

Abstract
The high pulmonary vascular resistance (PVR) of atelectatic, hypoxic, fetal lungs limits intrauterine pulmonary blood flow (PBF) to less than 10% of combined right and left ventricular output. At birth, PVR decreases precipitously to accommodate the entire cardiac output. The present review focuses on the role of endothelium-derived nitric oxide (NO), prostacyclin, and vascular smooth muscle potassium channels in mediating the decrease in PVR that occurs at birth, and in maintaining reduced pulmonary vasomotor tone during the neonatal period. The contribution of vasodilator and vasoconstrictor modulator activity to the pathophysiology of neonatal pulmonary hypertension is also addressed.
Keywords: nitric oxide, perinatal, potassium channels, prostacyclin, pulmonary hypertension
Introduction
During late fetal development, PVR is high and PBF is limited to less than 10% of combined ventricular output. This provides adequate nutrition and stimulus for growth to the lung, while optimizing flow to other fetal tissues and the placenta. At birth, mechanical distention of the lungs, increased oxygen tension, and increased shear stress result in a precipitous decrease in PVR and increase in PBF to 100% of cardiac output. Failure of this normal transition leads to persistent right-to-left shunting across fetal cardiovascular channels, resulting in profound hypoxemia and ultimately death. Even when PVR decreases normally at birth (Fig. 1), subsequent pulmonary vasoconstriction in response to hypoxia or other pressor stimuli can lead to a resumption of right-to-left shunting across fetal cardiovascular channels, with potentially fatal consequences. The present review addresses the contributions of NO, prostacyclin, and potassium channel activation to the normal transition from fetal to neonatal pulmonary hemodynamics and to the defense against postnatal pulmonary vasoconstriction.
Figure 1.
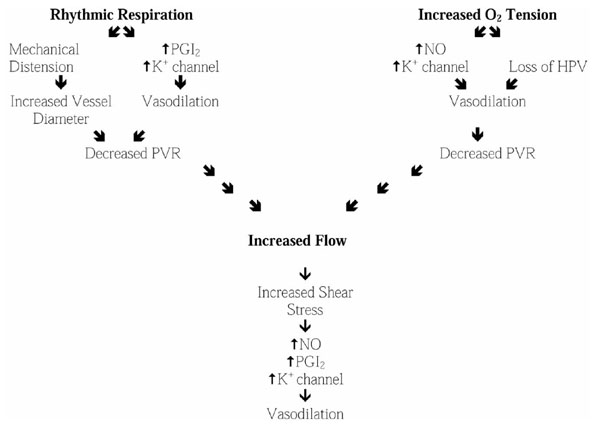
Birth-related stimuli that lead to decreased pulmonary vascular resistance. See text for details. PGI2, prostacyclin.
Pulmonary vascular resistance during fetal development
During early fetal development, PBF is limited by the paucity of pulmonary vessels. The number of fetal pulmonary vessels increases by an order of magnitude between mid-gestation and term. PVR remains high during the last trimester, however, and most of the right heart output is shunted across the ductus arteriosus and foramen ovale to the low-resistance systemic circuit. This is due in part to mechanical compression of pulmonary vessels by the fluid-filled, atelectatic lungs. In addition, fetal pulmonary vessels exhibit active tone.
In sheep, at 90–100 days of gestation (term 140 days) maternal hyperoxia increased fetal partial oxygen tension from approximately 20 to 175 mmHg, but had no effect on fetal PBF [1]. In contrast, after 115–120 days of gestation (ie at >75% term) an increase in fetal partial oxygen tension to 40–50 mmHg was associated with an almost 10-fold increase in PBF [1,2]. Thus, hypoxic pulmonary vasoconstriction (HPV) appears to develop during the third trimester when the number of pulmonary vessels increases.
Unlike the mature pulmonary circulation, the fetal pulmonary vasculature also appears to autoregulate flow through a myogenic response. This may explain why stimuli such as ductal compression, endothelium-dependent vasodilators, and increased oxygen tension cause only a transient increase in fetal PBF [3,4]. Finally, the balance between endogenous vasoconstrictor and vasodilator modulators contribute to the high PVR of the fetus.
Vasoconstrictor modulators in the fetus
Arachidonic acid is metabolized via the cyclo-oxygenase, lipoxygenase, or cytochrome P450-dependent epoxygenase pathways to both vasodilator and vasoconstrictor modulators. Whether the epoxygenase metabolites contribute to fetal vasomotor tone has not been established. The cyclo-oxygenase pathway is active in the fetus [5], however, and gives rise to both vasodilator prostaglandins and the vasoconstrictor thromboxane A2. The observation that thromboxane A2 inhibition caused fetal vasodilatation [6] provided evidence that thromboxane A2 contributes to basal PVR in the fetus. Lipoxygenase metabolites of arachidonic acid, particularly leukotriene D4, may also contribute to elevated fetal PVR [7], although the importance of this modulator in maintaining basal tone has been questioned [8]. More recently, several studies have suggested that the potent endothelium-derived contracting factor endothelin-1 plays a key role in maintaining high fetal pulmonary vasomotor tone. Endothelin-1 causes vasoconstriction by activating endothelin receptor subtype A (ETA) receptors, and ETA receptor blockade enhanced the increase in PBF seen during ductal compression in utero [9]. Furthermore, levels of endothelin-1 mRNA expression, endothelin-1 peptide, and ETA receptor mRNA expression are all highest at 125–130 days of gestation in ovine fetuses, and then decline as term approaches and the need for vasodilatation becomes paramount [10].
Vasodilator modulators in the fetus
The vasoconstrictor effects of hypoxia, myogenic tone, and pressor modulators are counterbalanced by several endogenous vasodilator modulators of vasomotor tone. Of these, NO and prostacyclin play particularly important roles in maintaining adequate PBF during fetal development and in mediating the precipitous decrease in PVR at birth. Endothelial, inducible, and neuronal nitric oxide synthase (NOS) have all been identified in fetal lungs. However, the present review focuses on the role of endothelium-derived NO, which is synthesized from L-arginine by endothelial NOS in the presence of calcium and other cofactors. NO diffuses from endothelial cells into adjacent pulmonary vascular smooth muscle cells, where it causes vasodilatation through several mechanisms. These include the classic NO-induced activation of guanylate cyclase, leading to increased levels of cGMP. The cGMP in turn stimulates production of a cGMP-dependent kinase that can cause vasodilatation through direct action on myosin phosphorylation. In addition, there is evidence that NO can directly or indirectly activate vascular smooth muscle potassium channels, leading to hyperpolarization and a decrease in cytosolic calcium in both the fetal [11] and mature pulmonary vasculature [12].
Immunohistochemical studies [13] have identified endothelial NOS as early as under one-third of term in lamb fetal lungs. Both expression of the endothelial NOS gene [14] and the NO-induced increase in cGMP concentration [15] appear to increase as term approaches. In addition, the endothelin receptor subtype B (ETB) receptor, which mediates vasodilatation through a NO-dependent mechanism, is most abundant at term and may explain the apparently paradoxic vasodilatation seen in response to endothelin-1 infusion in the late gestation fetus [10,16]. Other endothelium-dependent pulmonary vasodilators that act by increasing endothelial NOS activity cause acute vasodilatation in fetal pulmonary vessels, and in utero administration of NOS inhibitors increases fetal PVR and blocks endothelium-dependent vasodilatation [17,18,19]. Furthermore, authentic NO, NO donors, and cGMP analogs all cause vasodilatation of fetal lungs and isolated fetal vessels [2,18].
Vasodilator responses to physiologic as well as pharmacologic stimuli appear to be mediated by NO in the fetus. For example, endothelial NO synthesis was greater at elevated oxygen tension in fetal pulmonary arteries [15], and the increase in fetal lamb PBF caused by maternal hyperoxia was blocked by NOS inhibition [4]. Shear stress-induced vasodilatation in the fetus also appeared to be dependent on NO [20], although this might have been due to increased inducible as well as endothelial NOS activity.
Like the NOS isoforms, both constitutive and inducible cyclo-oxygenase (cyclo-oxygenase 1 and 2) are present in the ovine fetal lung [5]. Infusion of several cyclo-oxygenase metabolites of arachidonic acid (eg prostacyclin, and prostaglandins E1, E2, D2 and H2) causes vasodilatation of the high-vascular-resistance fetal pulmonary circulation. However, prostacyclin is the most potent vasodilator prostaglandin [8]. Prostacyclin acts on the vascular smooth muscle by activating adenylate cyclase. The increased cAMP subsequently causes smooth muscle relaxation either through a direct effect on myosin phosphorylation or by activating a potassium channel via a cAMP-dependent kinase, leading to vascular smooth muscle hyperpolarization [21]. Prostacyclin synthesis increases during the last trimester [22], and several endothelium-dependent vasodilators, including acetylcholine and bradykinin, act at least in part by enhancing prostacyclin synthesis in the fetus [23]. Prostacyclin does not appear to contribute to the vasodilatory effects of maternal hyperoxia [24], however, and cyclo-oxygenase inhibitors have little effect on basal PVR in the fetus, probably because they block both vasoconstrictor and vasodilator prostanoids.
Over the past two decades, calcium-dependent (KCa), ATP- dependent (KATP), and several voltage-dependent (KV) potassium channels have been identified on both pulmonary endothelial and vascular smooth muscle cells. Shear stress can activate endothelial potassium channels, leading to NO synthesis [25], which then causes vasodilatation as described above. Vascular smooth muscle cell potassium channel activation leads to hyperpolarization of the vascular smooth muscle and to a decrease in cytosolic calcium, which results in vasodilatation. These channels can be activated by NO, prostacyclin, and other endothelium-derived hyperpolarizing factors. Studies of isolated arteries and intact lambs [26] suggest that vascular smooth muscle KATP channels are present in fetal lambs, but inhibition of these channels appears to play little role in regulating basal pulmonary vasomotor tone. KCa channels are also present in vascular smooth muscle cells of the fetal pulmonary circulation, and there is evidence [11] that they mediate the NO-dependent vasodilatation that is seen in response to some endothelium-dependent vasodilators. KV channels (particularly KV2.1) have been implicated as sensors and mediators of HPV in mature lungs. There appears to be little KV2.1 activity in the fetal pulmonary circulation, however. Instead, KCa channels may play an important role in sensing and mediating fetal and neonatal HPV [27].
Changes in pulmonary vascular resistance at birth
At birth, PVR must decrease abruptly to accommodate 100% of cardiac output, thus allowing the lungs to assume their normal extrauterine gas exchange and metabolic functions. Several inter-related stimuli, including expansion of the lungs, increased oxygen tension and increased systemic vascular resistance, contribute to the decrease in PVR. Collectively, these stimuli, as well as the increase in levels of several endogenous vasoactive substances, lead to a marked increase in the ratio of vasodilator to vasoconstrictor modulators.
It has long been known that the initiation of rhythmic breathing causes vasodilatation, even in the absence of an increase in oxygen tension [28]. This is partly due to mechanical distension of the lungs, which increases vessel radius - a key physical determinant of vascular resistance. In addition, mechanical deformation of the lungs may directly enhance vasodilator modulator synthesis. Studies of neonatal animals found that ventilation caused an increase in prostacyclin synthesis [29] and cyclo-oxygenase inhibition prevented that normal decrease in PVR associated with rhythmic lung distension at birth [30,31]. NOS inhibition [32] and KCa channel inhibition [33] also blunt ventilation-induced pulmonary vasodilatation.
Increased oxygen tension at birth also reduces PVR, even in the absence of ventilation [28]. This is partly due to the loss of HPV. The mechanism of HPV remains uncertain, but several factors appear to contribute to the response. Recent studies of mature animal preparations [34,35] support the hypothesis that hypoxia causes ETA-mediated inhibition of a KV channel; this leads to vessel depolarization and calcium influx, resulting in vasoconstriction. The increase in oxygen at birth, together with the perinatal decrease in ETA receptor message, probably contributes to decreased HPV at birth. However, it is noteworthy that KCa rather than KV channels may play the depolarizing/hyperpolarizing role in response to changes in oxygen tension [27,36]. In addition to reducing HPV, the increased oxygen tension appears to enhance NO synthesis at birth [15]. A major role for NO in the transitional circulation is further supported by studies [19,32] that showed that NOS inhibition blunts the oxygen-induced decrease in PVR at birth.
Although the above paragraphs imply that oxygenation and ventilation have specific and direct effects on NO and prostacyclin synthesis, these stimuli, in conjunction with the recruitment and distension of the pulmonary vasculature by increased left atrial pressure, may act together through a flow-induced increase in shear stress. In the postnatal pulmonary circuit, increased shear stress in response to increased flow is a potent stimulus for endothelium-derived vasodilator modulator synthesis. This in turn establishes a positive feedback loop that enhances PBF until the increase in shear stress due to increased flow is offset by the decrease in shear stress due to increased vessel diameter. Distinguishing the role of shear stress, or indeed the effects of increased synthesis of other endogenous vasoactive substances (eg adenosine, bradykinin, etc), from the direct effects of oxygen and ventilatory movements remains an unfinished task.
Changes in pulmonary vascular resistance during neonatal development
Following the initial acute decrease in PVR at birth, there is a more gradual decline in resistance over the following days and weeks. Initially, this decrease in PVR reflects further recruitment and distension of the vascular bed, and spreading of the endothelial and vascular smooth muscle cells [37]. In addition, some studies [38] have identified a progressive decrease in arterial muscularization during the first few days of life. These developmental changes lead to a major decrease in PVR within days of birth [39]. Subsequently, lung growth and the increase in intra-alveolar vessel number lead to a more gradual reduction in PVR until adult levels are achieved.
During the early newborn period, however, an increase in PVR due to hypoxia or other pressor stimuli can lead to a resumption of right-to-left shunting across fetal cardiovascular channels. The resultant profound hypoxemia can lead to significant morbidity or death if pulmonary vasoconstriction is not reversed. Fortunately, despite evidence of increased pulmonary vascular muscularization in young newborn lungs, HPV appears to be more attenuated in younger than in older neonates [40,41,42,43]. Several factors may contribute to the neonatal defenses against pulmonary vasoconstriction. There is some evidence that hypoxia is not sensed as well by the younger newborn pulmonary vasculature [42], possibly because of the relative paucity of KV2.1 channels [27]. Alternatively, the relative immaturity of neonatal pulmonary vascular smooth muscle may impair contractility [44]. Finally, there is considerable evidence that modulators of vasomotor tone attenuate vasoconstriction more in younger than in older newborns.
Prostacyclin synthesis is enhanced by hypoxia in arteries from 1- to 2-week-old newborns, but not in arteries from older newborns [22]. Furthermore, prostacyclin concentrations are higher in the perfusate of hypoxic 1-day-old than in 1-month-old lamb lungs [42]. In addition, prostaglandins E1, E2 and D2 cause vasodilatation in hypoxic newborn lungs, but cause vasoconstriction in older animals [8]. Finally, cyclo-oxygenase inhibition enhances HPV more in lungs from lambs that are younger than 4 days old than in those from lambs older than 2 weeks [43]. Whether NO modulates pulmonary vasomotor tone more in younger than in older newborns is more controversial. In some studies of isolated vessels [18,45] endothelium-dependent vasodilatation was greater in arteries from younger than in those from older animals, whereas in others [46] it decreased with age. On the other hand, studies of isolated lungs suggest that both endothelium-dependent and -independent vasodilatation is greater in younger newborns [47], and NOS inhibition increased vasoconstriction more in lungs from younger than in those from older newborns [48].
Vasodilator modulators and the pathogenesis of neonatal pulmonary hypertension
Not only does acute inhibition of vasodilator modulators increase basal PVR and enhance vascular reactivity in normal newborn lungs, but also there is evidence that an imbalance between vasoconstrictor and vasodilator modulators may contribute to the pathogenesis of various forms of neonatal pulmonary hypertension. The syndrome of persistent pulmonary hypertension of the newborn (PPHN) is characterized by abnormally increased pulmonary vascular muscularization and severe neonatal pulmonary hypertension in the absence of other pulmonary or cardiac disease. Studies conducted during the 1970s and 1980s [49] found that chronic in utero cyclo-oxygenase inhibition could result in the anatomic and physiologic features of PPHN. More recently, a study of newborn lambs [50] showed that in utero infusion of a NOS inhibitor for 10 days mimicked the physiologic, but not the anatomic features of PPHN. In addition, chronic fetal ETB receptor inhibition, which results in unopposed ETA-mediated constriction, led to pulmonary hypertension [51]. Conversely, both acute and chronic intrauterine pulmonary hypertension due to ductal compression led to impaired endothelium-dependent vasodilatation [52,53] and reduced channel expression [54]. Chronic hypoxia KCa during the newborn period also leads to pulmonary hypertension, associated with decreased NOS protein and message, and impaired endothelium-dependent vasodilatation [55,56].
The pathophysiology of PPHN is not only dependent on a deficiency in the vasodilator modulators, but may also result from an excess of vasoconstrictor modulators. In one study of infants with PPHN [57], leukotriene C4 and leukotriene concentrations were higher than in D4 neonates without PPHN. Lung thromboxane A2 concentrations were also higher in an ovine model of PPHN than in control lambs [58]. Finally, serum endothelin-1 concentrations were higher in infants with PPHN [59].
Conclusion
Although modulators of pulmonary vasomotor tone appear to contribute to elevated fetal pulmonary vasomotor tone, the decrease in PVR at birth, and the defenses against pulmonary vasoconstriction during early life, many questions remain. Is there sufficient redundancy among modulator classes that the loss of one can be compensated for by an increase in another? Do the reported differences in modulator activity between arteries and veins mean that all modulators must be synthesized in order to achieve normal development [17,60]? What do apparent interspecies differences in modulator activity imply for the prevention and therapy of neonatal pulmonary hypertension in humans? Can the loss of modulator activity be identified and treated in utero? Future studies must address these and other questions in order to gain a better understanding of the physiology and pathophysiology of pulmonary vasomotor tone in the fetus and young neonate.
Abbreviations
ETA/B = endothelin receptor subtype A/B; HPV = hypoxic pulmonary vasoconstriction; KATP = ATP-dependent potassium channel; KCa = calcium- dependent potassium channel; KV = voltage dependent potassium channel; NO = nitric oxide; NOS = nitric oxide synthase; PBF = pulmonary blood flow; PPHN = persistent pulmonary hypertension of the newborn; PVR = pulmonary vascular resistance.
Acknowledgments
Acknowledgements
It has been impossible to cite in this review all of the important work investigating the control of fetal and neonatal PVR over the past 50 years. We would therefore like to apologize to and thank all of those investigators whose work has contributed to our understanding of the development of the pulmonary circulation, but which is not referenced here.
References
- Morin FC, Egan EA, Ferguson W, Lundgren CE. Development of pulmonary vascular response to oxygen. Am J Physiol. 1988;254:H542–H546. doi: 10.1152/ajpheart.1988.254.3.H542. [DOI] [PubMed] [Google Scholar]
- Kinsella JP, Ivy DD, Abman SH. Ontogeny of NO activity and response to inhaled NO in the developing ovine pulmonary circulation. Am J Physiol. 1994;267:H1955–H1961. doi: 10.1152/ajpheart.1994.267.5.H1955. [DOI] [PubMed] [Google Scholar]
- Storme L, Rairigh RL, Parker TA, Kinsella JP, Abman SH. In vivo evidence for a myogenic response in the fetal pulmonary circulation. Pediatr Res. 1999;45:425–431. doi: 10.1203/00006450-199903000-00022. [DOI] [PubMed] [Google Scholar]
- McQueston JA, Cornfield DN, McMurtry IF, Abman SH. Effects of oxygen and exogenous L-arginine on EDRF activity in fetal pulmonary circulation. Am J Physiol. 1993;264:H865–H871. doi: 10.1152/ajpheart.1993.264.3.H865. [DOI] [PubMed] [Google Scholar]
- Brannon TS, MacRitchie AN, Jaramillo MA, Sherman TS, Yuhanna IS, Margraf LR, Shaul P. Ontogeny of cyclooxygenase-1 and cyclooxygenase-2 gene expression in ovine lung. Am J Physiol. 1998;274:L66–L71. doi: 10.1152/ajplung.1998.274.1.L66. [DOI] [PubMed] [Google Scholar]
- Tod ML, Cassin S. Thromboxane synthase inhibition and perinatal pulmonary response to arachidonic acid. J Appl Physiol. 1985;58:710–716. doi: 10.1152/jappl.1985.58.3.710. [DOI] [PubMed] [Google Scholar]
- Soifer SJ, Loitz RD, Roman C, Heymann MA. Leukotriene end organ antagonists increase pulmonary blood flow in fetal lambs. Am J Physiol. 1985;249:H570–H576. doi: 10.1152/ajpheart.1985.249.3.H570. [DOI] [PubMed] [Google Scholar]
- Cassin S. Role of prostaglandins, thromboxanes, and leukotrienes in the control of the pulmonary circulation in the fetus and newborn. Semin Perinatol. 1987;11:53–63. [PubMed] [Google Scholar]
- Ivy D, Kinsella J, Abman S. Endothelin blockade augments pulmonary vasodilation in the ovine fetus. J Appl Physiol. 1996;81:2481–2487. doi: 10.1152/jappl.1996.81.6.2481. [DOI] [PubMed] [Google Scholar]
- Ivy D, LeCras T, Parker T, Zenge J, Jakkula M, Markham N, Kinsella J, Abman S. Developmental changes in endothelin expression and activity in the ovine fetal lung. Am J Physiol. 2000;278:L785–L793. doi: 10.1152/ajplung.2000.278.4.L785. [DOI] [PubMed] [Google Scholar]
- Saqueton CB, Miller RB, Porter VA, Milla CE, Cornfield DN. NO causes perinatal pulmonary vasodilation through K+-channel activation and intracellular Ca2+ release. Am J Physiol. 1999;276:L925–L932. doi: 10.1152/ajplung.1999.276.6.L925. [DOI] [PubMed] [Google Scholar]
- Yuan XJ, Tod ML, Rubin LJ, Blaustein MP. NO hyperpolarizes pulmonary artery smooth muscle cells and decreases the intracellular Ca2+ concentration by activating voltage-gated K+ channels. Proc Nat Acad Sci USA. 1996;93:10489–10494. doi: 10.1073/pnas.93.19.10489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbower AC, Tuder RM, Franklin WA, Pollock JS, Förstermann U, Abman SH. Maturation-related changes in endothelial nitric oxide synthase immunolocalization in developing ovine lung. Am J Physiol. 1994;267:L585–L591. doi: 10.1152/ajplung.1994.267.5.L585. [DOI] [PubMed] [Google Scholar]
- Kawai N, Bloch DB, Filippov G, Rabkina D, Suen HC, Losty PD, Janssens SP, Zapol WM, de la Monte S, Bloch KD. Constitutive endothelial nitric oxide synthase gene expression is regulated during lung development. Am J Physiol. 1995;268:L589–L595. doi: 10.1152/ajplung.1995.268.4.L589. [DOI] [PubMed] [Google Scholar]
- Shaul PW, Farrar MA, Magness RR. Pulmonary endothelial nitric oxide production is developmentally regulated in the fetus and newborn. Am J Physiol. 1993;265:H1056–H1063. doi: 10.1152/ajpheart.1993.265.4.H1056. [DOI] [PubMed] [Google Scholar]
- Tod ML, Cassin S. Endothelin-1-induced pulmonary arterial dilation is reduced by Nω-nitro-L-arginine in fetal lambs. J Appl Physiol. 1992;72:1730–1734. doi: 10.1152/jappl.1992.72.5.1730. [DOI] [PubMed] [Google Scholar]
- Gao Y, Zhou H, Raj JU. Heterogeneity in role of endothelium-derived NO in pulmonary arteries and veins of full-term fetal lambs. Am J Physiol. 1995;268:H1586–H1592. doi: 10.1152/ajpheart.1995.268.4.H1586. [DOI] [PubMed] [Google Scholar]
- Abman SH, Chatfield BA, Rodman DM, Hall SL, McMurtry IF. Maturational changes in endothelium-derived relaxing factor activity of ovine pulmonary arteries in vitro. Am J Physiol. 1991;260:L280–L285. doi: 10.1152/ajplung.1991.260.4.L280. [DOI] [PubMed] [Google Scholar]
- Abman SH, Chatfield BA, Hall SL, McMurtry IF. Role of endothelium-derived relaxing factor during transition of pulmonary circulation at birth. Am J Physiol. 1990;259:H1921–H1927. doi: 10.1152/ajpheart.1990.259.6.H1921. [DOI] [PubMed] [Google Scholar]
- Rairigh RL, Storme L, Parker TA, le Cras TD, Kinsella JP, Jakkula M, Abman S. Inducible NO synthase inhibition attenuates shear stress-induced pulmonary vasodilation in the ovine fetus. Am J Physiol. 1999;276:L513–L521. doi: 10.1152/ajplung.1999.276.3.L513. [DOI] [PubMed] [Google Scholar]
- Schubert R, Serebryakow V. Iloprost dilates rat small arteries: role of KATP- and KCa-channel activation by cAMP-dependent protein kinase. Am J Physiol. 1997;272:H1147–H1156. doi: 10.1152/ajpheart.1997.272.3.H1147. [DOI] [PubMed] [Google Scholar]
- Shaul PW, Farrar MA, Magness RR. Oxygen modulation of pulmonary arterial prostacyclin synthesis is developmentally regulated. Am J Physiol. 1993;265:H621–H628. doi: 10.1152/ajpheart.1993.265.2.H621. [DOI] [PubMed] [Google Scholar]
- Frantz E, Soifer SJ, Clyman RI, Heymann MA. Bradykinin produces pulmonary vasodilation in fetal lambs: role of prostaglandin production. J Appl Physiol. 1989;67:1512–1517. doi: 10.1152/jappl.1989.67.4.1512. [DOI] [PubMed] [Google Scholar]
- Morin FC, III, Egan EA, Norfleet WT. Indomethacin does not diminish the pulmonary vascular response of the fetus to increased oxygen tension. Pediatr Res. 1988;24:696–699. doi: 10.1203/00006450-198812000-00009. [DOI] [PubMed] [Google Scholar]
- Cooke JP, Rossitch E, Jr, Andon NA, Loscalzo J, Dzau VJ. Flow activates an endothelial potassium channel to release an endogenous nitrovasodilator. J Clin Invest. 1991;88:1663–1671. doi: 10.1172/JCI115481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theis JG, Liu Y, Coceani F. ATP-gated potassium channel activity of pulmonary resistance vessels in the lamb. Can J Physiol Pharmacol. 1997;75:1241–1248. doi: 10.1139/cjpp-75-10-11-1241. [DOI] [PubMed] [Google Scholar]
- Cornfield D, Saqueton C, Porter V, Herron J, Resnik E, Haddad IY, Reeve HL. Voltage-gated K+ channel activity in ovine pulmonary vasculature is developmentally regulated. Am J Physiol. 2000;278:L1297–L1304. doi: 10.1152/ajplung.2000.278.6.L1297. [DOI] [PubMed] [Google Scholar]
- Cassin S, Dawes GS, Mott JC, Ross BB, Strang LB. The vascular resistance of the fetal and newly ventilated lung of the lamb. J Physiol (Lond) 1964;171:61–79. doi: 10.1113/jphysiol.1964.sp007361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leffler CW, Hessler JR, Green RS. The onset of breathing stimulates pulmonary vascular prostacyclin synthesis. Pediatr Res. 1984;18:938–942. doi: 10.1203/00006450-198410000-00006. [DOI] [PubMed] [Google Scholar]
- Tod ML, Yoshimura K, Rubin LJ. Indomethacin prevents ventilation-induced decreases in pulmonary vascular resistance of the middle region in fetal lambs. Pediatr Res. 1991;29:449–454. doi: 10.1203/00006450-199105010-00008. [DOI] [PubMed] [Google Scholar]
- Velvis H, Moore PK, Heymann MA. Prostaglandin inhibition prevents the fall in pulmonary vascular resistance as a result of rhythmic distension of the lungs in fetal lambs. Pediatr Res. 1991;30:62–68. doi: 10.1203/00006450-199107000-00013. [DOI] [PubMed] [Google Scholar]
- Cornfield DN, Chatfield BA, McQueston JA, McMurtry IF, Abman SH. Effects of birth-related stimuli on L-arginine-dependent pulmonary vasodilation in ovine fetus. Am J Physiol. 1992;262:H1474–H1481. doi: 10.1152/ajpheart.1992.262.5.H1474. [DOI] [PubMed] [Google Scholar]
- Tristani-Firouzi M, Martin E, Tolarova S, Weir EK, Archer SL, Cornfield DN. Ventilation-induced pulmonary vasodilation at birth is modulated by potassium channel activity. Am J Physiol. 1996;271:H2353–H2359. doi: 10.1152/ajpheart.1996.271.6.H2353. [DOI] [PubMed] [Google Scholar]
- Weir EK, Archer SL. The mechanism of acute hypoxic pulmonary vasoconstriction: the tale of two channels. FASEB J. 1995;9:183–189. doi: 10.1096/fasebj.9.2.7781921. [DOI] [PubMed] [Google Scholar]
- Sham JS, Crenshaw BR, Jr, Deng LH, Shimoda LA, Sylvester JT. Effects of hypoxia in porcine pulmonary arterial myocytes: roles of K(V) channel and endothelin-1. Am J Physiol. 2000;279:L262–L272. doi: 10.1152/ajplung.2000.279.2.L262. [DOI] [PubMed] [Google Scholar]
- Cornfield D, Reeve H, Tolarova S, Weir E, Archer S. Oxygen causes fetal pulmonary vasodilation through activation of a calcium-dependent potassium channel. Proc Natl Acad Sci USA. 1996;93:8089–8094. doi: 10.1073/pnas.93.15.8089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haworth SG, Hall SM, Chew M, Allen KM. Thinning of fetal pulmonary arterial wall and postnatal remodelling: ultrastructural studies on the respiratory unit arteries of the pig. Virchows Arch Pathol Anat Histopathol. 1987;411:161–171. doi: 10.1007/BF00712740. [DOI] [PubMed] [Google Scholar]
- Michel RP, Gordon JB, Chu K. Development of the pulmonary vasculature in newborn lambs: structure-function relationships. J Appl Physiol. 1991;70:1255–1264. doi: 10.1152/jappl.1991.70.3.1255. [DOI] [PubMed] [Google Scholar]
- Haworth SG, Hislop AA. Normal structural and functional adaptation to extra-uterine life. J Pediatr. 1981;98:915–918. doi: 10.1016/s0022-3476(81)80587-2. [DOI] [PubMed] [Google Scholar]
- Owen-Thomas JB, Reeves JT. Hypoxia and pulmonary artery pressure in the rabbit. J Physiol (Lond) 1969;201:665–672. doi: 10.1113/jphysiol.1969.sp008779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durmowicz AG, Orton EC, Stenmark KR. Progressive loss of vasodilator responsive component of pulmonary hypertension in neonatal calves exposed to 4,570 m. Am J Physiol. 1993;265:H2175–H2183. doi: 10.1152/ajpheart.1993.265.6.H2175. [DOI] [PubMed] [Google Scholar]
- Clement de Clety S, Decell M, Tod M, Sirois P, Gordon J. Developmental changes in synthesis of and responsiveness to prostaglandins I2 and E2 in hypoxic lamb lungs. Can J Physiol Pharmacol. 1998;76:764–771. doi: 10.1139/cjpp-76-7-8-764. [DOI] [PubMed] [Google Scholar]
- Gordon JB, Hortop J, Hakim TS. Developmental effects of hypoxia and indomethacin on distribution of vascular resistances in lamb lungs. Pediatr Res. 1989;26:325–329. doi: 10.1203/00006450-198910000-00008. [DOI] [PubMed] [Google Scholar]
- Belik J, Halayko A, Rao K, Stephens NL. Pulmonary vascular smooth muscle: biochemical and mechanical developmental changes. J Appl Physiol. 1991;71:1129–1135. doi: 10.1152/jappl.1991.71.3.1129. [DOI] [PubMed] [Google Scholar]
- Liu SF, Hislop AA, Haworth SG, Barnes PJ. Developmental changes in endothelium-dependent pulmonary vasodilatation in pigs. Br J Pathol. 1992;106:324–330. doi: 10.1111/j.1476-5381.1992.tb14335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell DC, Tod ML, Gordon JB. Developmental changes in endothelium-dependent relaxation of pulmonary arteries: role of EDNO and prostanoids. J Appl Physiol. 1996;81:2013–2019. doi: 10.1152/jappl.1996.81.5.2013. [DOI] [PubMed] [Google Scholar]
- Gordon JB, Martinez FR, O'Donnell DC, Tod ML. Effects of hypoxia and vascular tone on endothelium-dependent and -independent responses in developing lungs. J Appl Physiol. 1995;79:824–830. doi: 10.1152/jappl.1995.79.3.824. [DOI] [PubMed] [Google Scholar]
- Perreault T, De Marte J. Maturational changes in endothelium-derived relaxations in newborn piglet pulmonary circulation. Am J Physiol. 1993;264:H302–H309. doi: 10.1152/ajpheart.1993.264.2.H302. [DOI] [PubMed] [Google Scholar]
- Levin DL. Effects of inhibition of prostaglandin synthesis on fetal development, oxygenation, and the fetal circulation. Semin Perinatol. 1980;4:35–44. [PubMed] [Google Scholar]
- Fineman JR, Wong J, Morin FC, III, Wild LM, Soifer SJ. Chronic nitric oxide inhibition in utero produces persistent pulmonary hypertension in newborn lambs. J Clin Invest. 1994;93:2675–2683. doi: 10.1172/JCI117281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivy D, Parker T, Abman S. Prolonged endothelin B receptor blockade causes pulmonary hypertension in the ovine fetus. Am J Physiol. 2000;279:L758–L765. doi: 10.1152/ajplung.2000.279.4.L758. [DOI] [PubMed] [Google Scholar]
- Storme L, Rairigh RL, Parker TA, Kinsella JP, Abman SH. Acute intrauterine pulmonary hypertension impairs endothelium-dependent vasodilation in the ovine fetus. Pediatr Res. 1999;45:575–581. doi: 10.1203/00006450-199904010-00018. [DOI] [PubMed] [Google Scholar]
- McQueston JA, Kinsella JP, Ivy DD, McMurtry IF, Abman SH. Chronic pulmonary hypertension in utero impairs endothelium-dependent vasodilation. Am J Physiol. 1995;268:H288–H294. doi: 10.1152/ajpheart.1995.268.1.H288. [DOI] [PubMed] [Google Scholar]
- Cornfield D, Resnick E, Herron J, Abman S. Chronic intra-uterine pulmonary hypertension decreases calcium-sensitive potassium channel mRNA expression. Am J Physiol. 2000;297:L857–L862. doi: 10.1152/ajplung.2000.279.5.L857. [DOI] [PubMed] [Google Scholar]
- Fike C, Kaplowitz M, Thomas C, Nelin L. Chronic hypoxia decreases nitric oxide production and endothelial nitric oxide synthase in newborn pig lungs. Am J Physiol. 1998;274:L517–L526. doi: 10.1152/ajplung.1998.274.4.L517. [DOI] [PubMed] [Google Scholar]
- Tulloh RM, Hislop AA, Boels PJ, Deutsch J, Haworth SG. Chronic hypoxia inhibits postnatal maturation of porcine intrapulmonary artery relaxation. Am J Physiol. 1997;272:H2436–H2445. doi: 10.1152/ajpheart.1997.272.5.H2436. [DOI] [PubMed] [Google Scholar]
- Stenmark KR, James SL, Voelkel NF, Toews WH, Reeves JT, Murphy RC. Leukotriene C4 and D4 in neonates with hypoxemia and pulmonary hypertension. N Engl J Med. 1983;309:77–80. doi: 10.1056/NEJM198307143090204. [DOI] [PubMed] [Google Scholar]
- Abman SH, Stenmark KR. Changes in lung eicosanoid content during normal and abnormal transition in perinatal lambs. Am J Physiol. 1992;262:L214–L222. doi: 10.1152/ajplung.1992.262.2.L214. [DOI] [PubMed] [Google Scholar]
- Allen SW, Chatfield BA, Koppenhafer SA, Schaffer MS, Wolfe RR, Abman SH. Circulating immunoreactive endothelin-1 in children with pulmonary hypertension. Am Rev Respir Dis. 1993;148:519–522. doi: 10.1164/ajrccm/148.2.519. [DOI] [PubMed] [Google Scholar]
- Steinhorn RH, Morin FC, III, Gugino SF, Giese EC, Russell JA. Developmental differences in endothelium-dependent responses in isolated ovine pulmonary arteries and veins. Am J Physiol. 1993;264:H2162–H2167. doi: 10.1152/ajpheart.1993.264.6.H2162. [DOI] [PubMed] [Google Scholar]