

Abstract
Deficiency of Coenzyme Q10 (CoQ10) in muscle has been associated with a spectrum of diseases including infantile-onset multisystemic diseases, encephalomyopathies with recurrent myobinuria, cerebellar ataxia, and pure myopathy. CoQ10 deficiency predominantly affects children, but patients have presented with adult-onset cerebellar ataxia or myopathy. Mutations in the CoQ10 biosynthetic genes, COQ2 and PDSS2, have been identified in children with the infantile form of CoQ10 deficiency; however, the molecular genetic bases of adult-onset CoQ10 deficiency remains undefined.
A lipid-soluble component of virtually all cell membranes, coenzyme Q10 (CoQ10) or ubiquinone is an isoprenylated benzoquinone. CoQ10 transports electrons from complexes I and II to complex III in the mitochondrial respiratory chain and is essential for the stability of complex III (Santos-Ocana et al, 2002). It is also an antioxidant (Villalba et al, 2000) and is involved in multiple aspects of cellular metabolism (Turunen et al, 2004).
Primary CoQ10 deficiency causes clinically heterogeneous diseases: 1) encephalomyopathy characterized by the triad of recurrent myoglobinuria, brain involvement and ragged-red fibers (Ogasahara et al, 1989; Sobreira et al, 1997; Boitier et al, 1998; DiGiovanni et al, 2001; Aure et al, 2004); 2) severe infantile multisystemic disease (Rötig et al, 2000; Rahman et al, 2001; Salviati et al, 2005); 3) cerebellar ataxia (Musumeci et al, 2001; Lamperti et al, 2003; Gironi et al, 2004; Artuch et al, 2006); 4) Leigh syndrome with growth retardation, ataxia and deafness (Van Mardergem et al, 2002); and 5) isolated myopathy (Lalani et al, 2005; Horvath et al, 2006). These disorders are transmitted as autosomal recessive traits and in most cases respond to CoQ10 supplementation. In most of the reported patients, the exact site and nature of the defects in the biosynthesis of CoQ10 have not yet been identified. Because ubiquinone biosynthesis is complex and not fully defined, identification of the molecular genetic defect is not straightforward (Figure 1).
Figure.
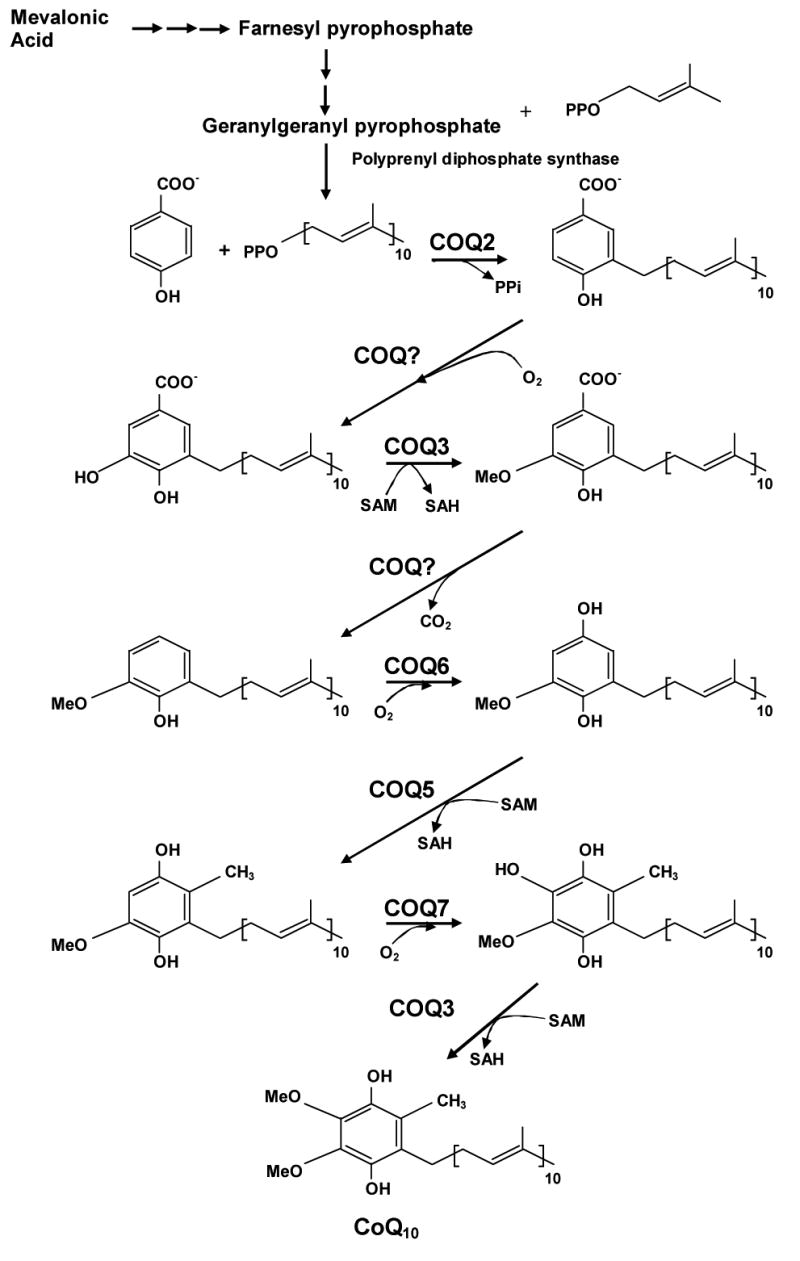
CoQ10 biosynthetic pathway with eight known biosynthetic enzymes denoted as polyprenyl diphosphosphate synthase (COQ1) and COQ2-8. CoQ10 is composed of a benzoquinone and a decaprenyl side chain. While the quinone ring is derived from amino acids tyrosine or phenylalanine, the isoprenoid side chain is produced by addition of isopentenyl pyrophosphate molecules to geranylgeranyl pyrophosphate (derived from mevalonate pathway) by decaprenyl diphosphate synthase. After para-hydroxybenzoate and decaprenyl pyrophosphate are produced, at least seven enzymes (encoded by COQ2-8) catalyze condensation, methylation, decarboxylation, and hydroxylation reactions to synthesize CoQ10
The pathogenic molecular defect has been identified in only 3 patients with the infantile form of primary CoQ10 deficiency: a homozygous mutation in the COQ2 gene, which encodes 4-para-hydroxybenzoate:polyprenyl transferase, in two siblings with nephropathy and encephalopathy (Salviati et al, 2006; Quinzii et al, 2006), and compound heterozygous mutations in the PDSS2 gene, which encodes subunit 2 of polyprenyl diphosphate synthase, the first enzyme of the CoQ10 biosynthetic pathway (Figure 1), in an infant with lactic acidosis, Leigh syndrome, and nephropathy (Lopez Garcia et al, 2006). The renal disease in all three cases manifested as steroid unresponsive nephrotic syndrome.
Among reported patients with presumed primary CoQ10 deficiency, 12 are adults (Table 1) (Musumeci et al, 2001, Van Maldergem et al, 2002; Lamperti et al, 2003 ; Gironi et al, 2004; Horvath et al, 2006). In some adult patients, onset of the disease was during childhood, but Lamperti and colleagues described four with adult-onset ataxia among a cohort of eighteen patients with cerebellar ataxia and low CoQ10 levels in muscle (Lamperti et al, 2003) while Gironi et al. and Horvath et al. reported four patients who presented at ages ranging from 29 to 39 years old (Gironi et al, 2004; Horvath et al, 2006).
Table.
CoQ10 levels in muscle of 12 adults patients
| Age (years) | Patient muscle CoQ10 levels | Control muscle CoQ10 levels | Reference | |
|---|---|---|---|---|
|
| ||||
| Patient 1 | 20 | 7.41 | 25±3.51 | Musumeci et al., 2001 |
| Patient 2 | 25 | 6.61 | ||
| Patient 3 | 24 | 7.11 | ||
| Patient 4 | 24 | 7.11 | ||
|
| ||||
| Patient 5 | 31 | 432 | 7932 | Van Maldergem et al., 2002 |
|
| ||||
| Patient 6 | 24 | 12.81 | 27.6±4.41 | Lamperti et al., 2003 |
| Patient 7 | 35 | 9.21 | ||
| Patient 8 | 27 | 8.71 | ||
| Patient 9 | 30 | 14.81 | ||
|
| ||||
| Patient 10 | 48 | 15.81 | 27.6 ±4.41 | Gironi et al., 2004 |
| Patient 11 | 35 | 13.51 | ||
|
| ||||
| Patient 11 | 33 | 0.63 | 2.7-73 | Horvath et al., 2006 |
| Patient 12 | 29 | 0.83 | ||
μg/gm fresh tissue (control values=mean ±standard deviation),
μg/gm protein (control value=mean), and
nmol/unit citrate synthase (control=range)
The adult patients with CoQ10 deficiency reported by Musumeci et al. and Lamperti et al. had phenotypes similar to children with the ataxic form of CoQ10 deficiency, namely, cerebellar ataxia and atrophy, associated with seizures in 37% of the patients, pyramidal signs, mental retardation, weakness and motor development delay. Notably, the 3 adult patients reported by Lamperti and colleagues did not respond to CoQ10 supplementation, whereas the 3 young adults described by Musumeci and colleagues showed dramatic improvements. All three affected siblings were wheelchair-bound, with alternating esotropia, severe limb ataxia with the slightest purposeful movement, peripheral neuropathies, and scoliosis. One had generalized seizures and another had dystonia. After the proband began CoQ10 supplementations at age 20 years, his strength and ataxia improved and he became able to walk a few steps. His siblings showed similar improvements. In addition, seizures in the affected sister disappeared on CoQ10 therapy and her anti-convulsant medication was discontinued (Musumeci et al, 2001). In these 3 patients, we demonstrated that CoQ10 deficiency was secondary to a stop codon mutation in the APTX gene, which is known to cause ataxia-oculomotor-aprataxia 1 (AOA1) (Quinzii et al, 2005, Date et al, 2001; Moreira et al, 2001). Results from measuring CoQ10 concentration in skeletal muscle from 12 additional patients from 6 different families with AOA1 confirmed this data (data not published). Intriguingly, both CoQ10 and cholesterol share a common biosynthetic pathway, therefore, in AOA1, altered levels of these molecules could be due to aberrant biosynthesis. There is no obvious link between aprataxin and regulation of CoQ10 synthesis or catabolism. Nevertheless, we did not detect mutations in APTX genes in other 13 patients with cerebellar ataxia and CoQ10 deficiency (Quinzii, DiMauro, Hirano, unpublished observation).
Van Maldergam and colleagues reported a 31-year-old woman and her older sister with the typical neuroradiological features of Leigh syndrome encephalopathy, growth retardation, infantilism, ataxia, deafness, and lactic acidosis, but unusually prolonged survival into adulthood. Both clinical and biochemical abnormalities improved remarkably with CoQ10 supplementation (Van Maldergam et al, 2002).
Gironi and colleagues described two brothers with hypergonadotropic hypogonadism and progressive cerebellar ataxia, which started in the fourth decade of life. The late onset of ataxia and associated low levels of testosterone distinguish these patients from those previously reported by Musumeci et al and Lamperti et al (Gironi et al, 2004). As the synthesis of steroid hormones starts with cholesterol, which has the same biosynthetic pathway as CoQ10, it is conceivable that CoQ10 and testosterone deficiencies may coexist; moreover, it is possible that hypergonadotrophic hypogonadism has been under-recognized as endocrine studies were not reported in described patients with the ataxic form of CoQ10 deficiency. Both patients responded to CoQ10 supplementation; they showed improved postural stability, gait and speech articulation, and testosterone returned to the normal range. The clinical improvements may have been related to increased muscle strength rather than amelioration of ataxia (Gironi et al, 2004). The lack of improvement of cerebellar functions with CoQ10 supplementation may be due to initiation of therapy after irreversible structural changes have occurred in the brain. Alternatively, insufficient tissue distribution of CoQ10, in particular its limited ability to cross the blood-brain barrier may explain the absence of therapeutic benefit in the CNS in these as well as in other patients subsequently reported (Aure et al, 2004, Lopez Garcia et al, 2006). Initial studies in rodents suggested that oral CoQ10 supplementation increased levels in plasma, spleen, and liver, but not CNS (Reahal et al, 1992, Zhang et al, 1996); however, subsequent publications demonstrated that long-term CoQ10 administration increased brain mitochondrial concentrations, particularly in aged rodents (Matthews et al, 1998, Kwong et al, 2002).
The most dramatic improvement after CoQ10 supplementation in adults has been associated with the pure myopathic form of CoQ10 deficiency (Lalani et al, 2005; Horvath et al, 2006). The clinical presentation of this variant appears to be homogeneous, with subacute (3 to 6 months) onset of exercise intolerance and proximal weakness affecting predominantly the hip and shoulder girdle muscles. Serum CK and lactate levels were markedly increased. Histologic examination of skeletal muscle revealed a lipid storage myopathy with subtle signs of mitochondrial dysfunction. Biochemical measurement of the respiratory chain enzymes showed reduced activities of complexes II and III (<50% of control mean) secondary to CoQ10 deficiency, as observed in all the variants of CoQ10 deficiency, and increased activity of citrate synthase, in keeping with mitochondrial proliferation. In the myopathic form, CoQ10 levels are low only in muscle, whereas in the infantile multi-systemic and in the cerebellar ataxic forms as well as in the patients described by Van Maldergam, CoQ10 is also reduced in fibroblasts.
CoQ10 deficiency can be also a secondary consequence of drugs, such as statins (3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors). Statins have been used for the treatment of hypercholesterolemia and coronary artery disease and for the prevention of stroke. Their mechanism of action is the inhibition of cholesterol synthesis at the level of mevalonic acid. The biosynthetic inhibition is not selective, because statins impair the synthesis of other compounds that share mevalonate as precursor, such as dolichols and CoQ10. For this reason, statin-related myopathy, manifesting as myalgia, muscle necrosis, and myoglobinura, has been hypothesized to be due to a partial deficiency of CoQ10 (Folkers et al., 1985; Rundek et al. 2004). Indirect support for this hypothesis comes from the first reported cases of CoQ10 deficiency, which presented as exercise intolerance, recurrent myoglobinuria, and encephalopathy (mental retardation and seizures) (Ogashara et al, 1989; Sobreira et al, 1997; Boitier et al, 1998; Di Giovanni et al, 2001; Aure et al, 2004). Several groups have studied the effects of statins on the blood concentration of CoQ10 in patients with hypercholesterolemia and healthy subjects and there are several reports showing that various statins partially decrease CoQ10 levels in blood of patients with hypercholesterolemia and controls, although the number of subjects studied and the severity of CoQ10 deficiency varied markedly (Folkers et al, 1985; Rundek et al, 2004). Recently, Lamperti et al. address the question of whether levels of CoQ10 were also decreased in muscles of patients with statin-related myopathy (Lamperti et al, 2006). The authors measured CoQ10 concentration and respiratory chain enzyme activities in biopsied muscle from 18 patients with statin-related myopathy. Moreover, they looked for evidence of mitochondrial myopathy or morphologic evidence of apoptosis using the TUNEL assay. Their studies revealed a mild decrease in muscle CoQ10 concentration without histochemical or biochemical evidence of mitochondrial myopathy or morphologic evidence of apoptosis in most patients (Lamperti et al, 2006).
Finally, it noteworthy that reduced levels of CoQ10 in blood and mitochondria have been reported in Parkinson disease (PD) by a number of investigators (Shults CW, 2005). These data, together with the implication of oxidative damage and mitochondrial dysfunction in Parkinson disease and other neurodegenerative diseases, such as amyotrophic lateral sclerosis (ALS), Huntington disease (HD), and Friedreich's ataxia (FRDA) have stimulated interest in potential therapeutic effects of CoQ10 as an antioxidant (Beal F, 2004). Initial small clinical trials have suggested beneficial effects in Parkinson disease and FRDA (Shults et al, 2002; Schapira, 2006). However, larger studies are necessary to better define the role of CoQ10 as primary or adjunctive therapy in neurodegenerative diseases.
The molecular bases and pathogenic mechanisms of the various primary and secondary forms of CoQ10 deficiency remain largely unknown. To date, primary CoQ10 deficiency has been genetically and biochemically proven just in few patients with infantile multi-systemic severe diseases, where nephropathy and encephalopathy seems to be the most consistent feature. However, CoQ10 deficiency should be considered in the differential diagnosis of subacute exercise intolerance and weakness and of all genetically undefined adult-onset cases of cerebellar ataxia, as well as in patients with AOA1, because CoQ10 supplementation seems to improve muscle weakness and other associated symptoms in some individuals. Further studies are likely to shed new insights into causes and to improve therapies for the multiple variants of CoQ10 deficiencies in adults and children.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Artuch R, Brea-Calvo G, Briones P, Aracil A, Galvan M, Espinos C, et al. Cerebellar ataxia with coenzyme Q10 deficiency: diagnosis and follow-up after coenzyme Q10 supplementation. J Neurol Sci. 246:153–8. doi: 10.1016/j.jns.2006.01.021. [DOI] [PubMed] [Google Scholar]
- Aure K, Benoist JF, Ogier de Baulny H, Romero NB, Rigal O, Lombes A. Progression despite replacement of a myopathic form of coenzyme Q10 defect. Neurology. 2004;63:727–729. doi: 10.1212/01.wnl.0000134607.76780.b2. [DOI] [PubMed] [Google Scholar]
- Beal MF. Mitochondrial dysfunction and oxidative damage in Alzheimer's and Parkinson's diseases and coenzyme Q10 as a potential treatment. J Bioenerg and Biomemb. 2004;36:381–386. doi: 10.1023/B:JOBB.0000041772.74810.92. [DOI] [PubMed] [Google Scholar]
- Boitier E, Degoul F, Desguerre I, Charpentier C, Francois D, Ponsot G, et al. A case of mitochondrial encephalomyopathy associated with a muscle coenzyme Q10 deficiency. J Neurol Sci. 1998;156:41–46. doi: 10.1016/s0022-510x(98)00006-9. [DOI] [PubMed] [Google Scholar]
- Date H, Onodera O, Tanaka H, Iwabuchi K, Uekawa K, Igarashi S, et al. Early-onset ataxia with ocular motor apraxia and hypoalbuminemia is caused by mutations in a new HIT superfamily gene. Nat Genet. 2001;29:184–188. doi: 10.1038/ng1001-184. [DOI] [PubMed] [Google Scholar]
- Di Giovanni S, Mirabella M, Spinazzola A, Crociani P, Silvestri G, Broccolini A, et al. Coenzyme Q10 reverses pathological phenotype and reduces apoptosis in familial CoQ10 deficiency. Neurology. 2001;57:515–518. doi: 10.1212/wnl.57.3.515. [DOI] [PubMed] [Google Scholar]
- Folkers K, Vadhanavikit S, Mortensen SA. Biochemical rationale and myocardial tissue data on the effective therapy of cardiomyopathy with coenzyme Q10. Proc Natl Acad Sci USA. 1985;82:901–4. doi: 10.1073/pnas.82.3.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gironi M, Lamperti C, Nemni R, Moggio M, Comi G, Guerini FR, et al. Late-onset cerebellar ataxia with hypogonadism and muscle coenzyme Q10 deficiency. Neurology. 2004;62:818–820. doi: 10.1212/01.wnl.0000113719.67643.b7. [DOI] [PubMed] [Google Scholar]
- Kwong LK, Kamzalov S, Rebin I, Bayne AC, Jana CK, Morris P, Forster MJ, Sohal RS. Effects of coenzyme Q(10) administration on its tissue concentrations, mitochondrial oxidant generation, and oxidative stress in the rat. Free Radic Biol Med. 2002;33:627–38. doi: 10.1016/s0891-5849(02)00916-4. [DOI] [PubMed] [Google Scholar]
- Horvath R, Schneiderat P, Schoser BG, Gempel K, Neuen-Jacob E, Ploger H, et al. Coenzyme Q10 deficiency and isolated myopathy. Neurology. 2006;66:253–255. doi: 10.1212/01.wnl.0000194241.35115.7c. [DOI] [PubMed] [Google Scholar]
- Lalani SR, Vladutiu GD, Plunkett K, Lotze TE, Adesina AM, Scaglia F. Isolated mitochondrial myopathy associated with muscle coenzyme Q10 deficiency. Arch Neurol. 2005;62:317–320. doi: 10.1001/archneur.62.2.317. [DOI] [PubMed] [Google Scholar]
- Lamperti C, Naini A, Hirano M, De Vivo DC, Bertini E, Servidei S, et al. Cerebellar ataxia and coenzyme Q10 deficiency. Neurology. 2003;60:1206–1208. doi: 10.1212/01.wnl.0000055089.39373.fc. [DOI] [PubMed] [Google Scholar]
- Lamperti C, Naini AB, Lucchini V, Prelle A, Bresolin N, Moggio M, Sciacco M, Kaufmann P, DiMauro S. Muscle coenzyme Q10 level in statin-related myopathy. Arch Neurol. 2005;62:1709–12. doi: 10.1001/archneur.62.11.1709. [DOI] [PubMed] [Google Scholar]
- Lopez LC, Schuelke M, Quinzii CM, Kanki T, Rodenburg RJ, Naini A, DiMauro S, Hirano M. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am J Hum Genet. 2006;79:1125–29. doi: 10.1086/510023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews RT, Yang L, Browne S, Baik M, Beal MF. Coenzyme Q10 administration increases brain mitochondrial concentrations and exerts neuroprotective effects. Proc Natl Acad Sci USA. 1998;95:8892–97. doi: 10.1073/pnas.95.15.8892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira MC, Barbot C, Tachi N, Kozuka N, Uchida E, Gibson T, et al. The gene mutated in ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein aprataxin. Nat Genet. 2001;29:189–193. doi: 10.1038/ng1001-189. [DOI] [PubMed] [Google Scholar]
- Musumeci O, Naini A, Slonim AE, Skavin N, Hadjigeorgiou GL, Krawiecki N, et al. Familial cerebellar ataxia with muscle coenzyme Q10 deficiency. Neurology. 2001;56:849–855. doi: 10.1212/wnl.56.7.849. [DOI] [PubMed] [Google Scholar]
- Ogasahara S, Engel AG, Frens D, Mack D. Muscle coenzyme Q deficiency in familial mitochondrial encephalomyopathy. Proc Natl Acad Sci USA. 1989;86:2379–2382. doi: 10.1073/pnas.86.7.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinzii C, Naini A, Salviati L, Trevisson E, Navas P, Dimauro S, et al. A Mutation in Para-Hydroxybenzoate-Polyprenyl Transferase (COQ2) Causes Primary Coenzyme Q10 Deficiency. Am J Hum Genet. 2006;78:345–349. doi: 10.1086/500092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinzii CM, Kattah AG, Naini A, Akman HO, Mootha VK, DiMauro S, et al. Coenzyme Q deficiency and cerebellar ataxia associated with an aprataxin mutation. Neurology. 2005;64:539–541. doi: 10.1212/01.WNL.0000150588.75281.58. [DOI] [PubMed] [Google Scholar]
- Rahman S, Hargreaves I, Clayton P, Heales S. Neonatal presentation of coenzyme Q10 deficiency. J Pediatr. 2001;139:456–458. doi: 10.1067/mpd.2001.117575. [DOI] [PubMed] [Google Scholar]
- Reahal S, Wrigglesworth J. Tissue concentrations of coenzyme Q10 in the rat following its oral and intraperitoneal administration. Drug Metab Dispos. 1992;20:423–27. [PubMed] [Google Scholar]
- Rotig A, Appelkvist EL, Geromel V, Chretien D, Kadhom N, Edery P, et al. Quinone-responsive multiple respiratory-chain dysfunction due to widespread coenzyme Q10 deficiency. Lancet. 2000;356:391–395. doi: 10.1016/S0140-6736(00)02531-9. [DOI] [PubMed] [Google Scholar]
- Rundek T, Naini A, Sacco R, Coates K, DiMauro S. Atorvastatin decreases the coenzyme Q10 level in the blood of patients at risk for cardiovascular disease and stroke. Arch Neurol. 2004;61:889–92. doi: 10.1001/archneur.61.6.889. [DOI] [PubMed] [Google Scholar]
- Salviati L, Sacconi S, Murer L, Zacchello G, Franceschini L, Laverda AM, et al. Infantile encephalomyopathy and nephropathy with CoQ10 deficiency: a CoQ10-responsive condition. Neurology. 2005;65:606–608. doi: 10.1212/01.wnl.0000172859.55579.a7. [DOI] [PubMed] [Google Scholar]
- Santos-Ocana C, Do TQ, Padilla S, Navas P, Clarke CF. Uptake of exogenous coenzyme Q and transport to mitochondria is required for bc1 complex stability in yeast coq mutants. J Biol Chem. 2002;277:10973–10981. doi: 10.1074/jbc.M112222200. [DOI] [PubMed] [Google Scholar]
- Schapira AH. Mitochondrial diseases. Lancet. 2006;1(368):70–82. doi: 10.1016/S0140-6736(06)68970-8. [DOI] [PubMed] [Google Scholar]
- Shults CW, Haas R. Clinical trials of coenzyme Q10 in neurological disorders. Biofactors. 2005;25:117–126. doi: 10.1002/biof.5520250113. [DOI] [PubMed] [Google Scholar]
- Sobreira C, Hirano M, Shanske S, Keller RK, Haller RG, Davidson E, et al. Mitochondrial encephalomyopathy with coenzyme Q10 deficiency. Neurology. 1997;48:1238–1243. doi: 10.1212/wnl.48.5.1238. [DOI] [PubMed] [Google Scholar]
- Turunen M, Olsson J, Dallner G, et al. Metabolism and function of coenzyme Q. Biochim Biophys Acta. 2004;1660:171–199. doi: 10.1016/j.bbamem.2003.11.012. [DOI] [PubMed] [Google Scholar]
- Van Maldergem L, Trijbels F, DiMauro S, Sindelar PJ, Musumeci O, Janssen A, et al. Coenzyme Q-responsive Leigh's encephalopathy in two sisters. Ann Neurol. 2002;52:7507–754. doi: 10.1002/ana.10371. [DOI] [PubMed] [Google Scholar]
- Villalba JM, Navas P, et al. Plasma membrane redox system in the control of stress-induced apoptosis. Antioxid Redox Signal. 2000;2000(2):213–30. doi: 10.1089/ars.2000.2.2-213. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Turunen M, Appelkvist EL. Restricted uptake of dietary coenzyme Q is in contrast to the unrestricted uptake of alpha-tocopherol into rat organs and cells. J Nutr. 1996;126:2089–97. doi: 10.1093/jn/126.9.2089. [DOI] [PubMed] [Google Scholar]