mechanism. Demonstration of a reduction in the ASK1 pThr845 level by co-expression of Cx43 supports an inhibitory role of this newly discovered protein: protein interaction possibly mediating the anti-apoptotic effect of Cx43. However, a site-directed- and/ or deletion mutants of Cx43 devoid of interacting with ASK1 must be first identified and studied to definitively prove that this protein: protein interaction is responsible for the protection against H2O2-induced apoptosis in C6 cells.
Most studies to date have examined Cx43 as a kinase substrate focusing on the effect of phosphorylation of Cx43 on gap junction communication. Our present study suggested that the Cx43-kinase interaction may have an effect on cellular apoptosis qualifying Cx43 as a signaling molecule in its own right modulating complex signaling pathways most likely to play a critical role in the biology of cells expressing high levels of Cx43 such as astrocytes. While further experiments are needed to fully elucidate the role of Cx43 as an intracellular signaling molecule, the differential sensitivity of Cx43-expressing normal astrocytes and C6 cells largely devoid of Cx43 to some apoptosis-inducers could be exploited to formulate a chemotherapeutic strategy targeting gliomas while sparing the normal brain.
Acknowledgments
We thank Drs. Jane Lin (New York Medical College) and Maiken Nedergaard (University of Rochester) for providing the C6-null and -Cx43 cell lines.
Grant Support: This work was partly supported by NIH RO1 GM071485 (JY).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Alexander DB, Goldberg GS. Transfer of biologically important molecules between cells through gap junction channels. Curr Med Chem. 2003;10:2045–2058. doi: 10.2174/0929867033456927. [DOI] [PubMed] [Google Scholar]
- [2].Saez JC, Berthoud VM, Branes MC, Martinez AD, Beyer EC. Plasma membrane channels formed by connexins: Their regulation and functions. Physiol Rev. 2003;83:1359–1400. doi: 10.1152/physrev.00007.2003. [DOI] [PubMed] [Google Scholar]
- [3].Andrade-Rozental AF, Rozental R, Hopperstad MG, Wu JK, Vrionis FD, Spray DC. Gap junctions: the “kiss of death” and the “kiss of life”. Brain Res Rev. 2000;32:308–315. doi: 10.1016/s0165-0173(99)00099-5. [DOI] [PubMed] [Google Scholar]
- [4].Nakase T, Fushiki S, Naus CC. Astrocytic gap junctions composed of connexin 43 reduce apoptotic neuronal damage in cerebral ischemia. Stroke. 2003;34:1987–1993. doi: 10.1161/01.STR.0000079814.72027.34. [DOI] [PubMed] [Google Scholar]
- [5].Kalvelyte A, Imbrasaite A, Bukauskiene A, Verselis VK, Bukauskas FF. Connexins and apoptotic transformation. Biochem Pharmacol. 2003;66:1661–1672. doi: 10.1016/s0006-2952(03)00540-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Plotkin LI, Manolagas SC, Bellido T. Transduction of cell survival signals by connexin-43 hemichannels. J Biol Chem. 2002;277:8648–8657. doi: 10.1074/jbc.M108625200. [DOI] [PubMed] [Google Scholar]
- [7].Furlan F, Lecanda F, Screen J, Civitelli R. Proliferation, differentiation and apoptosis in connexin43-null osteoblasts. Cell Comm Adhesion. 2001;8:367–371. doi: 10.3109/15419060109080755. [DOI] [PubMed] [Google Scholar]
- [8].Yin X, Gu S, Jiang JX. Regulation of lens connexin 45.6 by apoptotic protease, caspase-3. Cell Comm Adhesion. 2001;8:373–376. doi: 10.3109/15419060109080756. [DOI] [PubMed] [Google Scholar]
- [9].Iacobas DA, Urban-Maldonado M, Iacobas S, Scemes E, Spray DC. Array analysis of gene expression in connexin-43 null astrocytes. Physiol Genomics. 2003;15:177–190. doi: 10.1152/physiolgenomics.00062.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lin JH, Yang J, Liu S, Takano T, Wang X, Gao Q, Willecke K, Nedergaard M. Connexin mediates gap junction-independent resistance to cellular injury. J Neurosci. 2003;23:430–441. doi: 10.1523/JNEUROSCI.23-02-00430.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gaietta G, Deerinck TJ, Adams SR, Bouwer J, Tour O, Laird DW, Sosinsky GE, Tsien RY, Ellisman MH. Multicolar and electron microscopic imaging of connexin trafficking. Science. 2002;296:503–507. doi: 10.1126/science.1068793. [DOI] [PubMed] [Google Scholar]
- [12].Li W, Hertzberg EL, Spray DC. Regulation of connexin43-protein binding in astrocytes in response to chemical ischemia/ hypoxia. J Biol Chem. 2005;280:7941–7948. doi: 10.1074/jbc.M410548200. [DOI] [PubMed] [Google Scholar]
- [13].Cameron SJ, Malik S, Akaike M, Lerner-Marmarosh N, Yan C, Lee JD, Abe JI, Yang J. Regulation of epidermal growth factor-induced connexin 43 gap junction communication by big mitogen-activated protein kinase 1/ ERK5 but not ERK1/2 kinase activation. J Biol Chem. 2003;278:18682–18688. doi: 10.1074/jbc.M213283200. [DOI] [PubMed] [Google Scholar]
- [14].Matsukawa J, Matsuzawa A, Takeda K, Ichijo H. The ASK1-MAP kinase cascades in mammalian stress response. J Biochem. 2004;136:261–265. doi: 10.1093/jb/mvh134. [DOI] [PubMed] [Google Scholar]
- [15].Bukauskas FF, Jordan K, Bukauskiene A, Bennett MVL, Lampe PD, Laird DW, Verselis VK. Clustering of connexin 43-enhanced green fluorescent protein gap junction channels and functional coupling in living cells. Proc Natl Acad Sci USA. 2000;97:2556–2561. doi: 10.1073/pnas.050588497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kanemitsu MY, Loo LWM, Simon S, Lau AF, Eckhart W. Tyrosine Phosphorylation of Connexin 43 by v-Src Is Mediated by SH2 and SH3 Domain Interactions. J Biol Chem. 1997;272:22824–22831. doi: 10.1074/jbc.272.36.22824. [DOI] [PubMed] [Google Scholar]
- [17].Morita K, Saitoh M, Tobiume K, Matsuura H, Enomoto S, Nishitoh H, Ichijo H. Negative feedback regulation of ASK1 by protein phosphatase 5 (PP5) in response to oxidative stress. EMBO J. 2001;20:6028–6036. doi: 10.1093/emboj/20.21.6028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Doble BW, Ping P, Kardami E. The ε subtype of protein kinase C is required for cardiomyocyte connexin-43 phosphorylation. Circ Res. 2000;86:293–301. doi: 10.1161/01.res.86.3.293. [DOI] [PubMed] [Google Scholar]
- [19].Bowling N, Huang XD, Sandusky GE, Fouts RL, Mintze K, Esterman M, Allen PD, Maddi R, McCall E, Vlahos CJ. Protein kinase C-α and -ε modulate connexin-43 Phosphorylation in Human Heart. J Mol Cell Cardiol. 2001;33:789–798. doi: 10.1006/jmcc.2000.1349. [DOI] [PubMed] [Google Scholar]
- [20].Chang HY, Nishitoh H, Yang X, Ichijo H, Baltimore D. Activation of apoptosis signal-regulating kinase 1 (ASK1) by the adaptor protein Daxx. Science. 1998;281:1860–1863. doi: 10.1126/science.281.5384.1860. [DOI] [PubMed] [Google Scholar]
- [21].Liu Y, Min W. Thioredoxin promotes ASK1 ubiquitination and degradation to inhibit ASK1-mediated apoptosis in a redox activity-independent manner. Circ Res. 2002;90:1259–1266. doi: 10.1161/01.res.0000022160.64355.62. [DOI] [PubMed] [Google Scholar]
- [22].Liu H, Nishitoh H, Ichijo H, Kyriakis JM. Activation of apoptosis signal-regulating kinase 1 (ASK1) by tumor necrosis factor receptor-associated factor 2 requires prior dissociation of the ASK1 inhibitor thioredoxin. Mol Cell Biol. 2000;20:2198–2208. doi: 10.1128/mcb.20.6.2198-2208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhang R, He X, Liu W, Lu M, Hsieh JT, Min W. AIP1 mediates TNF-alpha-induced ASK1 activation by facilitating dissociation of ASK1 from its inhibitor 14-3-3. J Clin Invest. 2003;111:1933–1943. doi: 10.1172/JCI17790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Song JJ, Rhee JG, Suntharalingam M, Walsh SA, Spitz DR, Lee Y. Role of glutaredoxin in metabolic oxidative stress: glutaredoxin as a sensor of oxidative stress mediated by H2O2. J Biol Chem. 2002;277:46566–46575. doi: 10.1074/jbc.M206826200. [DOI] [PubMed] [Google Scholar]