

Abstract
The inflammatory response is critical to the development and progression of heart failure. Chemokines and their receptors are a distinct class of inflammatory modulators that may play a role in mediating myocardial dysfunction in heart failure. Levels of the chemokine CXCL12, also known as stromal cell-derived factor (SDF), and its receptor, CXCR4, are elevated in patients with heart failure, and we undertook this study to determine whether this chemokine system can directly affect cardiac function in the absence of leukocytes. Murine papillary muscles and adult rat cardiac myocytes treated with CXCL12, the only identified ligand of CXCR4, demonstrate blunted inotropic responses to physiologic concentrations of calcium. The negative inotropic effects on cardiac myocytes are accompanied by a proportional diminution of calcium transients. The effects are abrogated by AMD3100, a specific CXCR4 inhibitor. Overexpression of the receptor through adenoviral infection with a CXCR4 construct accentuates the negative inotropic effects of CXCL12 on cardiac myocytes during calcium stimulation. CXCR4 activation also attenuates beta-adrenergic-mediated increases in calcium mobilization and fractional shortening in cardiac myocytes. In electrophysiologic studies, CXCL12 decreases forskolin- and isoproterenol-induced voltage-gated L-type calcium channel activation. These studies demonstrate that activation of CXCR4 results in a direct negative inotropic modulation of cardiac myocyte function. The specific mechanism of action involves alterations of calcium channel activity on the membrane. The presence of functional CXCR4 on cardiac myocytes introduces a new target for treating cardiac dysfunction.
Keywords: Calcium, Chemokines, Chemokine receptors, Contractility, CXCL12, CXCR4, SDF
1. Introduction
Chemokines and their receptors are a relatively new class of inflammatory mediators that regulate inflammation by directing leukocytes to sites of injury. Chemokines and their receptors are up-regulated in patients with heart failure and therefore have been implicated in the development of this disease (reviewed in [1]). Contractile dysfunction is a prominent feature in heart failure, and there is an inverse relationship between chemokine levels and cardiac performance [2]. However, it is unclear whether these findings are merely coincidental or whether chemokines actually play a mechanistic role in mediating myocardial dysfunction. Furthermore, if chemokines mediate cardiac dysfunction, it is not clear whether the mechanism is via inflammatory cells or whether chemokines, through their receptors on the cardiac myocyte (CM) surface, can directly affect myocardial function.
The function of chemokines initially was thought to be limited to their migratory effects on inflammatory cells. However, as knowledge of the physiologic roles for chemokines and their receptors has markedly expanded, it is now appreciated that chemokine receptors mediate a myriad of important biological effects by activating their receptors on organs in a manner independent of inflammatory cells. CXCR4 exemplifies this concept. It is present on the mammalian heart and appears to play a critical role in cardiovascular development, in the mobilization of hematopoietic precursors and in vasculogenesis [3,4]. CXCR4 is also a co-receptor for HIV infection and can modulate Ca2+ channel function in neuronal cell lines [5].
CXCR4 is one of several chemokine receptors that are up-regulated in patients with heart failure [2,6,7]. To our knowledge, it is the only chemokine receptor that has been shown to be up-regulated in the failing myocardium of patients by direct staining [6]. Other studies examining chemokine receptors in heart failure have analyzed whole heart specimens to assess mRNA levels and therefore cannot localize the cellular source of the receptor given that the heart is composed of heterogeneous cell types. In early cardiac allograft rejection, where myocardial dysfunction can be significant without an obviously discernible cause (such as significant myocyte necrosis), CXCR4 expression is also elevated [8–11].
Given the increased expression of CXCR4 on dysfunctional myocardium in a variety of diseases and the specificity of CXCL12 (formerly known as stromal cell-derived factor (SDF-1)) for binding to CXCR4, this is an ideal model to demonstrate experimentally that a chemokine can directly affect myocardial function. The present study examines the effects of CXCR4 activation on myocardial contractility and explores the mechanisms at the cellular level. We demonstrate that CXCR4 negatively modulates myocardial contractility by binding its endogenous ligand, CXCL12. The mechanism of action involves an alteration of calcium (Ca2+) metabolism in response to β-adrenergic and Ca2+ stimulation in CM. These studies identify a potentially new class of receptors to target in the treatment of cardiac dysfunction.
2. Materials and methods
2.1. Papillary muscle contractility measurements
Papillary muscles (PM) from 10-week-old male FVBN-1 mice (Jackson Laboratories) were excised, loaded onto a force-transducer (Grass Instruments) and immersed in a circulating tissue bath at 37 °C. PM were field stimulated with platinum electrodes at 2× pacing threshold amplitude with a pacing frequency of 0.3 Hz and a stimulus duration of 0.3 ms. PM were then equilibrated in a modified Tyrode solution (containing the following in mM: K+, 5.9; Na+, 135.0; Cl−, 126.0; Ca2+, 1.0; HCO3−, 15.0; PO4−, 1.2; SO4−, 1.2; Mg2+, 1.2) and stretched to the length of maximal tension development. Following a 1-h equilibration, PM were subjected to stepwise increasing concentrations of extracellular calcium from a baseline of 1.0 mM to a maximal concentration of 5.0 mM. PM underwent this first Ca2+ challenge to establish baseline contractile function and confirm muscle viability. They were then re-equilibrated at baseline , treated for 5 min with either diluent (0.1% BSA) (control) or CXCL12 (125 ng/ml in 0.1% BSA) before undergoing a second Ca2+ challenge. Isometric tension data, measured by a force transducer, amplified and captured via Polyview data acquisition software (Grass Instruments), were expressed as % baseline tension at each concentration of Ca2+. The normalized data were subjected to a non-linear regression analysis, fitted to a dose response curve and log ED50 was calculated. To limit inter-animal variability, each PM served as its own control.
2.2. Cardiac myocyte isolation
Ca2+-tolerant rat ventricular CM were isolated via enzymatic dissociation, as described previously [12]. Briefly, rat hearts were perfused in a retrograde fashion for 5 min with oxygenated Krebs–Henseleit buffer (containing the following in mM: NaCl, 120.0; KCl, 4.7; KH2PO4, 1.2; MgSO4, 1.2 and HEPES, 25.0; pH 7.40 at 37 °C). The perfusate was then switched to an enzyme solution containing collagenase 0.7 mg/ml (Worthington Type II, 258 u/mg) and hyaluronidase 0.5 mg/ml (Sigma Type II, 667 u/mg), and the heart was perfused for another 10–15 min. Ventricular tissue was then finely minced and shaken gently in enzyme solution for 20–30 min. CM were filtered through a nylon mesh, collected and made Ca2+-tolerant over 15 min. CM were then re-suspended in ACCT medium consisting of DMEM (containing 2.0 mg/ml BSA, 50 IU/ml penicillin, 50 IU/ml streptomycin and the following in mM: L-carnitine, 0.4; creatine, 0.66; taurine, 0.62) and plated at a density of 2×104 cells/ml onto laminin-precoated coverslips (2 μg/cm2, Invitrogen).
2.3. Intracellular calcium and contractility measurements
Freshly isolated rat CM plated onto laminin-coated coverslips as described above were loaded with fura 2-AM (Molecular Probes), superfused with 1.2 mM Ca2+ Tyrode's solution, pH 7.40 at 37 °C, and electrically stimulated with a biphasic pulse (0.5 Hz, 50% above threshold). Cells included in the study were rod-shaped with a clear striation pattern and were quiescent in the absence of electrical stimulation. Contraction amplitude and intracellular Ca2+ transients were recorded online using a dual excitation spectrofluorometer and video-edge detection system (Ionoptix), as described previously [13].
During the Ca2+ stimulation protocol, CM were subjected to stepwise increases in from 1 mM to 8 mM. CM were treated with either diluent (0.1% BSA) (control) or CXCL12 (125 ng/ml) for 5, 8 and 60 min and assessed for fractional shortening and Ca2+ transients. Response to isoproterenol (ISO) was measured by increasing concentrations of ISO in a stepwise fashion from 0.001 μM to 10 μM. The concentration of was kept constant at 2.0 mM during the ISO stimulation.
The bicyclam AMD3100 was obtained from The National Institute of Health AIDS Research and Reference Reagent Program. During the blocking experiments, this compound was added to the chamber bathing the isolated myocytes to achieve a final concentration of 10 mM, prior to adding CXCL12.
2.4. Construction of recombinant adenoviruses and overexpression of CXCR4
The methods used to construct the recombinant adenovirus have been described previously by our group [14]. Briefly, the backbone vector, which contains most of the adenoviral genome (pAd.EASY1), was used and the recombination performed in Escherichia coli. CXCR4 cDNA was subcloned into the adenoviral shuttle vector (pAd.TRACK), which uses the cytomegalovirus long-terminal repeat as a promoter. pAd.TRACK also has a concomitantly expressed green fluorescent protein (GFP) under the control of a separate cytomegalovirus promoter. The adenoviruses were propagated in 293 cells. The titer of stocks used for these studies measured by plaque assays was 4×1010 pfu/ml for CXCR4-expressing adenovirus (Ad.CXCR4) with particle/pfu ratios of 4:1. These recombinant adenoviruses were tested for the absence of wild-type virus by polymerase chain reaction of the early transcriptional unit E1. Subsequently, CM were incubated with Ad.CXCR4 at a multiplicity of infection (MOI) of 100. Fractional shortening and Ca2+ transients then were measured as previously described for the uninfected CM.
2.5. Fluorescent immunostaining
Adult rat CM, isolated as described above, were fixed with 4% formaldehyde and double stained with an anti-sarcomeric α-actinin monoclonal antibody (Ab) (Sigma, 1:2000) and an anti-CXCR4 polyclonal Ab (eBioscience, 1:200). Primary antibodies were detected with either an anti-mouse-Cy3 secondary Ab for α-actinin or an anti-rabbit-Cy2 secondary Ab (Rockland, Gilbertsville, PA) for CXCR4. Human CM were isolated as previously described [15] and stained as described for the rat CM. Images were collected using a Leica DMI 6000 (Leica Microsystems) fluorescence microscope. Non-specific staining for CXCR4 was assessed by omission of the primary Ab and examination of the sections in the presence of the secondary Ab alone.
2.6. Whole-cell patch clamp measurements
Adult rat ventricular CM, isolated as described above, were suspended in a high potassium medium (PSS solution containing the following in mM: KCl, 50.0; glucose, 10.0; HEPES, 10.0; taurine, 20.0; MgCl2, 3.0; l-glutamic acid, 50.0; EGTA, 0.5; KH2PO4, 20.0) and stored at 4 °C. Isolated CM suspension was inoculated onto an acid-treated 12-mm round glass cover-slip placed at the bottom of the recording chamber. After the cells settled on the coverslip, viable CM were identified morphologically. To study inward Ca2+ currents using Ca2+ as the charge carrier, extracellular buffer (containing the following in mM: NaCl, 140.0; MgCl2, 0.5; CaCl2, 1.0; HEPES, 5.5; CsCl, 5.4; pH 7.4) was used. Whole-cell patch electrodes were pulled from Kimax glass pipette (Fisher Scientific Inc), with a series resistance of 1.0–1.5 MΩ when filled with the intracellular solution (containing the following in mM: TEA–Cl, 20.0; HEPES 5.0; EGTA, 5.0; CsCl, 130.0; ATP 5.0; GTP 0.01; pH 7.4). Whole-cell patch-clamp recordings were carried out with an EPC-9 amplifier and Pulse/PulseFit software package (HEKA Electronic). Data analyses were carried out with Pulsefit software.
2.7. Statistical Analysis
Experimental data were presented as the mean±SE. Student's t test was used in the analysis of paired and unpaired means. Unless indicated, all values were significant at p<0.05. Blocking experiments was performed on three consecutive CM and representative data are presented.
3. Results
3.1. CXCR4 activation decreases PM contractility
Control and CXCL-12-treated papillary muscles were subjected to a first Ca2+ challenge to establish baseline function. They were then equilibrated at baseline prior to a second Ca2+ challenge. Prior to the second Ca2+ challenge, control PM were treated for 5 min with either diluent or CXCL12. Control PM demonstrated similar Log ED50 during the second (1.5±0.13) and first (0.90±0.23) Ca2+ challenges (p>0.05) (Fig. 1A). In contrast, exposure to CXCL12 significantly blunted the PM response to Ca2+. After exposure to CXCL12, PM demonstrated a significantly higher Log ED50 during the second calcium challenge (2.4±0.14) when compared to the first calcium challenge (1.4±0.15) (p<0.05). There was no significant difference between the average cross-sectional areas of Control PM (0.08±0.04 mm2) and PM treated with CXCL12 (0.11±0.06 mm2)(p=0.20). Histologic examination demonstrated no significant inflammatory infiltrate (data not shown).
Fig. 1.
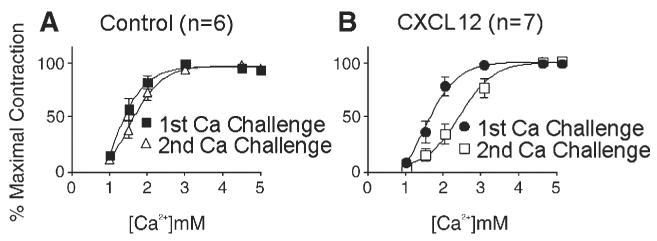
Activation of CXCR4 blunts PM response to Ca2+. Murine PM were subjected to two graded stepwise Ca2+ challenges separated by wash-out. PM exposed to diluent before the second Ca2+ challenge demonstrate similar responses when compared to the first challenge. (A) PM exposed to CXCL12 (125 ng/ml) demonstrated a blunted response during the second Ca2+ challenge (B).
3.2. CXCR4 decreases CM contractility in response to Ca2+ and isoproterenol (ISO)
To determine whether the blunted Ca2+ response of PM can be explained by an alteration of CM function, we examined the effect of CXCL12 on isolated rat CM. The average percent increase in shortening of CM treated for 5 min with CXCL12 was significantly decreased at 4 mM (4.5±0.4%) and at 8 mM (5.4±1.0%) when compared to Control CM at 4 mM (5.8±0.6%) and at 8 mM (6.7±0.9%) (p<0.05) (Fig. 2A). Similarly, the average peak Ca2+ transient of CM treated for 5 min with CXCL12 was significantly decreased at 4 mM (427±40 nM) and at 8 mM (583±97 nM) when compared to Control CM at 4 mM (601±94 nM) and at 8 mM (699±110 nM) (p<0.05) (Fig. 2B). Similar results were obtained after 10 min exposure to CXCL12 (Figs. 2A and B). At 1 mM and 2 mM , there was no difference in fractional shortening or Ca2+ transient between Control and CM exposed for either 5 or 10 min to CXCL12. Interestingly, CM exposed for 60 min to CXCL12 demonstrated no diminution in response to 4 mM and 8 mM compared to control. This suggested that CXCR4 may be down-regulated, or desensitized in response to prolonged agonist exposure, a common mechanism of chemokine receptor regulation [16] (Figs. 2A and B). To verify that CXCL12 also decreased contractility in response to a physiologically relevant positive inotrope, isoproterenol (ISO), CM were exposed to increasing ISO concentrations. Pretreatment with CXCL12 significantly decreased CM shortening in response to β-receptor stimulation with ISO (Fig. 2C). There was no difference at the lowest dose (0.001 μM) of ISO. These experiments demonstrate that the CXCR4-mediated decrement in contractility is not limited to Ca2+ stimulation.
Fig. 2.

Activation of CXCR4 decreases CM contractility. Adult rat CM were exposed to CXCL12 (125 ng/ml) for either 5, 10 or 60 min prior to a stepwise Ca2+ challenge (A, B) or to increasing ISO concentrations after a 5-min exposure to CXCL12 (C). In both experiments, control CM were exposed to diluent only. The average percent increase in shortening (A) and average peak Ca2+ transients (B) during the Ca2+ challenge were significantly lower in CM exposed for either 5 or 10 min to CXCL12. No difference in shortening or Ca2+ transient was detected in CM exposed for 60 min to CXCL12 (A and B). Percent increase in fractional shortening in the CM is shown as a function of increasing [ISO] (0.001–10 μM) (C). CXCL12 exposed CM demonstrated a significant decrease in shortening response. ISO experiments were performed in triplicate.
3.3. CXCR4 is present on the surface of CM
To confirm the expression of CXCR4 in adult CM, isolated rat and human CM were plated on slides and then stained with an anti-CXCR4 Ab and analyzed by fluorescent microscopy. As shown in Fig. 3 (top panel), CXCR4 was present on or near the surface of adult rat ventricular CM. The images were captured at various planes (data not shown) and the pattern of expression on the surface appeared linear (arrowheads, inset A) with some clustering of receptors. There was no detectable staining with omission of the CXCR4 Ab (inset B). Significant CXCR4 is detectable on human CM (bottom panel). Human CM appeared to display more localization of CXCR4 antigen at the Z-disks as distinguished by α-actinin staining.
Fig. 3.

CXCR4 is present on adult rat CM. Alpha-actinin (red), CXCR4 chemokine receptor (green) and DAPI-stained nuclei (blue) were visualized in rodent and human myocytes. A single rat CM was stained one h after isolation with a polyclonal anti-rat CXCR4 Ab (green) and an anti-α-actinin Ab (red) and examined by fluorescent microscopy (top panel). Higher magnification (inset A) reveals a punctate distribution of CXCR4 on the surface (arrowheads). The CXCR4 Ab was omitted and only the fluoresceinated anti-rabbit-Cy2 Ab was applied (inset B). Normal human cardiac myocyte also demonstrate a similar pattern of CXCR4 chemokine receptor staining (bottom panel).
3.4. CXCL12 effects on CM contractility are mediated through CXCR4
The effects of CXCL12 on CM function are mediated by CXCR4. Pre-incubation with the bicyclam AMD3100, a highly selective CXCR4 antagonist [17,18] completely abolished the negative effect of CXCL12 on CM shortening and Ca2+ transients (Figs. 4A and B). This suggests that the observed effects on CM are CXCR4 receptor mediated.
Fig. 4.
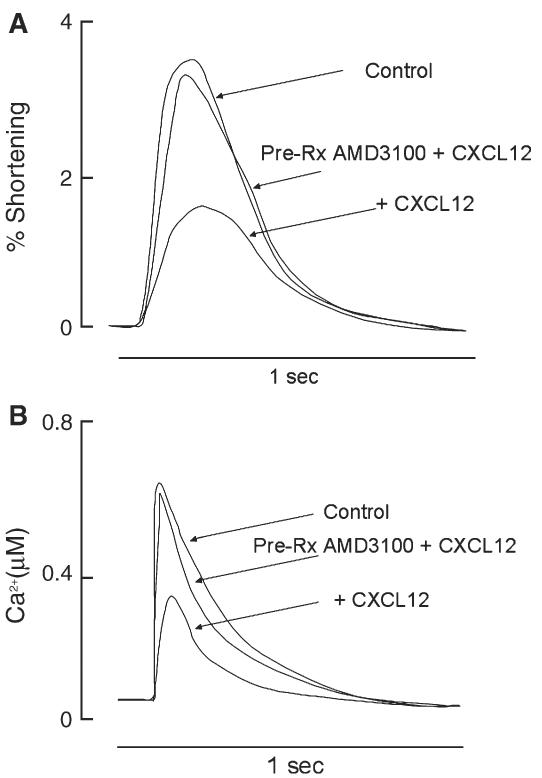
Effects of CXCL12 on CM are mediated through CXCR4. Adult rat CM were pretreated with AMD3100 for 1 h and CXCL12 (125 ng/ml) for 10 min prior to a graded stepwise Ca2+ challenge. A representative tracing for fractional shortening (A) and peak Ca2+ transients during stimulation (B) are presented. Experiments were performed in triplicate.
3.5. Infection of CM with an adenoviral CXCR4 vector increases its expression
To study the efficacy of adenoviral infection of CXCR4 on CM CXCR4 expression, CM were assessed by fluorescent microscopy at 24 (Fig. 5A) and 48 h (Fig. 5B) after infection with increasing concentrations (MOI) of Ad.CXCR4. CXCR4 was expressed in a concentration-dependent fashion at both 24 and 48 h post-infection. No significant toxicity was noted with Ad.CXCR4 infection up to 20 MOI. At this dose, there was no difference in the number of rod-shaped CM compared to a sample without infection.
Fig. 5.
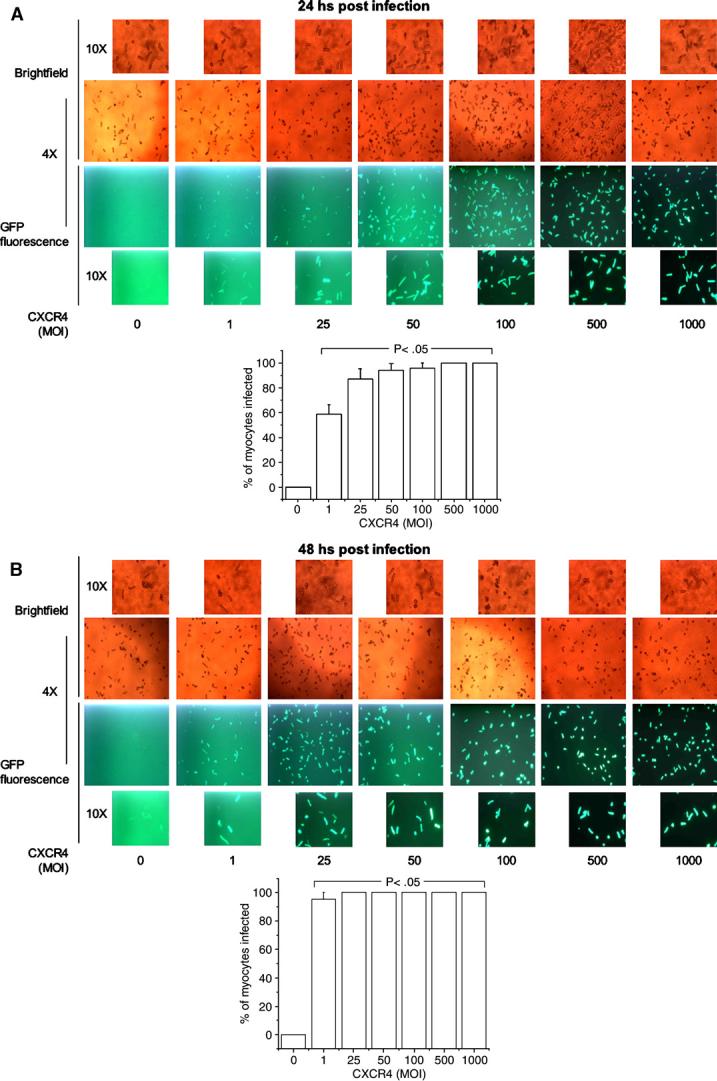
Adenoviral infection significantly increases expression of CXCR4 on rat CM. Adult rat CM were visualized to detect GFP fluorescence 24 (A) or 48 h (B) after infection with Ad.CXCR4. Co-expression of GFP demonstrates visually that CXCR4 gene is being expressed in the cells. A dose–response infection was performed in order to show significant increase in CXCR4 expression with increased MOI. The percentage of infected CM was determined as the number of GFP positives cells against the total cells counted in the bright-field. Different magnifications were used for optimal visualization of the cells. The bar graphs show that there was a significant increase in CXCR4-infected CM even at the lowest MOI.
3.6. Overexpression of CXCR4 increases the negative modulation of CM contractility
To further establish that the CXCL12-induced changes in myocyte function are CXCR4 mediated, CM expressing Ad.CXCR4 were studied for their response to CXCL12 exposure. CM expressing Ad.CXCR4 demonstrated accentuated decreases in fractional shortening and Ca2+ transients (Figs. 6A and B) in response to CXCL12 when compared to uninfected CM treated with CXCL12 (p<0.05). The effects of CXCR4 overexpression on fractional shortening and Ca2+ transients were statistically significant at all concentrations except 4 mM and 8 mM, where it reached near significance (p=0.06). The vector itself has been previously demonstrated to have no intrinsic effect on CM contractility [13].
Fig. 6.
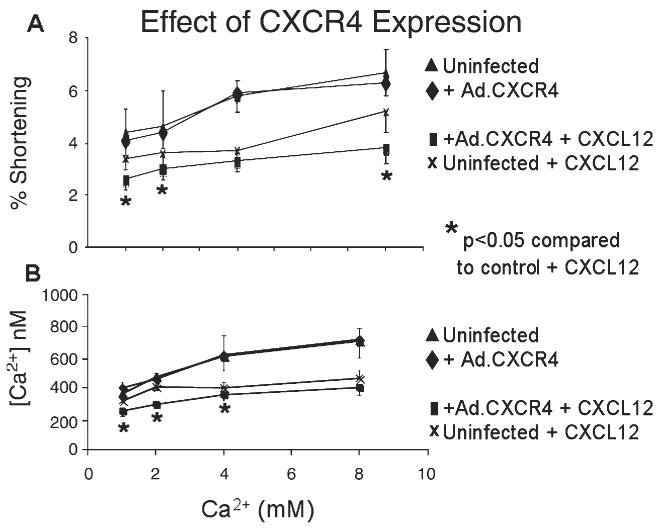
Overexpression of CXCR4 decreases CM contractility. Both CM infected with Ad.CXCR4 and control uninfected CM were exposed to CXCL12 (125 ng/ml) and then subjected to a graded stepwise Ca2+ challenge. The average percent shortening (A) at 37 °C at 1 Hz and average peak Ca2+ transients (B) at increasing external Ca2+ are presented. *p<0.05 vs. uninfected control in the presence of CXCL12.
3.7. CXCL12 attenuates cAMP-induced Ca2+ channel activation in CM
To study the possible mechanisms by which CXCR4 modulates CM function, we employed whole-cell patch clamp techniques to measure L-type Ca2+ channel activity of CM freshly isolated from rat left ventricles. With 1.0 mM CaCl2 in the bath, and a step voltage protocol from a holding potential of −50 mV, the inward current (Fig. 7A) displayed characteristic slow inactivating kinetics, sensitivity to cadmium (Cd) and an I-V plot with a peak inward current near 0 mV (Fig. 7B). These data fit the profile of voltage-gated L-type Ca2+ channels.
Fig. 7.
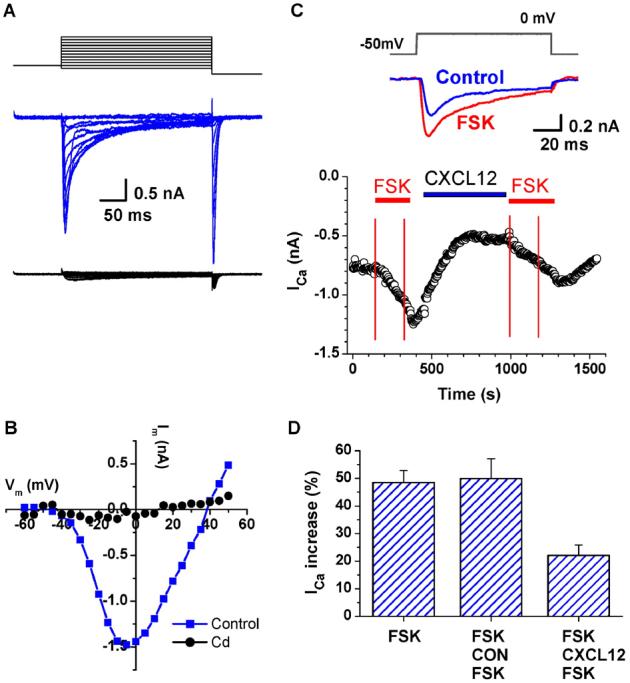
CXCL12 modulates forskolin (FSK)-induced Ca2+ channel activation in CM. Whole-cell recording of inward Ca2+ currents from isolated rat ventricular CM with 1 mM CaCl2 in the bath solution. L-type Ca2+ currents were activated with the step voltage clamp protocol shown in the top panel. Currents were mostly blocked by Cd (0.1 mM, lower panel in red) (A). I-V plot of the L-type Ca2+ current in control conditions (blue) and in 0.1 mM Cd (red) (B). Time course of Ca2+ current in response to the application of FSK (1 μm) and CXCL12 (100 ng/ml). The top panel illustrates the voltage protocol and typical current traces under each condition. The inward Ca2+ currents were activated by a repeating voltage step from −50 mV to 0 mV. Inward current values were measured at the inward peak. The bottom panel illustrates the relative changes of peak currents during 3-min applications of FSK (1 μM) indicated by the red horizontal lines. CM were exposed for approximately 5 min to either CXCL12 (FSK-CXCL12-FSK) or diluent (FSK-CON-FSK), as indicated by the blue horizontal line, before the second FSK application (C). Summary of the CXCL12 attenuation of the FSK-induced Ca2+ current stimulations is shown (n=5) (D).
To determine whether CXCL12 modulates the L-type Ca2+ channels, forskolin (FSK), a direct adenylyl cyclase activator, was used to stimulate Ca2+ currents. As expected, FSK alone increased Ca2+ currents in CM (n=5) by 45.6±4.5% (Fig. 7C, upper panel). To test the effects of CXCL12, in modulating the FSK-induced Ca2+ currents, initial FSK exposure was followed by either CXCL12 or diluent (control) prior to a second FSK challenge. Data from a typical experiment where CXCL12 was applied prior to the second FSK challenge are shown in Fig. 7C, lower panel. Treatment with CXCL12 markedly attenuated the Ca2+ current increase by FSK (FSK-CXCL12-FSK, 12.8±6.4%, n=12, p<0.01) compared to control (FSK-CON-FSK, 44.4±7.8%, n=5, p>0.05) (Fig. 7D).
To determine whether CXCL12 also causes a marked decrement in Ca2+ current increase in response to ISO, similar experiments were carried out as described for FSK. ISO, a β-adrenergic receptor (βAR) agonist, signals through the Gs protein pathway resulting in an increase intracellular cAMP through adenylate cyclase activation. Ca2+ channel activity in CM was significantly increased by ISO and was fully blocked by Cd (Fig. 8A). ISO alone increased Ca2+ currents by 223±42% (n=7) whereas pretreatment with CXCL12 (Fig. 8B) markedly attenuated the ISO-induced Ca2+ channel activity (68±14% (CXCL12+ISO, n=11, p<0.001)), as summarized in Fig. 8C.
Fig. 8.
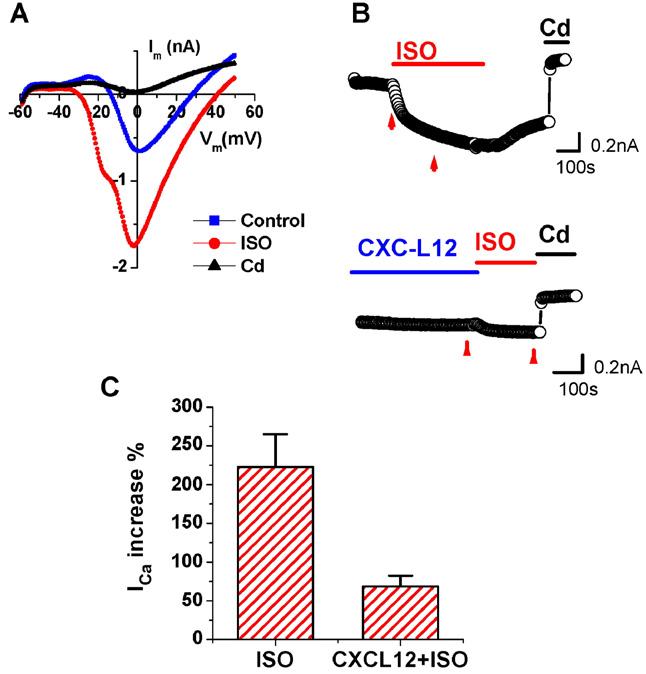
CXCL12 modulates β-adrenergic Ca2+ channel activation in CM. Whole-cell inward Ca2+ currents were recorded from isolated adult rat ventricular CM with 1 mM Ca2+ in the bath solutions. Ca2+ currents were activated with a protocol ramping from −50 mV to +50 mV from a holding potential of −60 mV. I-V plots of Ca2+ currents against membrane potentials recorded in control condition (blue), ISO (1 μM, red) and in CdCl2 (0.1 mM, black) (A). Representative time course record of the Ca2+ current from CM treated with ISO alone (1 μM, top panel), or pre-incubated with CXCL12 (100 ng/ml) for 5 min prior to ISO treatment (lower panel). Applications of ISO, (1 μM), CXCL12 (100 ng/ml) and CdCl2 (0.1 mM) are indicated by the bars. Data values for analysis were taken from the point before ISO and 200 s after ISO application, as indicated by the arrows in each panel B. Summary of the effects of CXCL12 on ISO-induced Ca2+ current increases (n=11) (C).
Of note, CXCL12 (100 ng/ml, 5–6 min) alone did not produce significant changes on the basal inward Ca2+ current before and after stimulation (−6.8±5.3%, n=11) (data not shown).
4. Discussion
Heart failure is a clinical syndrome characterized by the inability of the heart to produce an adequate cardiac output [19]. A key contributor to the dysfunction may be depressed myocardial contractility [20,21]. Studies that have examined the function of cardiac myocytes (CM) isolated from failing hearts suggest that a defect in CM contractility may contribute to the overall cardiac dysfunction [22]. The responsible agents or mechanisms, however, remain incompletely characterized.
Inflammatory molecules may be agents of myocardial depression in heart failure. The hypothesis that inflammation plays a role in cardiac dysfunction was first suggested from animal [23] and human studies of sepsis (reviewed by van der Poll et al. [24]). Sepsis-associated cardiac dysfunction was thought to be mediated, in part, by circulating or “humoral” myocardial depressants that were subsequently identified as predominantly inflammatory cytokines (including tumor necrosis factor-1alpha (TNF-1α), interleukin-1beta, (IL-1β) [25], IL-6 [26], IL-2 [27] and interferon-gamma (IFN-γ)). These ideas were supported by numerous animal [28–30] and by in vitro studies [25,31,32]. However, clinical trials (reviewed by Anker et al. [33]), that used “targeted” approaches to neutralize cytokines, such as TNF, were unsuccessful in treating patients with moderate to advanced heart failure.
Chemokines are a distinct class of inflammatory molecules that are also elevated in heart failure. This class of receptors is present on the myocardium and vascular smooth muscle cell types and is capable of mediating important biological processes in a manner that is independent of inflammatory cells [34]. Recently, several studies have demonstrated that circulating chemokine levels, including that of CXCL12, are elevated in animals and humans with cardiac dysfunction [35–37]. Therefore, chemokines have been proposed as mediators of cardiac dysfunction. However, the mechanism of chemokine-mediated cardiac dysfunction is unknown.
We hypothesized that the role of chemokines and their receptors in the cardiac response to inflammation may be quite distinct from that of cytokines' given the wide degree of structural, functional and regulatory differences between the two classes of molecules. Therefore, there is a rationale for assuming that treatment outcomes for cytokine-directed therapies of heart failure may not be predictive of chemokine receptor-directed modalities. Damas et al. [6] demonstrated that a variety of chemokine receptors was increased in the failing human myocardium compared to non-failing specimens. Although the CM is the predominant cell type in cardiac tissue, a potential drawback of this study is that the entire ventricle or atrium was processed and used for RNA protection assay analysis so that the relative contribution of each resident cell type to the elevated chemokine receptor levels could not be determined. Seino et al. [35] demonstrated that CCL2 (monocyte chemoattractant protein (MCP-1)) RNA levels are detectable in human myocardial biopsies and in TNF-α1-treated neonatal rat CM. These studies postulated that the mechanisms for chemokine effects on myocardial function were predominantly paracrine responses to circulating leukocytes [7,38]. Our study demonstrates that the chemokine CXCL12 can directly depress contractility of the myocardium.
In the present study, normal PM exposed to CXCL12 demonstrated a blunted response to Ca2+ stimulation. The concentrations of CXCL12 used in the experiments are similar to those used in measuring the physiologic effects of CXCL12 in a variety of cell types such as epithelial cells [39,40], lymphocytes [41] and neurons [42]. Histological examination of the PM demonstrated a paucity of inflammatory cells. Therefore, it was unlikely that inflammatory cells mediated the effect of CXCL12 on PM contractility. However, because the papillary muscle is composed of heterogeneous cell types, the effects of CXCL12 could not be ascribed to one particular cell type based on studies employing multi-cellular preparations.
The blunted Ca2+ response of papillary muscles is explained in part by a direct effect of CXCL12 on CM. CM exposed to CXCL12 demonstrated a significantly decreased shortening in response to both Ca2+ and ISO stimulation at higher concentrations. At lower concentrations of Ca2+ (1 mM and 2 mM) and ISO (0.001 μM) CXCL12 exposure did not cause decreased CM shortening. This was consistent with the observation that whereas the failing myocardium shows normal baseline function, the contractile reserve is diminished [43]. The lack of CXCL12 effect on CM shortening at normal and low ISO concentration imply that in patients with normal left ventricular function, CXL12 may not have a direct negative effect on contractility. However, in conditions such as heart failure where there is sympathetic activation, it is possible that the presence of CXCL12 induces a negative inotropic effect.
CXCL12 imparts its effects on CM through interaction with its exclusive receptor CXCR4. A highly specific CXCR4 antagonist, AMD3100, abrogated the depressed contractile response to Ca2+, and overexpression of CXCR4 by adenoviral infection of CM depressed the contractile response to Ca2+ treatment even further in the presence of CXCL12. Ligand-induced receptor internalization is a general regulatory mechanism in chemokine signaling. During Ca2+ stimulation, we observed that CM returned to normal contractile response after prolonged CXCL12 exposure, which is consistent with receptor desensitization. These observations demonstrate that the effect of CXCL12 is mediated through its interaction with CXCR4.
Immunofluorescent microscopy showed that there was a significant amount of CXCR4 on or near the surface of human and rat adult CM. The expression appeared to be linear and the receptors in certain sections appeared to be clustered, a finding consistent with reports of CXCR4 expression in other cell types [44]. Further studies are ongoing to determine the factors that regulate CXCR4 surface expression on CM. The immunohistochemical studies of CXCR4 on human CM demonstrated that CXCR4 expression on CM is not limited to murine species and therefore may be relevant to human diseases.
To determine the possible mechanisms by which CXCR4 activation decreases myocardial function, we examined Ca2+ mobilization within CM when exposed to CXCL12. During Ca2+ stimulation peak, Ca2+ transients were decreased in CM exposed to CXCL12 at supraphysiologic concentrations of (4–8 mM), but not at basal levels (1–2 mM). CXCL12 may modulate Ca2+ transients by altering L-type Ca2+ channel activity in response to either calcium or an adrenergic challenge. Patch clamp studies measuring L-type Ca2+ channel activity of individual CM demonstrated that CXCL12 decreased Ca2+ currents induced by either β-adrenergic with ISO or FSK stimulation but CXCR4 activation did not have a significant effect on basal activity of the Ca2+ currents. Together, these data suggested both an upstream effect of CXCR4 on L-type Ca2+ channels as well as a downstream effect that appears to be unrelated to β-adrenergic receptor stimulation. This is consistent with the findings of others wherein treatment of colonic epithelial cells with CXCL12 inhibited cAMP response only in the presence of FSK stimulation [45]. It appears that an important function of CXCR4 is to modulate cAMP-mediated events. In CM, this may have profound physiologic implications such as the modulation of contractility as demonstrated in the present studies.
A novel and exciting potential methodology to reverse myocardial remodeling associated with myocardial infarction [46] is regeneration of myocardium using bone marrow (BM)-derived mesenchymal cells, endothelial progenitor cells and endogenous cells [47–50]. Given its essential role in BM homing and recruitment [49,51], CXCL12 is a candidate to promote stem cells recruitment to the heart. Because CXCR4 may negatively impact myocardial function, strategies [52] that primarily employ up-regulation of CXCL12 may need to be examined more closely. Our results would caution that a therapeutic approach whereby myocardial CXCL12 is increased, in an effort to recruit circulating or endogenous stem cells in the setting of an ischemic injury, may result in a significant decrease in contractility.
In this study we have shown, for the first time to our knowledge, that CXCR4 is functional on CM. CXCR4 activation can directly inhibit myocardial contractile response to increases in Ca2+ and ISO concentrations through modulation of L-type Ca2+ channel activity. Manipulation of CXCR4 activity may serve as a new target in the treatment of cardiac dysfunction. Additionally, the discovery of functional chemokine receptors on CM may have broad implications on preexisting chemokine/chemokine receptor based stem cell therapies for cardiomyopathies.
Acknowledgments
We thank Dr. Federica del Monte for providing human cardiomyocytes and for providing insightful comments in manuscript preparation. This work was supported in part by grants from NIH: HL073458, HL054469 to ADS and HL057623, HL071763, HL078691 to RJH.
References
- 1.Aukrust P, Damas JK, Gullestad L, Froland SS. Chemokines in myocardial failure-pathogenic importance and potential therapeutic targets. Clin Exp Immunol. 2001;124(3):343–5. doi: 10.1046/j.1365-2249.2001.01527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aukrust P, Ueland T, Muller F, Andreassen AK, Nordoy I, Aas H, et al. Elevated circulating levels of C-C chemokines in patients with congestive heart failure. Circulation. 1998;97(12):1136–43. doi: 10.1161/01.cir.97.12.1136. [DOI] [PubMed] [Google Scholar]
- 3.Zou Y-R, Kottmann AH, Kuroda M, Taniuchi I, Littman DR. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature. 1998;393(6685):595. doi: 10.1038/31269. [DOI] [PubMed] [Google Scholar]
- 4.Tachibana K, Hirota S, Iizasa H, Yoshida H, Kawabata K, Kataoka Y, et al. The chemokine receptor CXCR4 is essential for vascularization of the gastrointestinal tract. Nature. 1998;393(6685):591–4. doi: 10.1038/31261. [DOI] [PubMed] [Google Scholar]
- 5.Oh SB, Endoh T, Simen AA, Ren D, Miller RJ. Regulation of calcium currents by chemokines and their receptors. J Neuroimmunol. 2002;123(1–2):66–75. doi: 10.1016/s0165-5728(01)00485-4. [DOI] [PubMed] [Google Scholar]
- 6.Damas JK, Eiken HG, Oie E, Bjerkeli V, Yndestad A, Ueland T, et al. Myocardial expression of CC- and CXC-chemokines and their receptors in human end-stage heart failure. Cardiovasc Res. 2000;47(4):778–87. doi: 10.1016/s0008-6363(00)00142-5. [DOI] [PubMed] [Google Scholar]
- 7.Damas JK, Gullestad L, Aass H, Simonsen S, Fjeld JG, Wikeby L, et al. Enhanced gene expression of chemokines and their corresponding receptors in mononuclear blood cells in chronic heart failure-modulatory effect of intravenous immunoglobulin. J Am Coll Cardiol. 2001;38(1):187–93. doi: 10.1016/s0735-1097(01)01335-3. [DOI] [PubMed] [Google Scholar]
- 8.Pyo RT, Wahler GM. Ventricular myocytes isolated from rejecting cardiac allografts exhibit a reduced beta-adrenergic contractile response. J Mol Cell Cardiol. 1995;27(2):773–6. doi: 10.1016/0022-2828(95)90083-7. [DOI] [PubMed] [Google Scholar]
- 9.DiSesa VJ, Masetti P, Diaco M, Schoen FJ, Marsh JD, Cohn LH. The mechanism of heart failure caused by cardiac allograft rejection. J Thorac Cardiovasc Surg. 1991;101(3):446–9. [PubMed] [Google Scholar]
- 10.Farivar AS, Mackinnon-Patterson BC, McCourtie AS, Ward PA, Mulligan MS. The role of CC and CXC chemokines in cardiac allograft rejection in rats. Exp Mol Pathol. 2005;78(3):171–6. doi: 10.1016/j.yexmp.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 11.Fahmy NM, Yamani MH, Starling RC, Ratliff NB, Young JB, McCarthy PM, et al. Chemokine and receptor-gene expression during early and late acute rejection episodes in human cardiac allografts. Transplantation. 2003;75(12):2044–7. doi: 10.1097/01.TP.0000069601.73079.94. [DOI] [PubMed] [Google Scholar]
- 12.Communal C, Singh M, Menon B, Xie Z, Colucci WS, Singh K. beta1 integrins expression in adult rat ventricular myocytes and its role in the regulation of beta-adrenergic receptor-stimulated apoptosis. J Cell Biochem. 2003;89(2):381–8. doi: 10.1002/jcb.10520. [DOI] [PubMed] [Google Scholar]
- 13.Hajjar RJ, Schmidt U, Kang JX, Matsui T, Rosenzweig A. Adenoviral gene transfer of phospholamban in isolated rat cardiomyocytes: rescue effects by concomitant gene transfer of sarcoplasmic reticulum Ca2+-ATPase. Circ Res. 1997;81(2):145–53. doi: 10.1161/01.res.81.2.145. [DOI] [PubMed] [Google Scholar]
- 14.del Monte F, Williams E, Lebeche D, Schmidt U, Rosenzweig A, Gwathmey JK, et al. Improvement in survival and cardiac metabolism after gene transfer of sarcoplasmic reticulum Ca (2+)-ATPase in a rat model of heart failure. Circulation. 2001;104(12):1424–9. doi: 10.1161/hc3601.095574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.del Monte F, Harding SE, Schmidt U, Matsui T, Kang ZB, Dec GW, et al. Restoration of contractile function in isolated cardiomyocytes from failing human hearts by gene transfer of SERCA2a. Circulation. 1999;100(23):2308–11. doi: 10.1161/01.cir.100.23.2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haribabu B, Richardson RM, Fisher I, Sozzani S, Peiper SC, Horuk R, et al. Regulation of human chemokine receptors CXCR4. Role of phosphorylation in desensitization and internalization. J Biol Chem. 1997;272(45):28726–31. doi: 10.1074/jbc.272.45.28726. [DOI] [PubMed] [Google Scholar]
- 17.Schols D, Este JA, Henson G, De Clercq E. Bicyclams, a class of potent anti-HIV agents, are targeted at the HIV coreceptor Fusin/CXCR-4. Antiviral Res. 1997;35(3):147. doi: 10.1016/s0166-3542(97)00025-9. [DOI] [PubMed] [Google Scholar]
- 18.Donzella GA, Schols D, Lin SW, Este JA, Nagashima KA, Maddon PJ, et al. AMD3100, a small molecule inhibitor of HIV-1 entry via the CXCR4 co-receptor. Nat Med. 1998;4(1):72–7. doi: 10.1038/nm0198-072. [DOI] [PubMed] [Google Scholar]
- 19.Alpert NR, Mulieri LA, Warshaw D. The failing human heart. Cardiovasc Res. 2002;54(1):1–10. doi: 10.1016/s0008-6363(02)00248-1. [DOI] [PubMed] [Google Scholar]
- 20.Bristow MR, Feldman AM. Changes in the receptor-G protein-adenylyl cyclase system in heart failure from various types of heart muscle disease. Basic Res Cardiol. 1992;87(Suppl 1):15–35. doi: 10.1007/978-3-642-72474-9_2. [DOI] [PubMed] [Google Scholar]
- 21.Mann DL. Basic mechanisms of disease progression in the failing heart: the role of excessive adrenergic drive. Prog Cardiovasc Dis. 1998;41(1 Suppl 1):1–8. doi: 10.1016/s0033-0620(98)80025-x. [DOI] [PubMed] [Google Scholar]
- 22.Davies CH, Davia K, Bennett JG, Pepper JR, Poole-Wilson PA, Harding SE. Reduced contraction and altered frequency response of isolated ventricular myocytes from patients with heart failure. Circulation. 1995;92(9):2540–9. doi: 10.1161/01.cir.92.9.2540. [DOI] [PubMed] [Google Scholar]
- 23.Kapadia S, Lee J, Torre-Amione G, Birdsall HH, Ma TS, Mann DL. Tumor necrosis factor-alpha gene and protein expression in adult feline myocardium after endotoxin administration. J Clin Invest. 1995;96(2):1042. doi: 10.1172/JCI118090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van der Poll T, van Deventer SJ. Cytokines and anticytokines in the pathogenesis of sepsis. Infect Dis Clin North Am. 1999;13(2):413–26. doi: 10.1016/s0891-5520(05)70083-0. ix. [DOI] [PubMed] [Google Scholar]
- 25.Gulick T, Chung MK, Pieper SJ, Lange LG, Schreiner GF. Interleukin 1 and tumor necrosis factor inhibit cardiac myocyte {beta}-adrenergic responsiveness. Proc Natl Acad Sci. 1989;86(17):6753–7. doi: 10.1073/pnas.86.17.6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Finkel MS, Hoffman RA, Shen L, Oddis CV, Simmons RL, Hattler BG. Interleukin-6 (IL-6) as a mediator of stunned myocardium. Am J Cardiol. 1993;71(13):1231–2. doi: 10.1016/0002-9149(93)90654-u. [DOI] [PubMed] [Google Scholar]
- 27.McGowan FX, Jr, Takeuchi K, del Nido PJ, Davis PJ, Lancaster JR, Jr, Hattler BG. Myocardial effects of interleukin-2. Transplant Proc. 1994;26(1):209–10. [PubMed] [Google Scholar]
- 28.Kubota T, McTiernan CF, Frye CS, Slawson SE, Lemster BH, Koretsky AP, et al. Dilated cardiomyopathy in transgenic mice with cardiac-specific overexpression of tumor necrosis factor-{alpha} Circ Res. 1997;81(4):627–35. doi: 10.1161/01.res.81.4.627. [DOI] [PubMed] [Google Scholar]
- 29.Bryant D, Becker L, Richardson J, Shelton J, Franco F, Peshock R, et al. Cardiac failure in transgenic mice with myocardial expression of tumor necrosis factor-{alpha} Circulation. 1998;97(14):1375–81. doi: 10.1161/01.cir.97.14.1375. [DOI] [PubMed] [Google Scholar]
- 30.Sivasubramanian N, Coker ML, Kurrelmeyer KM, MacLellan WR, DeMayo FJ, Spinale FG, et al. Left ventricular remodeling in transgenic mice with cardiac restricted overexpression of tumor necrosis factor. Circulation. 2001;104(7):826–31. doi: 10.1161/hc3401.093154. [DOI] [PubMed] [Google Scholar]
- 31.Chung MK, Gulick TS, Rotondo RE, Schreiner GF, Lange LG. Mechanism of cytokine inhibition of beta-adrenergic agonist stimulation of cyclic AMP in rat cardiac myocytes. Impairment of signal transduction. Circ Res. 1990;67(3):753–63. doi: 10.1161/01.res.67.3.753. [DOI] [PubMed] [Google Scholar]
- 32.Kapadia S, Torre-Amione G, Yokoyama T, Mann DL. Soluble TNF binding proteins modulate the negative inotropic properties of TNF-alpha in vitro. Am J Physiol. 1995;268(2 Pt 2):H517–25. doi: 10.1152/ajpheart.1995.268.2.H517. [DOI] [PubMed] [Google Scholar]
- 33.Anker SD, Coats AJ. How to RECOVER from RENAISSANCE? The significance of the results of RECOVER, RENAISSANCE, RENEWAL and ATTACH. Int J Cardiol. 2002;86(2–3):123–30. doi: 10.1016/s0167-5273(02)00470-9. [DOI] [PubMed] [Google Scholar]
- 34.Schecter AD, Berman AB, Taubman MB. Chemokine receptors in vascular smooth muscle. Microcirculation. 2003;10(3–4):265–72. doi: 10.1038/sj.mn.7800192. [DOI] [PubMed] [Google Scholar]
- 35.Seino Y, Ikeda U, Sekiguchi H, Morita M, Konishi K, Kasahara T, et al. Expression of leukocyte chemotactic cytokines in myocardial tissue. Cytokine. 1995;7(3):301. doi: 10.1006/cyto.1995.0037. [DOI] [PubMed] [Google Scholar]
- 36.Shioi T, Matsumori A, Kihara Y, Inoko M, Ono K, Iwanaga Y, et al. Increased expression of interleukin-1b and monocyte chemotactic and activating factor/monocyte chemoattractant protein-1 in the hypertrophied and failing heart with pressure overload. Circ Res. 1997;81(5):664–71. doi: 10.1161/01.res.81.5.664. [DOI] [PubMed] [Google Scholar]
- 37.Behr TM, Wang X, Aiyar N, Coatney RW, Li X, Koster P, et al. Monocyte chemoattractant protein-1 is upregulated in rats with volume-overload congestive heart failure. Circulation. 2000;102(11):1315–22. doi: 10.1161/01.cir.102.11.1315. [DOI] [PubMed] [Google Scholar]
- 38.Damas JK, Gullestad L, Ueland T, Solum NO, Simonsen S, Froland SS, et al. CXC-chemokines, a new group of cytokines in congestive heart failure-possible role of platelets and monocytes. Cardiovasc Res. 2000;45(2):428–36. doi: 10.1016/s0008-6363(99)00262-x. [DOI] [PubMed] [Google Scholar]
- 39.Akekawatchai C, Holland JD, Kochetkova M, Wallace JC, McColl SR. Transactivation of CXCR4 by the insulin-like growth factor-1 receptor (IGF-1R) in human MDA-MB-231 breast cancer epithelial cells. J Biol Chem. 2005;280(48):39701–8. doi: 10.1074/jbc.M509829200. [DOI] [PubMed] [Google Scholar]
- 40.Dwinell MB, Ogawa H, Barrett KE, Kagnoff MF. SDF-1/CXCL12 regulates cAMP production and ion transport in intestinal epithelial cells via CXCR4. Am J Physiol Gastrointest Liver Physiol. 2004;286(5):G844–50. doi: 10.1152/ajpgi.00112.2003. [DOI] [PubMed] [Google Scholar]
- 41.Yopp AC, Ochando JC, Mao M, Ledgerwood L, Ding Y, Bromberg JS. Sphingosine 1-phosphate receptors regulate chemokine-driven transendothelial migration of lymph node but not splenic T cells. J Immunol. 2005;175(5):2913–24. doi: 10.4049/jimmunol.175.5.2913. [DOI] [PubMed] [Google Scholar]
- 42.Dziembowska M, Tham TN, Lau P, Vitry S, Lazarini F, Dubois-Dalcq M. A role for CXCR4 signaling in survival and migration of neural and oligodendrocyte precursors. Glia. 2005;50(3):258–69. doi: 10.1002/glia.20170. [DOI] [PubMed] [Google Scholar]
- 43.Gudjonsson T, Rahko PS. Relation of “inotropic reserve” to functional capacity in heart failure secondary to ischemic or nonischemic cardiomyopathy. Am J Cardiol. 2002;89(9):1057–61. doi: 10.1016/s0002-9149(02)02275-0. [DOI] [PubMed] [Google Scholar]
- 44.Singer II, Scott S, Kawka DW, Chin J, Daugherty BL, DeMartino JA, et al. CCR5, CXCR4, and CD4 are clustered and closely apposed on microvilli of human macrophages and T cells. J Virol. 2001;75(8):3779–90. doi: 10.1128/JVI.75.8.3779-3790.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dwinell MB, Ogawa H, Barrett KE, Kagnoff MF. SDF-1/CXCL12 regulates cAMP production and ion transport in intestinal epithelial cells via CXCR4. Am J Physiol Gastrointest Liver Physiol. 2004;286(5):G844–50. doi: 10.1152/ajpgi.00112.2003. [DOI] [PubMed] [Google Scholar]
- 46.Limbourg FP, Drexler H. Bone marrow stem cells for myocardial infarction: effector or mediator? Circ Res. 2005;96(1):6–8. doi: 10.1161/01.RES.0000153667.26414.10. [DOI] [PubMed] [Google Scholar]
- 47.Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, et al. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003;114(6):763. doi: 10.1016/s0092-8674(03)00687-1. [DOI] [PubMed] [Google Scholar]
- 48.Kocher AA, Schuster MD, Szabolcs MJ, Takuma S, Burkhoff D, Wang J, et al. Neovascularization of ischemic myocardium by human bone-marrow-derived angioblasts prevents cardiomyocyte apoptosis, reduces remodeling and improves cardiac function. Nat Med. 2001;7(4):430–6. doi: 10.1038/86498. [DOI] [PubMed] [Google Scholar]
- 49.Hughes S. Cardiac stem cells. J Pathol. 2002;197(4):468–78. doi: 10.1002/path.1159. [DOI] [PubMed] [Google Scholar]
- 50.Britten MB, Abolmaali ND, Assmus B, Lehmann R, Honold J, Schmitt J, et al. Infarct remodeling after intracoronary progenitor cell treatment in patients with acute myocardial infarction (TOPCARE-AMI) mechanistic insights from serial contrast-enhanced magnetic resonance imaging. Circulation. 2003;108(18):2212–8. doi: 10.1161/01.CIR.0000095788.78169.AF. [DOI] [PubMed] [Google Scholar]
- 51.Jo D-Y, Rafii S, Hamada T, Moore MAS. Chemotaxis of primitive hematopoietic cells in response to stromal cell-derived factor-1. J Clin Invest. 2000;105(1):101–11. doi: 10.1172/JCI7954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abbott JD, Huang Y, Liu D, Hickey R, Krause DS, Giordano FJ. Stromal cell-derived factor-1alpha plays a critical role in stem cell recruitment to the heart after myocardial infarction but is not sufficient to induce homing in the absence of injury. Circulation. 2004;110(21):3300–5. doi: 10.1161/01.CIR.0000147780.30124.CF. [DOI] [PubMed] [Google Scholar]