

Abstract
Myostatin (GDF8) is a negative regulator of skeletal muscle growth and mice lacking myostatin show a significant increase in muscle mass and bone density compared to normal mice. In order to further define the role of myostatin in regulating bone mass we sought to determine if loss of myostatin function significantly altered the potential for osteogenic differentiation in bone marrow-derived mesenchymal stem cells in vitro and in vivo. We first examined expression of the myostatin receptor, the type IIB activin receptor (AcvrIIB), in bone marrow-derived mesenchymal stem cells (BMSCs) isolated from mouse long bones. This receptor was found to be expressed at high levels in BMSCs, and we were also able to detect AcvrIIB protein in BMSCs in situ using immunofluorescence. BMSCs isolated from myostatin-deficient mice showed increased osteogenic differentiation compared to wild-type mice; however, treatment of BMSCs from myostatin-deficient mice with recombinant myostatin did not attenuate the osteogenic differentiation of these cells. Loading of BMSCs in vitro increased the expression of osteogenic factors such as BMP-2 and IGF-1, but treatment of BMSCs with recombinant myostatin was found to decrease the expression of these factors. We investigated the effects of myostatin loss-of-function on the differentiation of BMSCs in vivo using hindlimb unloading (7 days tail suspension). Unloading caused a greater increase in marrow adipocyte number, and a greater decrease in osteoblast number, in myostatin-deficient mice than in normal mice. These data suggest that the increased osteogenic differentiation of BMSCs from mice lacking myostatin is load-dependent, and that myostatin may alter the mechanosensitivity of BMSCs by suppressing the expression of osteogenic factors during mechanical stimulation. Furthermore, although myostatin deficiency increases muscle mass and bone strength, it does not prevent muscle and bone catabolism with unloading.
Keywords: osteogenesis, AcvrIIB, unloading, stromal cell, muscle mass
Introduction
A number of recent studies have shown that muscle and bone mass are highly correlated in humans [1-4] as well as in laboratory animals [5-9]. These data have, in turn, led to the conclusion that muscle mass is a primary determinant of bone strength, and that a “muscle-bone mechanostat” exists [10]. The forces imposed upon bones and joints by muscles are significantly larger than those gravitational forces associated with body mass [11]. Hence, muscle strength should be a primary determinant of peak bone strain and, therefore, gross and ultrastructural bone morphology [12]. Decreased muscle mass is associated with osteopenia in patients with muscular dystrophy [13], and age-related muscle loss (sarcopenia) is also associated with bone loss [14], further corroborating the hypothesis that muscle size and strength are significant factors in the regulation of bone mass. Likewise, significant muscle loss occurs with exposure to microgravity [15], and the studies reviewed above suggest that a primary mechanism underlying bone loss in space is loss of muscle mass.
We have attempted to better define the basic mechanisms underlying muscle-bone interactions using a mouse knockout model, mice lacking myostatin, also known as GDF-8. Myostatin is a member of the transforming growth factor-β superfamily of secreted growth and differentiation factors that is a negative regulator of skeletal muscle growth. Myostatin null mice have approximately twice the skeletal muscle mass of normal mice [16], and cattle with a spontaneous mutation in the myostatin sequence also show a “double-muscled” phenotype [17]. More recently, a child with a naturally occurring mutation in the myostatin gene was also shown to have increased muscle mass and decreased subcutaneous fat [18]. Myostatin expression in skeletal muscle increases with weightlessness and unloading [19, 20], and exogenous administration of myostatin causes muscle wasting [21]. Thus, expression of myostatin with unloading and/or immobilization is believed to be a major factor in the progression of disuse atrophy [20]. Myostatin is expressed in skeletal muscle but it can also circulate as an endocrine factor [21]. Serum levels of myostatin-immunoreactive protein have been observed to increase with age [23], suggesting that altered myostatin signaling my play in role in the loss of muscle and bone mass that accompanies aging in musculoskeletal tissues. Myostatin is not thought to directly influence bone cells because the myostatin receptor, the type IIB activin receptor (AcvrIIB), is not expressed at significant levels in cortical bone or fracture callus [19]. However, myostatin has been observed to promote adipogenesis in multipotential mesenchymal cells, suggesting that loss of myostatin function may directly suppress adipocyte differentiation [24]. It is well known that mesenchymal stem cells within bone marrow can differentiate to form adipocytes or osteoblasts, and pharmacological agents such as statins that can inhibit marrow adipogenesis also increase osteogenesis [25]. Thus, myostatin may have directs effects on bone formation aside from its well-documented effects on muscle.
We sought to test the hypothesis that loss of myostatin function significantly alters the potential for osteogenic differentiation in bone marrow-derived mesenchymal stem cells in vitro and in vivo, in order to better understand the role of myostatin in regulating bone mass. We also investigated the effects of myostatin on the osteogenic response of BMSCs to mechanical loading in vitro, and we examined the effects of unloading on stromal cell differentiation in myostatin-deficient mice in vivo. Our results indicate that loss of myostatin function increases the osteogenic differentiation of BMSCs in vitro, but that the osteogenic effect is ablated with unloading.
Materials and Methods
Detection of Myostatin Receptor (AcvrIIB) Expression in BMSCs
RNA was extracted from BMSCs of adult wild-type and myostatin-deficient mice at four months of age for detection of AcvrIIB expression using RT-PCR analysis. Bone marrow was harvested from mouse femora and tibiae by flushing with α-MEM, dispersing to single-cell suspension, and plating in flasks. After a 2-hr incubation at 37°C in a 5% CO2 atmosphere, the media containing non-adherent cells was removed. In order to eliminate hematopoietic cells from the adherent cell population, hematopoietic (granulocytes, macrophages, myeloid-derived dendritic cells, natural killer cells, B-1 cells, B lymphocytes, T lymphocytes, and macrophage progenitors) cells were isolated and removed using magnetic beads conjugated to monoclonal antibodies against mouse CD-11b and CD45 (BD Pharmingen). After seeding in culture, total RNAs were extracted from the 8th passage of the adherent BMSCs by homogenization in Trizol, RNA extraction with chloroform, precipitation with isopropanol, and purification using the Quiagen RNeasy kit. RNAs were also collected from BMSCs of wild-type and myostatin-deficient mice after two days culture in osteogenic medium (regular growth medium plus 50 μM ascorbic acid-2-phosphate, 5 mM ß-glycerophosphate, and 100 nM dexamethasone). Total RNA was converted to cDNA by reverse transcription. PCR amplification on samples equivalent to 0.5 μg total RNA were carried out with primer sequences (Table 1) for the type IIB activin receptor (AcvrIIB), osteocalcin, and the adipogenic marker PPAR gamma. Specificity was verified by Southern hybridization analysis, and PCR-amplified DNA samples were run on 2% agarose gels stained with ethidium bromide with a PCR program of 35 cycles of 95°C×30s, 52°C ×30s, and 72°C ×30.
Table 1.
Primer sequences used for analyses of gene expression bone marrow stromal cells
| Gene | Sequence | Product
Size (bp) |
Accession
Number |
|
|---|---|---|---|---|
| AcvrIIB | F | 5’-GAT ACC CAT GGA CAG GTT GG-3’ | 262 | gi∣6680633 |
| R | 5’-TAA TCG TGG GCC TCA TCT TC-3’ | |||
| PPARγ | F | 5’-GGG TGA AAC TCT GGG AGA TT-3’ | 437 | NM_011146 |
| R | 5’-ATG CTT TAT CCC CAC AGA C-3’ | |||
| OC | F | 5’- CCAT GAG GAC CATC TTT CTG CTC A -3’ | 233 | NM_007541 |
| R | 5’- TAG CTC GTC ACA AGC AGG GTT AAG -3’ | |||
| BMP-2 | F | 5’- TGT TTG GCC TGA AGC AGA GA -3’ | 83 | L25602 |
| R | 5’- TGA GTG CCT GCG GTA CAG AT -3’ | |||
| IGF-1 | F | 5’- CAG ACA GGA GCC CAG GAA AG -3’ | 110 | NM184052 |
| R | 5’- AAG TGC CGT ATC CCA GAG GA -3’ | |||
| OSF-2 | F | 5’-CTG CTT CAG GGA GAC ACA CC-3’ | 96 | D13664 |
| R | 5’-AAC GGC CTT CTC TTG ATC GT-3’ | |||
| 18S | F | 5’-AGT GCG GGT CAT AAG CTT GC-3’ | 90 | V00851 |
| R | 5’-GGG CCT CAC TAA ACC ATC CA-3’ | |||
| GADPH | F | 5’-CAT GGC CTC CAA GGA GTA AGA-3’ | 105 | M32599 |
| R | 5’-GAG GGA GAT GCT CAG TGT TGG-3’ | |||
| RUNX-2 | F | 5’-GGA AAG GCA CTG ACT GAC CTA-3’ | 103 | NM009820 |
| R | 5’-ACA AAT TCT AAG CTT GGG AGG A-3’ |
Localization of AcvrIIB protein in BMSCs was performed using fluorescent immunohistochemistry. BMSCs were isolated and cultured as described above and then fixed in cold 2% paraformaldehyde. Cells were stained with rabbit anti-human polyclonal antibody to AcvrIIB (Santa Cruz Biotechnology H-70) diluted 1:100 and then in FITC-conjugated goat anti-rabbit IgG diluted 1:300. Cells were then stained in DAPI (1 mg/ml diluted 1:1000 in PBS) and mounted in aqueous medium.
Osteogenic Culture and Treatment of BMSCs
Bone marrow was flushed from six CD-1 mice and six myostatin-deficient mice on a CD-1 background [8], 8 weeks of age, and adherent BMSCs isolated as described above. To induce osteogenic differentiation the cells were allowed to grow in growth media (DMEM, 10% FBS, with P/S) for 7 days and then treated with osteogenic supplement (OS; regular growth medium plus 50 μM ascorbic acid-2-phosphate, 5 mM ß-glycerophosphate, and 100 nM dexamethasone) for 12 days and stained for alkaline phosphatase (ALP). For ALP staining, cells were rinsed 3 times with double distilled H2O, fixed in 4% paraformaldehyde, and stained using 1-StepTM NBT/BCIP stain solution (PIERCE ,Rockford, IL,USA). Cells were also cultured for 21 days in osteogenic medium and stained for mineralized nodule formation using either 1% alizarin red or Von Kossa staining. Staining intensity in dishes was quantified by scanning the dishes on a flatbed scanner, converting images to grayscale, and then measuring the average pixel intensity in each dish where a value of 0 is black and 250 is white. In a separate experiment, bone marrow was flushed from the long bones of 6 myostatin-deficient mice, also 8 weeks of age, and adherent cells cultured in osteogenic medium (as described above) in the presence or absence of 0.1μM recombinant mouse myostatin (R&D Systems, Minneapolis, MN).
Mechanical Stimulation of Mesenchymal Stem Cells In Vitro
To determine if recombinant myostatin could alter the response of BMSCs to mechanical stimulation, adherent stromal cells were collected from the femora of Mstn-/- mice (n=3) for loading in a custom designed bioreactor (Fig. 1). Cells were removed from femora by incubation for 30 min on a rotator table in 0.2% collagenase (type II, Worthington Biochemical, Lakewood NJ) using Dulbecco’s modified Eagle minimal essential medium (DMEM) supplemented with 4.8 mmol/L CaCl2 and 40 mmol/L HEPES buffer (Invitrogen, Carlsbad, CA). The cells were washed 3x in phosphate buffered saline (PBS, Invitrogen), then allowed to attach overnight. Non-adherent cells were discarded, and the adherent cells were detached with 0.25% trypsin in HBSS (Mediatech, Herndon, VA), counted by haemocytometer and assayed with trypan blue for viability, then plated at 2×104 cells/cm2 in 6-well tissue culture plates (Becton Dickinson, Franklin Lakes, NJ) and grown for 7 days with 3 times/week changes of DMEM medium containing 50 IU/mL penicillin, 50 ug/mL streptomycin (Invitrogen), 0.3 mg/ml l-glutamine (Mediatech, Herndon, VA), and 10% fetal calf serum (Hyclone, Logan, UT). At Day 7 the cells were re-trypsinized and plated at 3×104/cm2 onto 22×22 mm cover slips (Fisher Scientific, Atlanta, GA), and allowed another 3 days to acclimate to the surface.
FIG. 1.
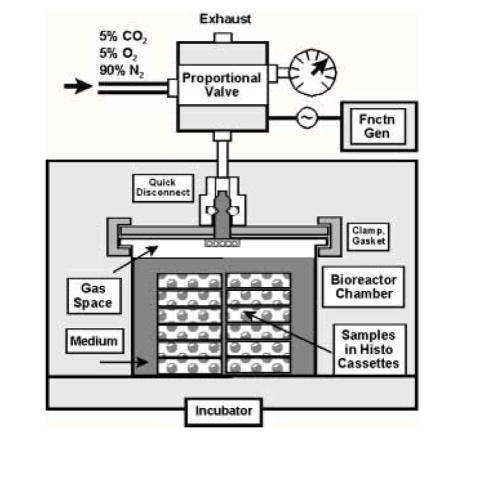
Custom bioreactor for application of hydrostatic compressive stress (pressure). Functionality of this design relies on mixed gases from a pressure cylinder to provide stress, pH balance, and support aerobic metabolism. The cyclic signal is controlled by a function generator input (upper right) to the proportional solenoid valve. Cells were plated onto glass cover slips which were positioned in histology cassettes to allow transmission of the stress to the samples.
At Day 10, recombinant mouse myostatin (R&D Systems, Minneapolis, MN) was added to the duplicate experimental and control cultures at concentrations of 0, 10, and 100 ng/ml. The following day the cover slip cultures were transferred to 30×30 mm histology cassettes and placed in a custom bioreactor for application of cyclic hydrostatic compressive stress (cHSC). This system (Fig. 1) consists of: (1) a vessel, 76 mm in diameter and 60 mm deep, made of 1/8” stainless steel, based on quick-clamp weld tube construction and sealed with high-pressure bolted clamps (fabricated by Douglas Fluid & Integration Technology, Prosperity, SC); (2) a proportional solenoid valve (model QB1-TFEE-500, ProportionAir, McCordsville, IN), into which a cyclic signal is input from (3) a function generator (model FG2, Beckman Industrial, Brea, CA); and (4) a gas cylinder mixed with 5% CO2, 5% O2, and 90% N2. The gas cylinder provides three functions: stress application, pH balance, and support for aerobic metabolism. The filtered gas is transferred through the proportional valve and into the pressure vessel in the incubator through a 1/8” copper tube with quick-disconnect fittings. In this study, our test parameters consisted of 1 atmosphere of pressure (14.7 psi; 101 kPa; 760 mm Hg) applied at 0.5 Hz for 3 h/day. The magnitude of pressure approximates very high levels intramedullary pressure arising from bone bending stress, based on previous estimates using turkey ulnae [26]. The frequency of pressurization minimized transient differential solubilization of the various gases, such that their partial pressures theoretically were not substantially affected. To make a gross assessment of the balance of the solubilized gases, pH was monitored before and after testing and found to remain within a range of 7.3-7.4 pH.
Stress was applied on 3 consecutive days. Between stimulation periods, the cover slip cultures were transferred back to the myostatin-containing medium in the 6-well plates in which they were previously incubated. Fresh myostatin-containing medium was given after stimulation on the second day. Following stimulation on the third day, RNA was isolated using TRIzol® (Invitrogen, Carlsbad, CA). The samples were assayed for absorbance at 260 nm (Helios-Gamma, Thermo Spectronic, Rochester, NY), then reverse transcribed using iScript reagents from Bio-Rad on a programmable thermal cycler (PCR-Sprint, Thermo Electron, Milford, MA). 50 ng complementary deoxyribonucleic acid (cDNA) was amplified in each real-time polymerase chain reaction using a Bio-Rad iCycler, ABgene reagents (distributed by Fisher) and custom designed primers and probes specific to the mouse genome (Table 1). PCR products were sequenced and BLAST-searched to confirm specificity. Melt curves indicated single peaks. For graphical purposes, data are presented as the delta-delta-Ct values, meaning the net cycle difference between target and housekeeping genes (delta-Ct) at a specified optical density above the noise threshold in the comparison of experimental and control samples (delta-delta-Ct). The actual ratio of experimental to control gene expression is obtained by raising the number 2 to the power of the delta-delta-Ct value. The genes included: bone morphogenetic protein BMP-2; insulin-like growth factor IGF-1; the transcription factor Runx-2, and the osteoblast specific factor (OSF)-2 (periostin).
Tail Suspension Experiments
We investigated the effects of hindlimb unloading on muscle mass and BMSC differentiation in 20 male wild-type (CD-1) mice, 15-16 weeks of age, and 20 male Mstn-/- mice, also 15-16 weeks of age, on the CD-1 background [8] using a tail suspension protocol over a 7-day period as approved by IACUC protocol 02-05-141. A 7-day suspension period was selected because tail suspension protocols of 5 days or more have been shown to produce demonstrable changes in bone metabolism in rodents [27-28]. Mouse tails were glued within a closed velcro sleeve and the sleeve was attached to a line leading to a spool mounted above the cage. The mice were suspended at an angle so that their hind toes were just above the cage floor and the line length allowed them to move freely about the cage using their forepaws. The cage floor was covered with bedding as well as a layer of mesh so that the mice could gain traction with their forepaws when moving to and from their food and water sources. A water bottle was placed in each cage close to each mouse along with two petri dishes of water on the cage floor. All mice were given food (Harlan Tek Lad Rodent Diet) ad libitum. Mice were monitored approximately twice a day by laboratory staff.
Tissue Collection
The 40 mice included for the tail-suspension experiments were separated into four treatment groups: 10 wild-type cage control mice (WT-CON), 10 suspended wild-type mice (WT-SUSP), 10 cage control myostatin-deficient mice (KO-CON), and 10 suspended myostatin-deficient mice (KO-SUSP). After 7 days mice were sacrificed by CO2 overdose as approved by Medical College of Georgia IACUC. Mice were weighed and the left iliopsoas, quadriceps femoris, and triceps surae (gastrocnemius and soleus muscles) dissected free and weighed to the nearest 0.01 g. Femora were fixed in 10% buffered formalin and stored in 70% ETOH for histological analysis.
Bone histomorphometry
The left distal femur was cut across the distal metaphysis and the distal end decalcified in 4%EDTA, embedded in paraffin, sectioned at 5 microns in the transverse plane, and sections stained for tartrate-resistant alkaline phosphatase activity (TRAP; Sigma kit, St. Louis) as a marker of osteoclasts. Osteoclasts were identified on trabeculae as TRAP-positive cells resting on scalloped resorptive pits, counted, and expressed as number of osteoclasts per bone perimeter (N.Oc/B.Pm) after Parfitt et al. [29]. Alternate sections were stained with hematoxylin and eosin (H&E) and toluidine blue. Sections stained with H&E were used for measuring adipocyte density (N.At/Ma.Ar), in which adipocytes were counted over a 0.50 mm2 area. Osteoblast surface was measured as the length of trabecular and endocortical bone surfaces covered with osteoblasts, and osteoblast surface (Ob.S) was expressed relative to the total bone surface (BS) as Ob.S/BS following Parfitt et al. [29]. Trabecular BV/TV was calculated from sections stained with H&E.
Statistical Analysis
The collected data were analyzed using a series of one-way ANOVAs seeking to identify statistically significant differences among the groups. When significant differences were detected the group means were compared using Fisher’s least significant difference (LSD) test for post-hoc, pairwaise comparisons. Two-factor ANOVA with treatment and genotype as the two factors was also used to investigate treatment effects and significant treatment*genotype interactions.
Results
Detection of AcvrIIB expression and in vitro differentiation of BMSCs
RT-PCR results show clearly that the myostatin receptor, AcvrIIB, is expressed at significant levels in bone-marrow derived mesenchymal stem cells from both wild-type and myostatin-deficient mice (Fig. 2a). The expression of AcvrIIB in BMSCs is not altered relative to GAPDH when BMSCs are cultured in either adipogenic or osteogenic medium. Immunofluorescent staining for AcvrIIB protein in BMSCs confirms the PCR findings, and demonstrates that the receptor for myostatin is expressed in BMSCs of wild-type and myostatin-deficient mice (Fig. 2b). BMSCs from myostatin-deficient mice cultured in osteogenic medium show significantly increased staining for alkaline phosphatase after 12 days in culture (Fig. 3a,b) and significantly increased alizarin red staining after 21 days in culture (Fig. 3c, d). In contrast, treatment of BMSCs from myostatin-deficient mice with recombinant myostatin does not alter the osteogenic differentiation of BMSCs (Fig. 4a). However, mechanical stimulation of BMSCs from myostatin-deficient mice increases the expression of osteogenic factors such as OSF-2 (periostin), BMP-2, IGF-1, and RUNX-2 (Fig. 4b), and pre-treatment of these cells with recombinant myostatin decreases the expression of these osteogenic factors with mechanical stimulation (Fig. 4b).
FIG. 2.
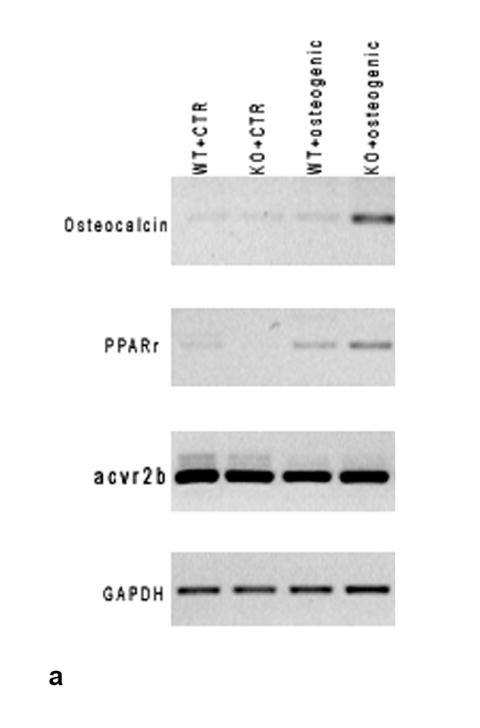
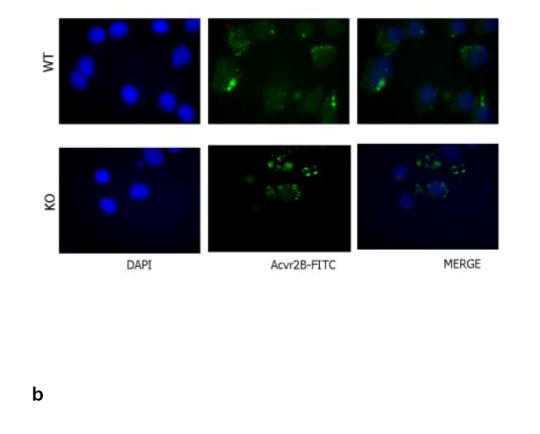
a. RT-PCR results showing expression of the myostatin receptor (AcvrIIB) in BMSCs of wild-type and myostatin-deficient mice. Expression of the receptor is not altered during osteogenic culture of BMSCs. b. Immunofluorescent staining of BMSCs cultured on coverslips showing nuclear staining (DAPI, blue) and FITC-staining (green) of AcvrIIB in both wild-type and myostatin-deficient mice.
FIG. 3.
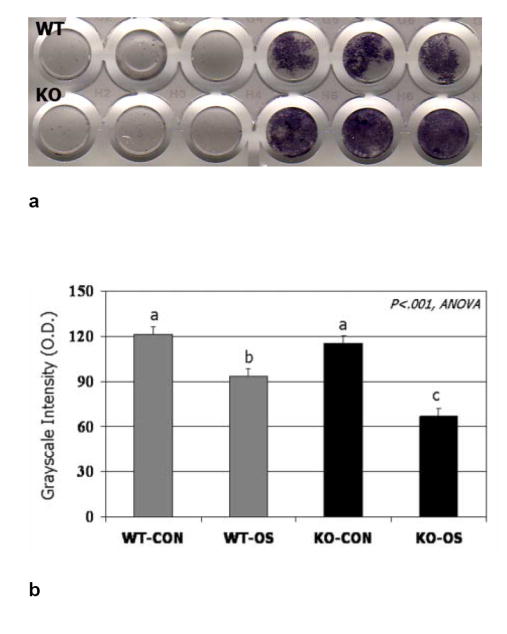
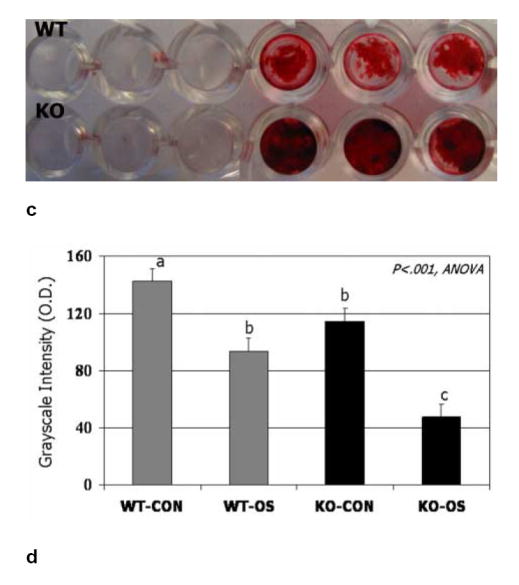
BMSCs from wild-type and myostatin-deficient mice cultured in osteogenic medium stained after 12 days for alkaline phosphatase (ALP) activity (a) and after 21 days using alizarin red (c). Quantification shows significantly darker (low O.D. values) staining for both ALP (b) and alizarin red (d) in BMSCs from knockout mice cultured in osteogenic medium (KO-OS) compared to BMSCs from wild-type mice cultured in osteogenic medium (WT-OS). Means with different superscripts differ significantly (P<.05) in pairwise comparisons.
Fig. 4.
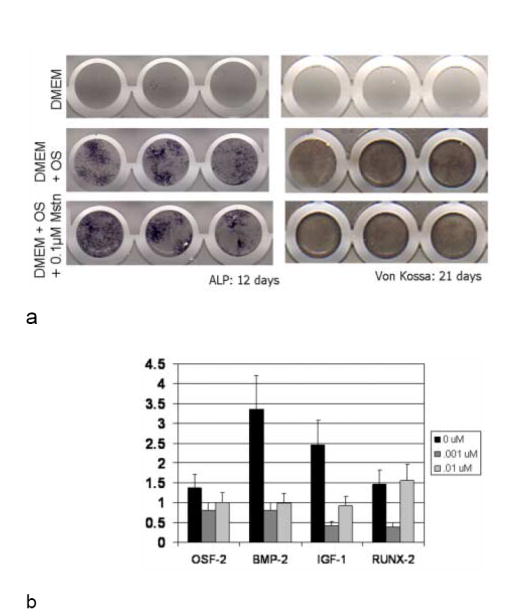
a. BMSCs from myostatin-deficient mice cultured in osteogenic medium and stained for alkaline phosphatase (ALP) and Von Kossa to detect mineralized nodule formation show similar staining in the presence and absence of recombinant myostatin. Different columns of dishes represent three sample replicates. 4b. Gene expression of Mstn-/- adherent stromal cells after 3 days of cyclic hydrostatic compressive stress stimulation, consisting of 1 atmosphere at 0.5 Hz for 3 h/day. 24 h prior to testing, and between tests, cultures were incubated with 0, 10, or 100 ng/ml myostatin. Data (mean+/-SEM) are shown relative to unstressed controls which had been incubated with the same amount of myostatin. Loading increased the expression of osteogenic factors, but the addition of myostatin mitigated this trend of upregulation.
Effects of hindlimb unloading on the differentiation of BMSCs in vivo
Tail suspension induced significant changes in the muscles and bone marrow cell populations of wild-type and myostatin-deficient mice. Two-factor ANOVAs indicate significant differences between treatment groups and genotypes for all four body composition parameters measured. The wild-type mice lost approximately 5.5% of their body mass over the 7 days whereas the myostatin-deficient mice lost about 6.5% of their body mass during the 7 day treatment period. The muscles of the tail suspended myostatin-deficient mice were significantly (P<0.001) larger than those of tail suspended wild-type mice, and the data show that the wild-type mice lost approximately 15% of their quadriceps mass and over 10% of their triceps surae mass during the 7 days (Table 2). In contrast, the myostatin-deficient mice only lost about 7% of their quadriceps mass and 8% of their triceps surae mass, and did not differ significantly from ground control knockout mice in either variable. The normal and myostatin-deficient mice both lost over 20% of their iliopsoas mass with tail suspension (Table 2). Tail suspension decreased osteoblast surface in mice of both genotypes but the decrease was only significant for the myostatin-deficient mice, in which osteoblast surface was almost totally ablated (Fig. 5, 6). Number of marrow adipocytes did not differ significantly between suspended wild-type mice and their loaded congeners, whereas suspended myostatin-deficient mice showed a 200% increase (P=.001) in marrow adipocyte number compared to non-suspended myostatin-deficient mice (Fig. 5, 6). Tail suspension significantly increased the number of osteoclasts on the trabecular bone surface in the distal femur of both normal (P<.05) and myostatin-deficient (P<.01) mice (Fig. 5, 6). Trabecular BV/TV in the femora of mice did not decline significantly with 7 days of tail suspension in mice of either genotype (P=0.45).
Table 2.
Mean (S.D.) values for body and muscle mass measurements of ground control wild-type mice (Ground), tail suspended wild-type mice (Suspended), ground control myostatin-deficient mice (Mstn-/- Ground), and tail suspended myostatin-deficient mice (Mstn-/- Suspended)
| Parameter | Wild-type
(Ground) |
Wild-type
(Suspended) |
Mstn-/- (Ground) |
Mstn-/- (Suspended) |
|---|---|---|---|---|
| Body mass (g)*† | 38.5 (4.7)a | 36.3 (3.5)a | 42.0 (1.4)b | 39.3 (4.3)a |
| Quadriceps mass (g) *† | 0.28 (.03)a | 0.24 (.02)a | 0.39 (.05)b | 0.36 (.04)b |
| Iliopsoas mass (g) *† | 0.15 (.01)a | 0.11 (.01)b | 0.22 (.04)c | 0.17 (.03)a |
| Triceps surae mass (g) *† | 0.18 (.02)a | 0.16 (.01)a | 0.25 (.03)b | 0.23 (.04)b |
Significant difference between ground control and tail-suspended mice by two-factor ANOVA (p<.05).
Significant difference between wild-type and myostatin-deficient mice by two-factor ANOVA (p<.05).
Means identified by different superscript letters within any row are significantly different at P<0.05 by Fisher’s LSD test.
FIG. 5.
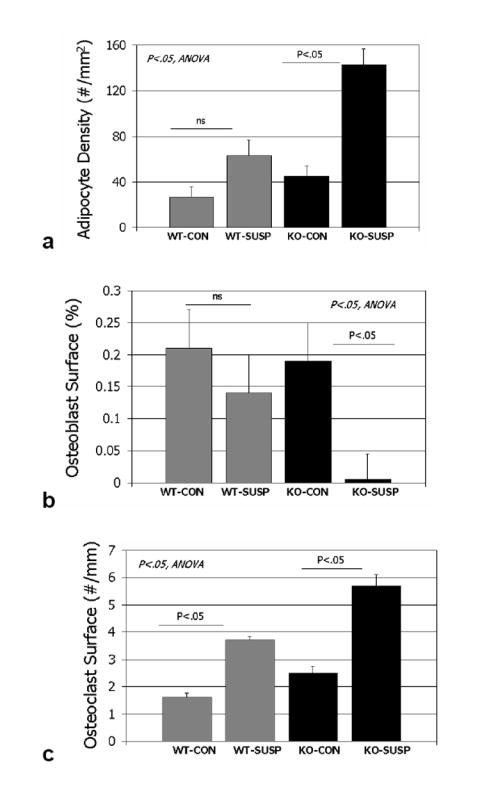
Histomorphometric data for wild-type cage control (wt-con), wild-type tail-suspended (wt-susp), myostatin-deficient cage control (ko-con), and myostatin-deficient tail-suspended (ko-susp). Histomorphometric variables are collected from the distal femur.
FIG. 6.
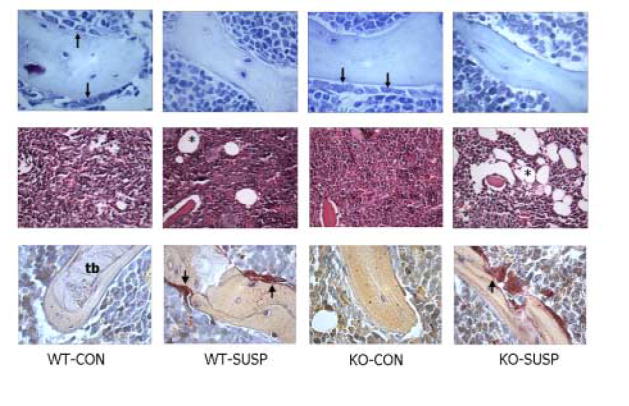
Marrow tissue stained with alcian blue (top row), hematoxylin and eosin (middle row) and for tartrate-resistant acid phosphatase (bottom row) in wild-type cage control (wt-con), wild-type tail-suspended (wt-susp), myostatin-deficient cage control (ko-con), and myostatin-deficient tail-suspended (ko-susp) mice. Note the large number of adipocytes (asterisks) in marrow tissue from the tail-suspended myostatin-deficient animal (middle row), and decrease in osteoblasts (arrows, top row) and increase in osteoclasts (arrows, bottom row) with tail suspension. t=trabecular bone, X200, scale bar = 0.10 mm.
Discussion
Although the myostatin receptor (AcvrIIB) is not detected at significant levels in cortical bone, or in osteoblasts during fracture repair [23], our results indicate that this receptor is expressed in bone marrow-derived mesenchymal stem cells (BMSCs) isolated from long bones of wild-type and myostatin-deficient mice. These findings are consistent with previous work showing that the type II activin receptor is also expressed in proliferating chondrocytes and osteoblasts in the tibiae of neonatal rats [30]. Furthermore, we have demonstrated that loss of myostatin function significantly increases the osteogenic differentiation of BMSCs, although treatment with recombinant myostatin does not significantly attenuate the osteogenic response. It is possible that the recombinant myostatin added to the cultures is inactive because of proteins secreted by the osteoblasts or in the media, but this is unlikely. These results therefore suggest that other factors are involved in mediating the effects of myostatin deficiency on the osteogenic differentiation of BMSCs ex vivo. It is likely that the increased muscle mass of the myostatin-deficient mice alters the mechanical environment of stromal cells in the marrow compartment, possibly through changes in perfusion pressure associated with increased blood flow to the limbs [31], which may in turn increase fluid flow and the expression of osteogenic factors in BMSCs [32]. If stromal cells of myostatin-deficient mice are adapted to a different intramedullary strain environment than those of normal mice then stromal cells of the myostatin-deficient animals may show a greater tendency toward osteogenic differentiation. Our in vitro experiments using compressive loading of BMSCs suggest that mechanical stimulation increases the expression of osteogenic factors in BMSCs, but that pre-treatment with myostatin decreases the expression of these factors with mechanical loading. These data suggest that signaling through AcvrIIB may alter mechanosensitivity of bone marrow stromal cells, which is consistent with other studies [e.g., 33] showing that altered BMP signaling inhibits shear-induced proliferation of osteoblasts.
Bone marrow adipogenesis and bone resorption are known to increase with disuse and hindlimb unloading [34-36], and so we expected that the tail suspended mice in our study would show increased bone marrow adipogenesis and osteoclast number compared to normally loaded mice. Mice of both genotypes showed significant increases in osteoclast number with unloading, but our experiments also show that mice lacking myostatin differed from tail suspended wild-type mice in having a much larger population of bone marrow adipocytes after tail suspension. Myostatin has been observed to promote adipogenesis in multipotential mesenchymal cells, suggesting that loss of myostatin function may directly suppress adipocyte differentiation [24]. Other research, however, suggests that myostatin signals through a transforming growth factor β-like pathway involving the type-IIB activin receptor to block adipogenesis in vitro, and myostatin is also thought to be a potent inhibitor of adipocyte differentiation [37]. Treatment of preadipocytes with myostatin in vitro inhibits adipocyte differentiation [38], and systemic administration of myostatin in vivo induces loss of fat and muscle [21]. As noted above, it is likely that the BMSCs of mice lacking myostatin are adapted to a different intramedullary strain environment than those of normal mice due to the increased muscle mass of the former compared to the latter. Removal of these strains with unloading may therefore produce a greater adipogenic response in the myostatin-deficient mice than normal animals. In addition, the in vitro loading experiments raise the possibility that absence of myostatin alone itself may increase the mechanosensitivity of BMSCs.
Humans accumulate adipocytes in bone marrow with age, and women with osteoporosis are known to have a significantly greater number of bone marrow adipocytes than women with high bone mass [39]. Mesenchymal stem cells within bone marrow can differentiate to form adipocytes or osteoblasts. It has therefore been suggested that conditions favoring adipocyte differentiation will have adverse effects on bone formation because precursor cells are directed towards the adipocyte lineage rather than the osteoblast lineage [40-41]. This hypothesis, referred to as the “clonal switch” hypothesis [40], is supported by the fact that the rate of bone formation is inversely correlated with adipocyte number in bone tissue biopsies of adult men and women [42]. On the other hand, troglitazone-induced bone marrow adipogenesis causes no change in trabecular bone volume [43], and the accumulation of marrow fat following ovariectomy in rats occurs after bone has already been lost [44]. The latter studies therefore suggest that there may not be a clear cause-and-effect relationship between adipogenesis and bone loss, and that fat simply fills space in the medullary cavity once bone is lost. The rapid and significant decrease in osteoblast surface, and corresponding increase in marrow adipogenesis, with hindlimb unloading in myostatin-deficient mice supports the hypothesis that marrow adipogenesis occurs at the expense of bone formation.
McMahon et al. [45] recently found that myostatin-deficient (Mstn-/-) mice exposed to 7 days of tail suspension lost approximately 13% of their body weight and 17% of their quadriceps muscle mass. Our results indicate that, over the same period of tail suspension, myostatin-deficient mice lost only 6.5% of their body mass and 7% of their quadriceps mass. Furthermore, our findings demonstrate that myostatin-deficient mice lost less quadriceps muscle mass with unloading than normal mice, both in absolute terms and relative to body mass. The pattern of body mass reduction observed in our study is consistent with other studies [e.g., 28] of tail suspended rodents showing <10% reduction of body mass after 7 days of hindlimb unloading. The significant loss of iliopsoas mass with unloading in both normal and myostatin-deficient mice is surprising, since this muscle is composed primarily of fast-twitch (type II) muscle fibers in rodents (46) and fast-twitch fibers appear to be more resistant to disuse atrophy than slow-twitch (type I) fibers (47). This suggests considerable heterogeneity in the response of mammalian fast-twitch muscle fibers to unloading and disuse. Furthermore, although previous studies have found that increased myostatin expression with disuse was associated with skeletal muscle atrophy, loss of myostatin function may attenuate loss of muscle mass with hindlimb unloading in muscles having a large number of type I fibers, such as the quadriceps femoris and triceps surae, but loss of myostatin function has no protective effect on the loss of iliopsoas mass with unloading, despite the fact that this muscle is composed primarily of type II (fast-twitch) fibers. Furthermore, although myostatin deficiency increases muscle mass and bone strength, it does not prevent muscle and bone catabolism with unloading.
Acknowledgments
This work was supported by grants from the National Institutes of Health to MWH (AR 049717) and CMI (DK 058680), and by a grant from the National Space Biomedical Research Institute (NSBRI) through a NASA cooperative agreement NCC 9-58 to C.M. Isales. We are grateful to G. Warren, Georgia State University, for assistance with the tail suspension protocol.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Doyle F, Brown J, Lachance C. Relation between bone mass and muscle weight. Lancet. 1970;1:391–393. doi: 10.1016/s0140-6736(70)91520-5. [DOI] [PubMed] [Google Scholar]
- 2.Madesen OR, Schaadt O, Bliddal H, Egsmoose C, Syvest J. Relationship between quadriceps strength and bone mineral density of the proximal tibia and distal forearm in women. J Bone Min Res. 1993;8:1439–1444. doi: 10.1002/jbmr.5650081205. [DOI] [PubMed] [Google Scholar]
- 3.Schönau E, Werhan E, Schniedermaier U, Mokow E, Schiessl H, Schneidhauer K, Michalk D. Influence of muscle strength on bone strength during childhood and adolescence. Horm Res. 1996;45:63–66. doi: 10.1159/000184834. [DOI] [PubMed] [Google Scholar]
- 4.Snow-Harter C, Bouxsein M, Lewis B, Charette S, Weinstein P, Marcus R. Muscle strength as a predictor of bone mineral density in young women. J Bone Miner Res. 1990;5:589–595. doi: 10.1002/jbmr.5650050608. [DOI] [PubMed] [Google Scholar]
- 5.Banu J, Wang L, Kalu D. Effects of increased muscle mass on bone in male mice overexpressing IGF-1 in skeletal muscles. Calcif Tissue Intl. 2003;73:196–201. doi: 10.1007/s00223-002-1072-z. [DOI] [PubMed] [Google Scholar]
- 6.Hamrick MW, Pennington C, Byron C. Bone modeling and disc degeneration in the lumbar spine of mice lacking GDF8 (myostatin) J Orthop Res. 2003;21:1025–1032. doi: 10.1016/S0736-0266(03)00105-0. [DOI] [PubMed] [Google Scholar]
- 7.Hamrick MW. Increased bone mineral density in the femora of GDF8 knockout mice. Anat Rec. 2003;272A:388–391. doi: 10.1002/ar.a.10044. [DOI] [PubMed] [Google Scholar]
- 8.Hamrick M, Samaddar T, Pennington C, McCormick J. Increased muscle mass with myostatin deficiency improves gains in bone strength with exercise. Journal of Bone & Mineral Research. 2006;21:477–483. doi: 10.1359/JBMR.051203. [DOI] [PubMed] [Google Scholar]
- 9.Kaye M, Kusy RP. Genetic lineage, bone mass, and physical activity in mice. Bone. 1995;17:131–135. doi: 10.1016/s8756-3282(00)00164-2. [DOI] [PubMed] [Google Scholar]
- 10.Rauch F, Bailey DA, Baxter-Jones A, Mirwald R, Faulkner R. The muscle-bone unit during the pubertal growth spurt. Bone. 2004;34:771–5. doi: 10.1016/j.bone.2004.01.022. [DOI] [PubMed] [Google Scholar]
- 11.Paul JP. Joint kinetics. In: Sokoloff L, editor. The Joints and Synovial Fluid. II. New York: Academic Press; 1980. pp. 139–176. [Google Scholar]
- 12.Burr DB. Muscle strength, bone mass, and age-related bone loss. J Bone Miner Res. 12;1997:1547–1551. doi: 10.1359/jbmr.1997.12.10.1547. [DOI] [PubMed] [Google Scholar]
- 13.Aparicio L, Jurkovic M, DeLullo J. Decreased bone mineral density in ambulatory patients with Duchenne muscular dystrophy. J Pediatr Orthop. 2002;22:179–181. [PubMed] [Google Scholar]
- 14.Hamrick MW, Ding K-H, Pennington C, Chao Y-J, Wu YD, Howard B, Immel D, McNeil P, Borlongan C, Bollag W, Curl W, Yu JC, Isales CM. Age-related loss of muscle mass and bone strength in mice is associated with a decline in physical activity and serum leptin. Bone. 2006;39:845–853. doi: 10.1016/j.bone.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 15.LeBlanc A, Rowe R, Schneider V, Evans H, Hedrick T. Regional muscle loss after short duration spaceflight. Aviat Space Environ Med. 1995;66:1151–4. [PubMed] [Google Scholar]
- 16.McPherron AC, Lawler AM, Lee S-J. Regulation of skeletal muscle mass in mice by a new TGF-ß superfamily member. Nature. 1997;387:83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- 17.McPherron AC, Lee S-J. Double muscling in cattle due to mutations in the myostatin gene. Proc Natl Acad Sci. 1997;94:12457–12461. doi: 10.1073/pnas.94.23.12457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schuelke M, Wagner K, Stolz L, Hubner C, Riebel T, Komen W, Braun T, Tobin J, Lee S-J. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med. 2004;350:2682–2688. doi: 10.1056/NEJMoa040933. [DOI] [PubMed] [Google Scholar]
- 19.Lalani R, Bhasin S, Byhower F, Tarnuzzer R, Grant M, Shen R, Asa S, Ezzat S, Gonzalez-Cadavid N. Myostatin and insulin-like growth factor-1 and –2 expression in the muscle of rats exposed to the microgravity environment of the NeuroLab space shuttle flight. J Endocrinol. 2000;167:417–428. doi: 10.1677/joe.0.1670417. [DOI] [PubMed] [Google Scholar]
- 20.Wehling M, Cai B, Tidball J. Modulation of myostatin expression during modified muscle use. FASEB J. 2000;14:103–110. doi: 10.1096/fasebj.14.1.103. [DOI] [PubMed] [Google Scholar]
- 21.Zimmers TA, Davies MV, Koniaris LG, Haynes P, Esquela AF, Tomkinson KN, McPherron AC, Wolfman NM, Lee SJ. Induction of cachexia in mice by systemically administered myostatin. Science. 2002;296:1486–1488. doi: 10.1126/science.1069525. [DOI] [PubMed] [Google Scholar]
- 22.Yarasheski K, Bhasin S, Sinha-Hikim I, Pak-Loduca J, Gonzalez-Cadavid N. Serum myostatin-immunoreactive protein is increased in 60-92 year old women and men with muscle wasting. J Nutr Health Aging. 2002;6:343–8. [PubMed] [Google Scholar]
- 23.Shuto T, Sarkar G, Bronk J, Matsui N, Bolander M. Osteoblasts express types I and II activin receptors during early intramembranous and endochondral bone formation. J Bone Miner Res. 1997;12:403–411. doi: 10.1359/jbmr.1997.12.3.403. [DOI] [PubMed] [Google Scholar]
- 24.Artaza J, Bhasin S, Magee T, Reisz-Porszasz S, Shen R, Groome N, Fareez M, Gonzalez- Cadavid N. Myostatin inhibits myogenesis and promotes adipogenesis in C3H 10T(1/2) mesenchymal multi-potent cells. Endocrinol. 2005;146:3547–57. doi: 10.1210/en.2005-0362. [DOI] [PubMed] [Google Scholar]
- 25.Akune T, Ohba S, Kamekure S, Yamaguchi M, Chung UI, Kubota N, Terauchi Y, Harada Y, Azuma Y, Azuma Y, Nakamura K, Kadowaki T, Kawaguchi H. PPAR gamma insufficiency enhances osteogenesis through osteoblast formation from bone marrow progenitors. J Clin Invest. 2004;113:846–855. doi: 10.1172/JCI19900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qin Y-H, Kaplan T, Saldanha A, Rubin C. Fluid pressure gradients, arising from oscillations in intramedullary pressure, is correlated with the formation of bone and inhibition of intracortical porosity. J Biomech. 2003;36:1427–37. doi: 10.1016/s0021-9290(03)00127-1. [DOI] [PubMed] [Google Scholar]
- 27.Barou O, Lafage-Proust MH, Martel C, Thomas T, Tirode F, Laroche N, Barbier A, Alexandre C, Vico L. Bisphosphonate effects in rat unloaded hindlimb bone loss model: three- dimensional and microcomputed tomographic, histomorphometric, and densitometric analyses. JPET. 1999;291:321–328. [PubMed] [Google Scholar]
- 28.Grano M, Mori G, Minielli V, Barou O, Colucci S, Gianelli G, Alexandre C, Zallone A, Vico L. Rat hindlimb unloading by tail suspension reduces osteoblast differentiation, induces IL-6 secretion, and increases bone resorption in ex vivo cultures. Calcif Tissue Intl. 2002;70:176–185. doi: 10.1007/s00223-001-2034-6. [DOI] [PubMed] [Google Scholar]
- 29.Parfitt AM, Drezner M, Glorieux F, Kanis J, Malluche H, Meunier P, Ott S, Recker R. Bone histomorphometry: standardization of nomenclature, symbols, units. J Bone Miner Res. 1987;2:595–610. doi: 10.1002/jbmr.5650020617. [DOI] [PubMed] [Google Scholar]
- 30.Funaba M, Ogawa K, Abe M. Expression and localization of activin receptors during endochondral bone development. Eur J Endocrinol. 2001;144:63–71. doi: 10.1530/eje.0.1440063. [DOI] [PubMed] [Google Scholar]
- 31.Raitakari M, Nuutila P, Ruotsalainen U, Teras M, Eronen E, Laine H, Raitakari O, Iida H, Knuuti M, Yki-Jarvinen H. Relationship between limb and muscle blood flow in man. J Physiol. 1996;496:543–549. doi: 10.1113/jphysiol.1996.sp021705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kreke M, Huckle W, Goldstein A. Fluid flow stimulates expression of osteopontin and bone sialoprotein by bone marrow stromal cells in a temporally dependent manner. Bone. 2005;36:1047–55. doi: 10.1016/j.bone.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 33.Lau K, Kapur S, Kesavan C, Baylink D. Up-regulation of the Wnt, estrogen receptor, insulin-like growth factor 1, and bone morphogenetic proteins in C57BL/6J osteoblasts as opposed to C3H/HeJ osteoblasts in part contributes to the differential anabolic response to fluid shear. J Biol Chem. 2006;281:9576–9588. doi: 10.1074/jbc.M509205200. [DOI] [PubMed] [Google Scholar]
- 34.Minaire P, Meunier P, Edouard C, Berbard J, Courpron J, Bourret J. Quantitative histological data on disuse osteoporosis. Calcif Tissue Res. 1974;13:371–382. doi: 10.1007/BF02547214. [DOI] [PubMed] [Google Scholar]
- 35.Ahdjoudj S, Lasmoles F, Holy X, Zerath E, Marie P. Transforming growth factor beta-2 inhibits adipocyte differentiation induced by skeletal unloading in rat bone marrow stroma. J Bone Miner Res. 2002;17:668–677. doi: 10.1359/jbmr.2002.17.4.668. [DOI] [PubMed] [Google Scholar]
- 36.Wronski T, Morey-Holton E, Jee W. Skeletal alterations in rats during spaceflight. Adv Space Res. 1981;1:135–140. doi: 10.1016/0273-1177(81)90254-4. [DOI] [PubMed] [Google Scholar]
- 37.Rebbapragada A, Benchabane H, Wrana J, Celeste A, Attisano L. Myostatin signals through a transforming growth factor β-like signaling pathway to block adipogenesis. Mol Cell Biol. 2003;23:7230–7242. doi: 10.1128/MCB.23.20.7230-7242.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim H, Liang L, Dean R, Hausman D, Hartzell D, Baile CA. Inhibition of preadipocyte differentiation by myostatin treatment in 3T3-L1 cultures. Biochem Biophys Res Comm. 2001;281:902–906. doi: 10.1006/bbrc.2001.4435. [DOI] [PubMed] [Google Scholar]
- 39.Justesen J, Stenderup K, Ebbesen E, Mosekilde L, Steiniche T, Kassem M. Adipocyte tissue volume in bone marrow is increased with aging and in patients with osteoporosis. Biogerontol. 2001;2:165–171. doi: 10.1023/a:1011513223894. [DOI] [PubMed] [Google Scholar]
- 40.Nuttall M, Gimble J. Is there a therapeutic opportunity to either prevent or treat osteopenic disorders by inhibiting marrow adipogenesis? Bone. 2000;27:177–184. doi: 10.1016/s8756-3282(00)00317-3. [DOI] [PubMed] [Google Scholar]
- 41.Jilka R. Osteoblast progenitor cell fate and age-related bone loss. J Musculoskel Neuron Interact. 2002;2:581–583. [PubMed] [Google Scholar]
- 42.Verma S, Rajaratnam J, Denton J, Hoyland J, Byers R. Adipocytic proportion of bone marrow is inversely related to bone formation in osteoporosis. J Clin Pathol. 2002;55:693–698. doi: 10.1136/jcp.55.9.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tornvig L, Mosekilde L, Justesen J, Falk E, Kassem M. Troglitazone treatment increases bone marrow adipose tissue volume but does not affect trabecular bone volume in mice. Calcif Tissue Intl. 2001;69:46–50. doi: 10.1007/s002230020018. [DOI] [PubMed] [Google Scholar]
- 44.Martin RB, Zissimos S. Relationships between marrow fat and bone turnover in ovariectomized and intact rats. Bone. 1991;12:123–131. doi: 10.1016/8756-3282(91)90011-7. [DOI] [PubMed] [Google Scholar]
- 45.McMahon C, Popovich L, Oldham J, Jeanplong F, Smith H, Kambadur R, Sharma M, Maxwell L, Bass J. Myostatin deficient mice lose more skeletal muscle mass than controls during hindlimb suspension. Am J Physiol Endocrinol Metab. 2003;285:E82–87. doi: 10.1152/ajpendo.00275.2002. [DOI] [PubMed] [Google Scholar]
- 46.Hamalainen N, Pette D. The histochemical profiles of fast fiber types IIB, IID, and IIA in skeletal muscles of mouse, rat, and rabbit. J Histochem Cytochem. 1993;41:733–743. doi: 10.1177/41.5.8468455. [DOI] [PubMed] [Google Scholar]
- 47.Thomason DB, Booth FW. Atrophy of the soleus muscle by hindlimb unweighting. J Appl Physiol. 1990;68:1–12. doi: 10.1152/jappl.1990.68.1.1. [DOI] [PubMed] [Google Scholar]