

Abstract
The anion nitrite (NO2−) constitutes a biochemical reservoir for nitric oxide (NO). Nitrite reduction to NO may be catalyzed by hemoglobin, myoglobin or other metal-containing enzymes and occurs at increasing rates under conditions of physiologic hypoxia or ischemia. A number of laboratories have now demonstrated in animal models the ability of nitrite to provide potent cytoprotection following focal ischemia-reperfusion (IR) injury of the heart, liver, brain, and kidney. While the mechanism of nitrite-mediated cytoprotection remains to be fully characterized, the release of nitrite-derived NO following IR appears to be central to this mechanism. The evidence of nitrite-mediated cytoprotection against IR injury in multiple animal models opens the door to potential therapeutic opportunities in human disease. Here we review the mechanisms for nitrite formation in blood and tissue, its metabolic equilibrium with NO, nitrate, and NO-modified proteins, the evidence supporting nitrite-mediated cytoprotection, and the potential mechanisms driving cytoprotection, and we explore the opportunities for the therapeutic application of nitrite for human disease.
Keywords: nitric oxide, ischemia, reperfusion
1. Introduction
The anion nitrite was previously considered physiologically inert; a mere end product of nitric oxide (NO) metabolism [1]. Increasing evidence now suggests that nitrite lies at the center of a complicated hypoxia-sensitive redox and signaling chemistry [2, 3]. Under conditions of hypoxia and ischemia (low oxygen tension and acidosis), nitrite reduction to NO may be catalyzed by deoxyhemoglobin and deoxymyoglobin [4–9] and potentially other proteins with heme prosthetic groups, such as xanthine oxidoreductase, components of the mitochondrial electron transport chain, and nitric oxide synthase [10–14]. Thus NO generated by NOS (NOS) during normoxia may be chemically stored in a nitrite reservoir and re-generated during hypoxia and ischemia when NOS function is limited. In this context, nitrite can be considered an “ischemic NO buffer,” maintaining hypoxic-ischemic NO homeostasis. Interestingly, the different nitrite reductase “enzyme” systems are operant along a range of physiological and pathological hypoxia, with hemoglobin reducing nitrite at an oxygen tension from 60 mmHg down to 20 mmHg, myoglobin active below 4 mm Hg, and xanthine oxidase and acidic reduction reducing nitrite at zero oxygen and low pH [7, 9, 13]. This allows for graded nitrite reduction to NO along the circulating and metabolic oxygen gradient.
The numerous reports of NO-mediated cytoprotection following ischemia-reperfusion (IR) [15–19] injury have led to an exploration of the role for nitrite in this setting. Nitrite has been demonstrated to provide cytoprotection in a number of animal models of focal IR injury [20–25]. In studies attempting to investigate the mechanism of this protection, formation of nitrite-derived NO appears critical. Although the mechanism whereby nitrite mediates its protection against IR injury is currently unknown, the proposed putative mechanisms parallel hypotheses explaining NO-mediated cytoprotection [15, 19]. In addition to its ability to produce NO, nitrite may signal via NO-independent pathways, such as via the intermediacy of nitrous acid or N2O3 formation, opening the door to unique post-translational cytoprotective mechanisms for nitrite [26, 27].
This review will summarize what is presently known about the sources and mechanisms of nitrite formation in blood and tissue and its chemical equilibrium with NO and nitrate. We will summarize the existing studies demonstrating nitrite-mediated cytoprotection in the setting of IR injury and suggest potential mechanisms for this effect. Finally, we will discuss the potential therapeutic uses for nitrite and summarize human clinical trials being planned to test the efficacy of nitrite in the prevention of IR injury.
2. Nitrite as a biological reservoir of NO
2.1. Nitrite concentrations in plasma and the red blood cell
As depicted in Figure 1, nitrite exists in the center of a set of oxidation-reduction reactions and can be reduced to the highly biologically active free radical NO or oxidized to the abundant anion nitrate [3]. Plasma nitrite levels appear to be conserved across a number of mammalian species [28]. In humans, typical plasma nitrite levels range from 121 to 350 nM and lower levels have been associated with endothelial dysfunction and increasing coronary artery disease risk factors [1, 28–30].
Figure 1. The NO-nitrite-nitrate pool.
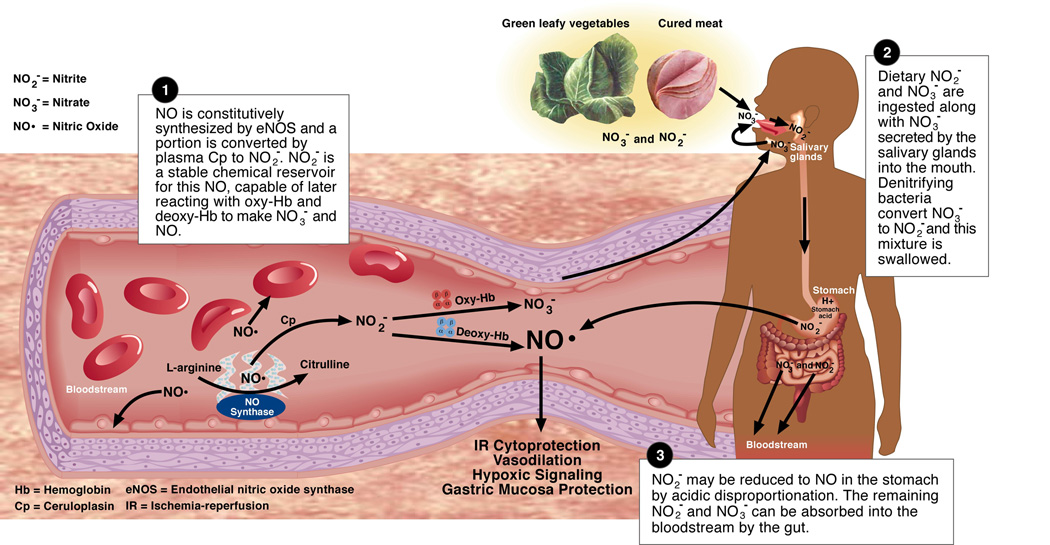
Nitrite (NO2−) maintains a chemical equilibrium with its reduction product NO and its oxidation product nitrate (NO3−). These reactions take place mainly in the bloodstream and the GI tract.
Recent evidence suggests that nitrite is present within erythrocytes (288 ± 47 nM) in concentrations roughly double those found in plasma (121 ± 9) [30]. The expected diffusion equilibrium between plasma and intracellular nitrite concentrations cannot explain this disparity suggesting an active process of transporting nitrite into the erythrocyte and a relative (and surprising) stability within the erythrocyte. In horse erythrocytes the anion exchanger AE1 (Band III) [31] was shown to be involved in nitrite uptake, but later studies in human and pig cells did not support this mechanism [32, 33]. Our group has recently discovered that nitrite binds to methemoglobin as a ferric-nitrite complex (Fe(III)-NO2−) under physiological concentrations of nitrite, suggesting a mechanism for the observed high erythrocyte nitrite levels (Basu et al., manuscript in review). At present, the mechanisms regulating nitrite compartmentalization in plasma versus the erythrocyte are still unclear. The implication of this finding is that measurement of only plasma nitrite levels may fail to reflect the full extent of nitrite bioavailability in the blood.
2.2. Nitrite uptake in the gastrointestinal tract and de-novo formation in blood
Although nitrite is present in food (e.g. cured meats), an equally important source of ingested nitrite in the diet originates from nitrate. Nitrate is actively concentrated in saliva and reduced to nitrite by bacteria in the posterior crypts of the human tongue [34]. Nitrite is also formed by the oxidation of NO catalyzed by ceruloplasmin [35]. We discuss below the contributions of dietary intake and NO oxidation to plasma nitrite.
Dietary pathway
In a recent overview, the range of daily dietary nitrate intake were between 53–350 mg/day and for nitrite between 0– 20 mg/day [36]. In the USA, 80% of dietary nitrate originates from vegetables, less than 15% from cured meats, and 5% from other sources [37]. This study calculated an ingestion of 106 mg/day of nitrate and 13 mg/day of nitrite for the average US resident in 1972 [37]. However, Fassett [38] has reported a daily nitrate intake of 300 mg. In the UK, daily dietary nitrate exposure in 1997 was estimated to be 52 mg, approximately 70% of which was derived from vegetables [39]. However, nitrate intake was estimated to be almost double for people consuming the whole range of vegetables commonly eaten in the UK [39]. Similar values of approximately 77 mg/day were calculated from a survey of 10,000 people in Finland [40]. Nitrite intake in this study was estimated around 5 mg/day [40].
Nitrate is converted to nitrite by oral flora bacteria which, in contrast to human tissue, possess nitrate reductase enzymes [34]. Two-thirds of the nitrite in the diet entering the stomach derives from salivary nitrate reduction and slightly less than one-third comes from cured meats [37]. Ingested nitrate not converted to nitrite is quickly taken up by the gut into the bloodstream. Nitrate is present in substantially larger concentrations in human plasma than nitrite: approximately 25–35 uM [1, 29] vs. 150–350 nM [1, 28–30]. Twenty-five percent of circulating nitrate is concentrated and secreted by the salivary glands into saliva at concentrations of 2–8 mM [34]. Approximately 20% of this secreted salivary nitrate is converted to nitrite by bacteria [34, 41]. After an average continental breakfast, salivary nitrate and nitrite levels were 74 ± 50 ppm (1.19 ± 0.81 mM) and 9 ± 5 ppm (0.20 ± 0.11 mM) respectively [34]. In this way 5% of the ingested nitrate gets converted to nitrite, making it a significant source of nitrite relative to directly ingested nitrite.
Since acidic disproportionation is a known mechanism of nitrite reduction (to NO) [42], the use of antacids such as histamine-2 receptor antagonists or proton pump inhibitors may result in less NO production in the stomach [43]. Experimental dietary restriction of nitrate and nitrite in rats reportedly did not lower plasma nitrite levels but did appear to deplete nitrite in the tissues [26]. This result has only been partially replicated in our lab in mice, where we have observed a decrease not only in tissue nitrite levels but also a 50% reduction in plasma nitrite levels following dietary nitrate and nitrite restriction (Raat, NJH et al, unpublished results).
The clinical significance of dietary nitrite has recently been investigated. Ingested nitrite is reduced to NO in the stomach and appears to play a central role in host defense by killing stomach bacteria, and in mucosal cytoprotection by increasing local mucus production, increasing stomach blood flow and conferring direct cellular cytoprotection [41, 44]. The clinical relevance of this mechanism was recently demonstrated in critically ill, intubated patients in whom gastric formation of nitric oxide was almost completely abolished and could be restored by intra-gastric infusion of a nitrite solution [45]. Nitrate supplementation of drinking water resulted in protection against non-steroidal anti-inflammatory drug-induced ulceration in an experimental rat model [44]. Lundberg and colleagues also recently demonstrated that dietary nitrate ingestion is associated with a direct reduction in diastolic and mean arterial blood pressure and an increase in plasma nitrate and nitrite levels [46]. Since the Mediterranean diet has been shown to modulate cardiovascular risk and is an especially robust source of nitrate and therefore nitrite, this group has speculated that the higher nitrate content in this diet might be responsible for its favorable effects [3, 46, 47]. This effect of nitrate and nitrite on gastric blood flow and systemic blood pressure is consistent with the newly discovered physiological and pharmacological vasodilatory effects of nitrite [2, 6, 48].
De novo nitrite formation in blood
Traditionally auto-oxidation of NO was thought to be the major route of nitrite production. In blood however, the third order overall reaction of NO with oxygen (k = 106 M−2s−1), which forms nitrite, is kinetically unfavorable in comparison to the second order reaction of NO with hemoglobin (k = 107M−1s−1), which yields nitrate [49, 50]. These considerations suggested the existence of metal-based enzymatic pathways for nitrite formation from NO. Indeed, we recently found that the plasma protein ceruloplasmin functions as an NO oxidase, catalyzing the one electron oxidation of NO to NO+, which is hydrated to form nitrite in plasma [35]. In these studies, depletion of ceruloplasmin from plasma was shown to decrease plasma nitrite levels after the addition of NO, while supplementation of blood with ceruloplasmin increased plasma nitrite yield after NO addition. Ceruloplasmin knockout mice had significantly lower basal plasma nitrite levels than wild-type mice (0.53 ± 0.03 vs. 0.79 ± 0.08 µM) as well as decreased plasma NO oxidase activity. The basal plasma nitrite levels in mice correlated to their plasma NO oxidase activity (R²=0.62, p=0.01) demonstrating that ceruloplasmin-dependent NO oxidation is responsible for a substantial portion of basal nitrite levels [35].
Consistent with a biological function of nitrite formed from ceruloplasmin-dependent oxidation of NO, ceruloplasmin knockout mice, after being subjected to hepatic IR, sustained significantly more liver injury than wild type mice as measured by an increase in the levels of plasma ALT. While this increased susceptibility could be due to a number of factors including the iron overload present in these mice, treatment of the ceruloplasmin deficient mice with intraperitoneal nitrite during ischemia restored ALT levels to that of wild type animals undergoing similar liver IR suggesting that the increased injury was due to the decreased basal nitrite concentration [35].
Ceruloplasmin, being an acute phase reactant, is upregulated in a number of pathological situations, such as hypoxia and IR injury [51, 52]. An increased ceruloplasmin concentration may be particularly important after IR or ischemic preconditioning, when NOS protein expression and activity is also increased. In the presence of this increased NO synthesis, increased ceruloplasmin dependent NO oxidation could be essential in converting the NO generated into a stable endocrine storage form-- namely nitrite.
2.3. Mechanisms of nitrite reduction to NO
Nitrite reduction to NO occurs by non-enzymatic acidic disproportionation [53, 54] and enzymatic reduction by xanthine oxidoreductase [13]. However, these mechanisms require low pH and extremely low oxygen tensions and are thus not likely to play a significant role in NO formation under more physiologic conditions. This is because, in the presence of oxygen xanthine oxidoreductase generates superoxide, which will consume any NO formed from the enzyme in a diffusion-limited reaction forming peroxynitrite.
An alternative mechanism was suggested by human physiological studies conducted over the last six years at the NIH. In the human circulation, we consistently measured artery-to-vein gradients in plasma nitrite, suggesting that this anion was being metabolized during A–V transit [48]. Further supporting this hypothesis, 80 ppm NO gas inhalation was associated with a measurable increase in peripheral blood flow despite NOS inhibition, and was associated with a significant increase in plasma nitrite [55]. To determine whether nitrite exhibited vasodilatory effects in the human circulation we infused nitrite at near-physiological concentrations into the human forearm [6]. We observed nitrite-dependent vasodilation during nitrite infusions into the resting human forearm which was augmented during exercise stress [6]. Increases in nitrite in blood correlated with the formation of NO-modified hemoglobin both in vivo and in vitro [6, 56]. Most importantly, the amount of NO-hemoglobin (mostly iron-nitrosylated hemoglobin; Fe(II)-NO) that formed during A–V transit increased as oxygen saturation of the hemoglobin decreased, suggesting an association between NO formation and hemoglobin deoxygenation. Investigations of these findings led to the hypothesis that nitrite reduction by deoxyhemoglobin, previously characterized by Brooks and colleagues [57] and later by Doyle and colleagues [8], could occur under physiologic conditions according to equation 1.
| (1) |
Some of the NO formed from this reaction would then be trapped on vicinal hemes of unreacted deoxyhemoglobin to form iron-nitrosyl hemoglobin (equation 2) which was measured as a ‘dosimeter” of the reaction.
| (2) |
While the oxygen, proton, and CO2 binding allosteric properties of hemoglobin have been well characterized for over 100 years, the allosteric nature of the reaction with nitrite had not been previously characterized. Kinetic analysis of reaction (1) has demonstrated that the rate of deoxyhemoglobin-dependent nitrite reduction is determined by the allosterically regulated quaternary structural conformation of hemoglobin [4, 7] such that the maximal rate of nitrite reduction and NO production occurs around the p50 of hemoglobin, when 50% of the heme sites are ligated with oxygen [7]. This phenomenon is determined by two opposing processes: 1) the ability of nitrite to bind T (deoxy) state hemoglobin (whose iron center is oxygen free) and 2) the lower heme reduction potential of R (oxy) state hemoglobin (making it a better electron donor) [4, 7].
Crawford and colleagues used an in vitro aortic ring system to simultaneously measure vessel tone and oxygen concentration in the presence of nitrite and red blood cells [6, 56]. In the absence of red blood cells, the addition of nitrite to pre-constricted aortic rings caused vasodilation only at high (greater than 10 uM) concentrations, consistent with previous data [58]. However, in the presence of deoxygenated red blood cells (0.3% hematocrit) physiological concentrations of nitrite (< 1uM) mediated vasodilation of aortic rings. The onset of significant vasodilation began around the p50 of hemoglobin, consistent with kinetic observations of nitrite reduction. The observation that iron-nitrosyl hemoglobin and S-nitroso-hemoglobin can be produced with nitrite infusions into the brachial artery of an exercising forearm [6] (where low oxygen tensions and acidosis were present) provides additional confirmation that nitrite is in fact being converted to NO within the circulation.
3. Nitrite mediated cytoprotection following IR injury
In the context of tissue ischemia, nitrite given immediately before or during reperfusion has been demonstrated to protect against IR injury in a number of animal models [3, 20–23] (Table 1). These studies examined focal (single organ) IR injury, generally modeled by the occlusion of the vascular supply of an organ of interest for a period of time, then releasing the occlusion to permit reperfusion. The exception to this is a study which tested the effects of nitrite in a rat Langendorff isolated rat heart model subjected to total heart ischemia and reperfusion [23]. Nitrite was given prior ischemia in only 2 studies [22, 23] and in the remainder was given midway through ischemia [20, 59] or just before reperfusion [21, 25].
Table 1. Animal models of IR injury testing nitrite therapy.
A summary of seven studies that evaluate the potential therapeutic role for nitrite after focal IR injury.
Abbreviations: H&E = hematoxylin & eosin staining, TUNEL = terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling, TTC = 2,3,5-triphenyltetrazolium chloride, AST = aspartate transaminase, ALT = alanine transaminase, MPO = myeloperoxidase, ATP = adenosine triphosphate, BUN = blood urea nitrogen, PTIO = 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazole-1-oxyl 3-oxide, L-NAME = N -nitro-L-arginine methyl ester, (−) BOF-4272 = sodium 8-(3-methoxy-4-phenylsulfinylphenyl)pyrazolo[1,5- ]-1,3,5-triazine-4-olate monohydrate, eNOS (−/−) = endothelial nitric oxide synthase knockout mice, HO-1 (−/−) = heme oxygenase-1 knockout mice, ODQ = [1H-[1,2,4]Oxadiazole [4,3-a]quinoxalin-1-one] MRI = magnetic resonance imaging.
| First Author Name and Reference | Year of Publication | Animal Model Used | Placebo Used | Dose of Nitrite, Route and Timing of Administration | Timing of Ischemia and Reperfusion | Outcome Measures of Cytoprotection | Inhibitors Used |
|---|---|---|---|---|---|---|---|
| Web, A[23] | 2004 | Rat Isolated Heart IR | saline | 0.025 - 2.5 uM constant infusion for 15min prior to ischemia onset | 60min ischemia and 30min reperfusion | p-nitroblue tetrazolium; left ventricular developed pressure and diastolic blood pressure | PTIO, L-NAME, allopurinol, BOF-4272 |
| Duranski, MR[20] | 2005 | Mouse Liver and Heart IR | nitrate or saline | 2.4 - 1920 nmol intraperitoneal (liver) or intraventricular (heart) midway through the ischemic time | Liver: 45min hepatic artery/portal vein occlusion, 5th reperfusion; Heart: 30min left main coronary artery occlusion, 24h reperfusion | H&E and TUNEL (liver), TTC (heart); AST/ALT (liver) | PTIO, L-NAME, ODQ, ZnDBGG, eNOS(−/−), HO-1(−/−) |
| Lu, P[22] | 2005 | Rat Liver IR | none (sham) | All rats given 2 mg/kg (29 nmol/g) nitrite | 40min hepatic artery/portal vein occlusion, 3h (labs) or 7d (path) reperfusion | H&E; 7d mortality; ALT MPO and ATP | PTIO, L-NAME, allopurinol |
| Basireddy, M [61] | 2006 | Rat Kidney IR | nitrate or saline | 1.2 nmol/g intraperitoneal prior to nephrectomy or midway during ischemia; 0.12 - 12 nmol/g intravenous prior to nephrectomy | R nephrectomy and 45min occlusion of L renal pedicle, 24-48h (labs) or 6d (path) reperfusion | H&E; BUN and creatinine | None - No treatment effect observed |
| Jung, KH [21] | 2006 | Rat Brain IR | nitrate | 48 - 48,000 nmol intravenous at the time of reperfusion | 90min occlusion of middle cerebral artery, 3h (LDF) or 24h (TTC) or 7d (neurological testing) reperfusion | TTC; laser doppler flow; neurological testing | PTIO |
| Tripatra, P [25] | 2007 | Rat Kidney IR | saline | 0.12 nmol/g topically applied to the kidneys or given intravenous 1 min prior to reperfusion | 60min bilateral renal ischemia, 6h reperfusion | H&E; BUN, creatinine, creatinine clearance, fractional urinary sodium excretion, AST | PTIO, allopurinol |
| Gonzalez, FM [24] | 2007 (not published) | Canine Heart IR | saline | 0.20 µmol/min/kg for 20 min followed by 0.17 µmol/min/kg for 40 min iv OR 0.20 µmol/min/kg for 5 min ending at reperfusion | 2h left anterior descending occlusion, 6h reperfusion | TTC, TUNEL; microspheres and MRI | None |
When compared to saline or nitrate controls, nitrite provides consistent cytoprotection in liver [20, 22], heart [3, 20, 23], brain [21] and kidney [25]. All of these studies save one examining nitrite in a canine myocardial IR model [3, 24] have been published in peer reviewed journals. In one study, nitrite given intraperitoneal or intravenously prior to or during left renal IR did not confer cytoprotection based on histopathology scoring and changes in BUN and creatinine [59]. This finding was recently corroborated by another group who did however note substantial cytoprotection when the nitrite was topically applied directly onto the kidney prior to reperfusion [25]. This group has hypothesized that the route of administration and the effective dose in the organ at the time of reperfusion may be the cause for this disparity.
In the studies where cytoprotection has been noted, it has been characterized by multiple methods including the use of direct pathologic measurements (hematoxylin-eosin staining [22, 25], 2,3,5-triphenyltetrazolium chloride (TTC) staining [20, 21] or p-nitroblue tetrazolium [23] staining), through measures of cardiac [3, 20, 23], brain [21] and kidney [25] physiological function or by quantifying plasma markers of tissue injury [20, 22, 23, 25]. All measures showed consistent cytoprotection by nitrite with more than a fifty percent reduction in infarction volume of all organs evaluated. In the two studies where a thorough dose effect curve was constructed, the “optimal cytoprotective dose” was remarkably similar—approximately 48 nmol in an adult mouse [20] or 480 nmol in the 10-fold larger adult rat [21]. The blood concentration associated with optimal protection was approximately 10 uM but even levels less than 200 nM were associated with protection [20].
In summary, these studies suggest that nitrite given prior to reperfusion prevents IR injury in heart, liver and brain, while a conclusion on the effect on kidney remains uncertain. To date, there is no evidence of nitrite protection against global IR injury, such as occurs in cardiac arrest or cardiopulmonary bypass.
3.1 Potential mechanisms of cytoprotection mediated by nitrite-derived NO
The mechanism of nitrite-mediated cytoprotection in these animal studies remains unclear but the leading hypothesis is that nitrite-derived NO mediates the protective effect. Measurement of NO production in vivo is difficult, yet a number of findings in the provide evidence that NO is in fact generated by nitrite reduction and mediates the cytoprotection observed. All the aforementioned published animal studies of nitrite cytoprotection have demonstrated a loss of protection when animals were pretreated with the NO scavenger 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazole-1-oxyl 3-oxide (PTIO) [20–23, 25] suggesting the importance of NO in the mechanism of cytoprotection. In a rat model of kidney IR, Okamoto and colleagues demonstrated using electron paramagnetic resonance (EPR) spectroscopy and N-15 labeled nitrite that the NO formed in blood is nitrite-derived [60]. Nitrite therapy was associated with increases in cyclic guanosine monophosphate (cGMP) levels [21] and the inhibition of soluble guanylate cyclase (sGC) using 1H-[1,2,4] oxadiazole [4,3-a]quinoxalin-1-one (ODQ) abolished cytoprotection [20]. The Zweier lab has also measured nitrite-derived xanthine oxidase catalyzed NO formation by EPR, chemiluminescence and using an electrochemical detector and demonstrated increasing NO formation under acidic conditions [13]. In a Langendorff model of myocardial ischemia, this group demonstrated the formation of iron-nitrosyls (heme-NO) in myocardium under ischemic conditions which was associated with increasing cGMP levels [61]. Pretreatment with inhibitors of NOS [20, 22, 23] and the use of endothelial NOS (eNOS) knockout mice [20] did not inhibit cytoprotection, proving that the nitrite effect is NOS-independent. Additional studies using heme oxygenase-1 inhibitors and knock-out mice revealed that the nitrite effect is independent of heme oxygenase-1 [20].
The pathway by which nitrite forms NO in hypoxic tissue remains to be determined. Two groups suggest in their studies the involvement of xanthine oxidoreductase in the reduction of nitrite to NO on the basis of reduced efficacy after treatments with allopurinol, a xanthine oxidase inhibitor [22, 23, 25]. As described above, xanthine oxidoreductase requires significant hypoxia and acidosis (ie ischemic conditions) to reduce nitrite to NO. Considering these requirements, xanthine oxidoreductase is unlikely to contribute to the nitrite/NO mediated regulation of physiological blood flow, but during prolonged ischemia the enzyme may be an important catalytic source of nitrite-derived NO [12, 13]. It remains to be determined if deoxyhemoglobin [2, 4–7, 62–64] plays a role in nitrite-mediated NO formation during IR. The fact that nitrite was protective in a hemoglobin-free buffer perfused isolated heart IR model suggests that hemoglobin is not necessary. We have considered the possibility that in the heart, myoglobin can serve this function and have recently demonstrated that deoxymyoglobin has nitrite reductase activity which is faster than hemoglobin and can modulate mitochondrial respiration [7, 9]. Studies with the myoglobin (−/−) mouse model will be necessary to answer this question definitively. Other potential mechanisms of tissue dependent-nitrite reduction include cytochrome c oxidase or ubiquinol in the mitochondrion [11, 65], eNOS [14] and cytochrome P450 [26].
A number of mechanisms have been advanced to explain the cytoprotective effects of NO following IR injury (Figure 2). Since a thorough discussion of NO-mediated cytoprotection against IR injury is beyond the scope of this review, we refer the reader to recent reviews of the subject [15, 19, 66] and provide a summary of the major theories. The timescale of these events, which is in the order of seconds to minutes, is sufficiently rapid to explain the cytoprotection against IR injury seen in the recent reports utilizing nitrite [20, 23].
Figure 2. Mechanisms of nitrite-derived NO mediated cytoprotection.
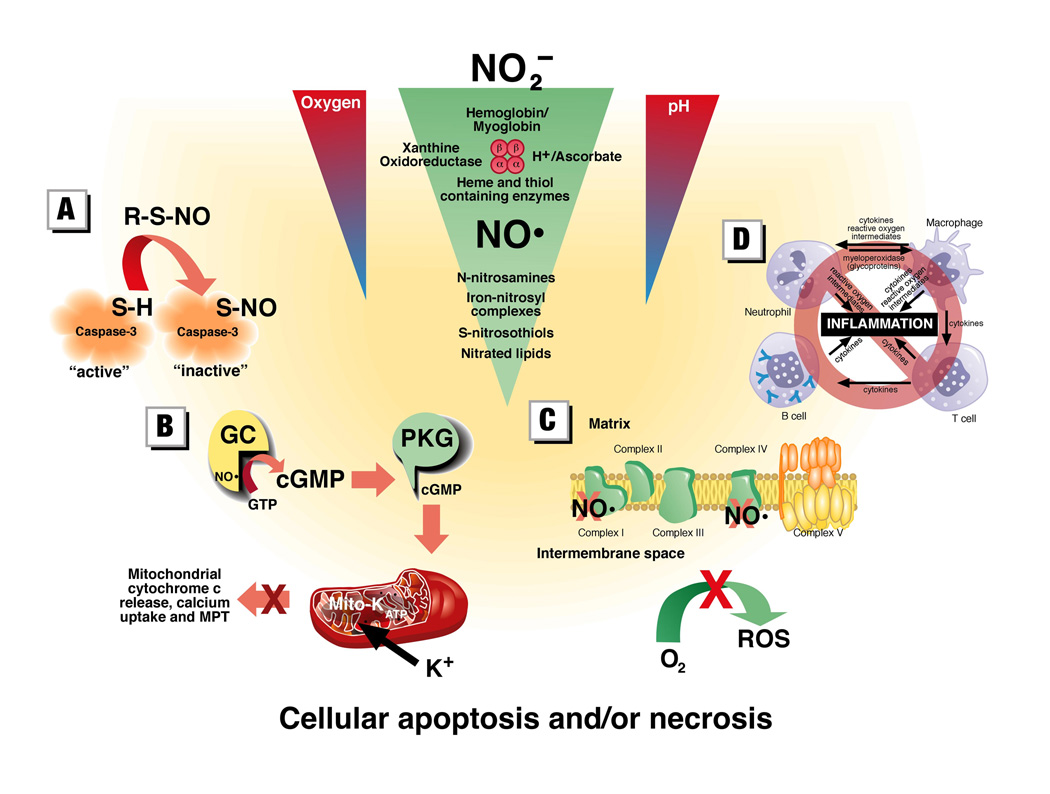
Nitrite may be reduced to NO by a variety of metal containing enzyme systems and the NO or the NO-modified proteins and lipids may in turn mediate cytoprotection against IR injury through any a variety of mechanisms. These mechanisms include: (A) S-nitrosation (and potentially N-nitrosation) of critical proteins involved in the signaling of apoptosis; (B) opening of mitochondrial KATP channels via the classic NO – cyclic guanidine monophosphate (cGMP) – protein kinase G (PKG) pathway which protects against cell death by preventing mitochondrial cytochrome c release, calcium overload and the opening of the mitochondrial permeability transition (MPT) pore; (C) Inhibition of complex I which reduces the direct production of reactive oxygen species (ROS) or the inhibition of complex IV which slows mitochondrial respiration and oxygen depletion during ischemia and thereby may minimize ROS production; (D) NO and NO-modified proteins have been associated with a variety of anti-inflammatory effects which minimize tissue injury after IR.
Pharmacologic agents purported to be specific for the opening or closing of mitochondrial ATP-dependent potassium channels (KATP channels) have been demonstrated to recreate or prevent the protection noted from ischemic preconditioning. Although this response has been characterized repeatedly, the actual identification of a protein channel has yet to occur and the specificity of the drugs activating or inhibiting their opening has been debated, leading to controversy regarding this mechanism. This topic was recently critically reviewed [67] and will therefore only be summarized. NO dependent activation of protein kinase G through the soluble guanylate cyclase-cGMP pathway has been shown to open mitochondrial KATP channels in at least two separate in vitro cardiomyocyte models of NO-mediated protection from IR injury [16, 68]. Opening of the mitochondrial KATP channels appears to result in a mild increase in reactive oxygen species generation and a modest dose-dependent depolarization of the mitochondria, both of which are believed to signal anti-apoptotic adaptations [16, 69, 70]. Opening of KATP channels has been associated with reduced calcium accumulation in the mitochondria [71] and prevention of the opening of the mitochondrial permeability transition pore [72, 73] and subsequent apoptosis. In isolated mitochondria, Korge and colleagues demonstrated mitochondrial KATP channel activation not only reduced calcium accumulation but also prevented cytochrome c loss from the intermembrane space, thereby preventing apoptosis [74].
We have recently demonstrated that during hypoxia nitrite can enter respiring cardiomyocytes and be reduced to NO by endogenous myoglobin to inhibit mitochondrial respiration [9], potentially by binding cytochrome c oxidase [75–79]. This nitrite and myoglobin dependent inhibition of respiration is concentration dependent, with near physiological levels of nitrite (2.5 uM) resulting in significant inhibition of myocytic respiration [9]. Binding of NO to complex IV, which is not associated directly with electron leaking during ischemia, is not known to reduce the burst of reactive oxygen species upon reoxygenation and indeed may increase this production by backing up electrons along the transport chain [80, 81]. However, the overall reversible slowing in the rate of mitochondrial respiration during ischemia and then reoxygenation may permit for preserved oxygen gradients during ischemia and a more controlled resumption of respiration during reoxygenation thereby having a beneficial effect in terms of minimizing free radical injury [82–84].
Tripatara and colleagues recently demonstrated lower levels of nitrotyrosine staining in nitrite-treated mice who clinically had cytoprotection against renal IR [25]. Although it appears paradoxical to have less nitrated tissue in nitrite treated animals, the authors hypothesized that nitrite-generated NO competes for electrons needed to form superoxide, thereby limiting formation of damaging reactive nitrogen species such as peroxynitrite, which is formed by reaction of NO with superoxide [85]. Nitrotyrosine, however, is an imperfect marker of peroxynitrite and endogenously formed peroxynitrite appears to have multiple roles in vivo including acting as an oxidant capable of inflicting IR injury, as a regulator of important physiologic functions and as a potential intermediate in cytoprotective S-nitrosation of thiols (reviewed in [86–88]).
Inhaled NO [89] and NO donors [90, 91] have been associated with decreased IR injury in murine and canine myocardial IR models and isolated rat hearts, respectively. In these studies, improved heart contractile function was correlated with decreased polymorphonuclear leukocyte (PMN) binding to IR damaged endothelium [90] and decreased myocardial PMN infiltration [89]. In an isolated heart model, IR injury was only noted when PMNs were added to the perfusate [91] strongly suggesting that inflammation is a key mediator of IR injury and NO cytoprotection. However, controversy remains over whether the inflammation noted with IR and prevented by NO is a cause or effect of necrosis/apoptosis induced by other means.
3.2 Other mechanisms of nitrite signaling and cytoprotection
It has been proposed that nitrite may mediate signaling, including the S-nitrosation of proteins and the regulation of gene expression, independent of intermediate NO production [26]; but the applicability of this NO-independent signaling to IR cytoprotection remains to be proven. Nitrite may mediate signaling by the post-translational iron nitrosylation of metal centers [4, 7, 92, 93], S-nitrosation of protein thiols [20, 26, 92] and N-nitrosation of protein amines [92, 93]. S-nitrosation and iron-nitrosylation have been associated with nitrite dependent cytoprotection following IR [20], but a cause and effect relationship remains unproven. It is unclear precisely which proteins are modified by nitrite or whether NO is a required intermediate, but modification of key proteins in the apoptotic cascade may be a means by which nitrite exerts its cytoprotective effect.
This inhibition of apoptosis by NO-dependent nitrosation has been most clearly demonstrated in the case of S-nitrosation. An example of one such modified protein is caspase 3, which is activated by the upstream caspases 8 and 9, and is a key pathway by which cells induce apoptosis [94]. S-nitrosation of caspase-3 has been linked to its inactivation, which would result in the inhibition of apoptotic cell death [95, 96]. S-nitrosation of the L-type calcium channel has been shown to reduce cytosolic calcium and reduce IR injury in an isolated mouse heart model; this has been mechanistically associated with eNOS activation and NOS-dependent gender differences in IR injury [97]. Whether nitrite serves as an intermediate between NO synthases, NO and IR cytoprotection in the latter study will require further investigation.
Small amounts of reactive oxygen species (ROS) generation, particularly in mitochondria, have been determined to be a necessary component of mitochondrial signaling in cytoprotection [70, 98–100]. However, the large burst of ROS generated after reperfusion is believed to be a major mechanism whereby cellular injury and necrosis occurs [101, 102]. It has been suggested that a transient, reversible inhibition of the mitochondrial electron transport chain (ETC) could minimize this injury [83, 84]. S-nitrosation and inhibition of complex I of the ETC has been documented to occur with S-nitrosoglutathione [103, 104]. Although in these reports the inhibition of complex I appeared to result in more ROS formation under basal non-ischemic conditions [103, 104], complex I inhibition in the setting of IR injury, using rotenone [105] amobarbital [106, 107] and S-nitroso-2-mercaptopropionyl-glycine [84] has been associated with cytoprotection. We have recently found that nitrite can potently S-nitrosate and inhibit complex I in vitro and in vivo; this inhibition resulted in reduced ROS on reperfusion, decreased calcium influx and a reduction in cytochrome c release. This mechanism is associated with cytoprotection after IR injury in both the heart and the liver (Shiva, S, et al., under review; and [3]). Complex I inhibition may also prevent opening of the mitochondrial permeability transition pore which often precedes apoptosis [84, 108] and appears to make mitochondria more tolerant to calcium overload [84].
4. Nitrite Therapeutics
To date, no human clinical trials of nitrite therapy for organ IR have been conducted; however, the pre-clinical data from multiple independent laboratories, in four tissues (heart, liver, brain, and kidney) and in four species (mouse, rat, dog, primate) suggests promise for human therapeutic application. The challenges to developing reperfusion therapeutics have recently been highlighted by Bolli and colleagues [109]. These include reproducibility across species and institutions, knowledge of timing and organ at risk, and safety of agents. The current data on nitrite suggests that these challenges have been met. Yet this promise must be tempered with the knowledge that over 30 years of pre-clinical success in protecting against IR injury has failed to translate into meaningful clinical interventions [109].
In addition, the use of nitrite as a therapy for IR injury in humans may have some important limitations. Hypotension due to nitrite mediated vasodilation [6] may worsen outcomes in a situation where hypoperfusion is already present. The production of NO in the setting of reperfusion may worsen oxidative and nitrosative stress both directly, in a dose dependent manner [15], or by formation of highly reactive intermediates such as peroxynitrite [88, 110]. It is unclear whether a therapy like nitrite will remain effective in humans with other co-morbidities, such as diabetes mellitus and renal insufficiency, in which biological processes such as eNOS uncoupling could adversely chemically “interact” with nitrite and produce unexpected consequences. Such unexpected effects of the NOS-NO axis has been observed in conditions associated with NO donor therapy and NOS uncoupling [111, 112].
The animal data suggest nitrite therapy would be most effective in the scenario where a patient presents to medical attention during the ischemic insult and nitrite is given prior to reperfusion therapy. Table 2 summarizes human diseases in which an opportunity for therapy with nitrite may exist.
Table 2. Human diseases where nitrite therapy may be beneficial.
A list of human diseases where the pathophysiology includes IR injury and nitrite therapy prior to reperfusion may be beneficial. To date, no human clinical trials evaluating nitrite therapy have been performed.
| Focal Ischemia | Global Ischemia |
|---|---|
| Myocardial infarction | Cardiac arrest |
| Stroke | Cardiopulmonary bypass |
| Pulmonary embolism | Trauma |
| Mesenteric ischemia | |
| Budd-Chiari syndrome | |
| Peripheral vascular disease | |
| Solid organ transplantation |
Given the myocardial protection noted across three species (mouse, rat and dog), nitrite given prior to reperfusion of blocked coronary vessels in the setting of myocardial infarction may have cardioprotective effects. In this scenario, nitrite could be infused at the time of presentation to the ER (ie during ischemia) and prior to reperfusion by either percutaneous intervention or thrombolysis. This would closely mimic the canine and mouse models of myocardial ischemia were nitrite therapy has been successful [20, 24]. The rat brain IR model [21], where nitrite demonstrated clear cytoprotection, was designed to model a large middle cerebral artery stroke and suggests an additional potential role for nitrite given prior to thrombolysis for stroke. Another clinical scenario mimicking the focal IR modeled in animal studies would be that of solid organ transplantation (eg liver or heart). In this setting, there is a predictable ischemic period after organ harvest during which nitrite therapy could be given prior to anastamosis and reperfusion.
Although animal models demonstrating nitrite’s efficacy in global IR injury, such as following cardiopulmonary bypass or cardiopulmonary arrest, are lacking, this remains an area where nitrite’s effects on multiple organs may modulate recovery. Additionally, nitrite-derived NO may have anti-inflammatory [23, 89–91, 113, 114] and anti-thrombotic [115, 116] properties which could be beneficial in situations such as cardiopulmonary bypass. In this scenario, cellular injury may be occurring through the pathways described above as well as through ischemia and reperfusion. Cardiac arrest is another example of global IR injury where the salutatory effects of NO on inflammation and thrombosis may provide additional benefit to its potential anti-apoptotic properties.
5. Conclusion
Nitrite represents the largest known chemical pool of bioavailable NO in the circulation and tissues. It is synthesized and ingested and may be reduced to yield NO during physiological hypoxia and pathological ischemia. This reduction is mediated by deoxyhemoglobin, deoxymyoglobin, xanthine oxidoreductase and by acidic reduction. Nitrite has now been found to confer cytoprotection against IR injury in a number of animal models. Although the mechanisms of this phenomenon are still being characterized, the reproducibility of this effect in multiple animal IR models suggests nitrite as a novel potential therapy for human ischemic diseases.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lauer T, Preik M, Rassaf T, Strauer BE, Deussen A, Feelisch M, et al. Plasma nitrite rather than nitrate reflects regional endothelial nitric oxide synthase activity but lacks intrinsic vasodilator action. Proc Natl Acad Sci USA. 2001;98:12814–12819. doi: 10.1073/pnas.221381098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gladwin MT, Crawford JH, Patel RP. The biochemistry of nitric oxide, nitrite, and hemoglobin: role in blood flow regulation. Free Radic Biol Med. 2004;36:707–717. doi: 10.1016/j.freeradbiomed.2003.11.032. [DOI] [PubMed] [Google Scholar]
- 3.Gladwin MT, Schechter AN, Kim-Shapiro DB, Patel RP, Hogg N, Shiva S, et al. The emerging biology of the nitrite anion. Nat Chem Biol. 2005;1:308–314. doi: 10.1038/nchembio1105-308. [DOI] [PubMed] [Google Scholar]
- 4.Huang KT, Keszler A, Patel N, Patel RP, Gladwin MT, Kim-Shapiro DB, et al. The reaction between nitrite and deoxyhemoglobin. Reassessment of reaction kinetics and stoichiometry. J Biol Chem. 2005;280:31126–31131. doi: 10.1074/jbc.M501496200. [DOI] [PubMed] [Google Scholar]
- 5.Herold S, Rehmann FJ. Kinetics of the reactions of nitrogen monoxide and nitrite with ferryl hemoglobin. Free Radic Biol Med. 2003;34:531–545. doi: 10.1016/s0891-5849(02)01355-2. [DOI] [PubMed] [Google Scholar]
- 6.Cosby K, Partovi KS, Crawford JH, Patel RP, Reiter CD, Martyr S, et al. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat Med. 2003;9:1498–1505. doi: 10.1038/nm954. [DOI] [PubMed] [Google Scholar]
- 7.Huang Z, Shiva S, Kim-Shapiro DB, Patel RP, Ringwood LA, Irby CE, et al. Enzymatic function of hemoglobin as a nitrite reductase that produces NO under allosteric control. J Clin Invest. 2005;115:2099–2107. doi: 10.1172/JCI24650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doyle MP, Pickering PA, DeWeert TM, Hoekstra JW, Pater D. Kinetics and mechanism of the oxidation of human deoxyhemoglobin by nitrites. J Biol Chem. 1981;256:12393–12398. [PubMed] [Google Scholar]
- 9.Shiva S, Huang Z, Grubina R, Sun J, Ringwood LA, MacArthur P, et al. Deoxymyoglobin is a Nitrite Reductase that Generates NO and Regulates Mitochondrial Respiration. Circ Res. 2007;100 doi: 10.1161/01.RES.0000260171.52224.6b. in press. [DOI] [PubMed] [Google Scholar]
- 10.Millar TM, Stevens CR, Benjamin N, Eisenthal R, Harrison R, Blake DR. Xanthine oxidoreductase catalyses the reduction of nitrates and nitrite to nitric oxide under hypoxic conditions. FEBS Lett. 1998;427:225–228. doi: 10.1016/s0014-5793(98)00430-x. [DOI] [PubMed] [Google Scholar]
- 11.Kozlov AV, Staniek K, Nohl H. Nitrite reductase activity is a novel function of mammalian mitochondria. FEBS Lett. 1999;454:127–130. doi: 10.1016/s0014-5793(99)00788-7. [DOI] [PubMed] [Google Scholar]
- 12.Godber BL, Doel JJ, Sapkota GP, Blake DR, Stevens CR, Eisenthal R, et al. Reduction of nitrite to nitric oxide catalyzed by xanthine oxidoreductase. J Biol Chem. 2000;275:7757–7763. doi: 10.1074/jbc.275.11.7757. [DOI] [PubMed] [Google Scholar]
- 13.Li H, Samouilov A, Liu X, Zweier JL. Characterization of the magnitude and kinetics of xanthine oxidase-catalyzed nitrite reduction. Evaluation of its role in nitric oxide generation in anoxic tissues. J Biol Chem. 2001;276:24482–24489. doi: 10.1074/jbc.M011648200. [DOI] [PubMed] [Google Scholar]
- 14.Gautier C, van Faassen E, Mikula I, Martasek P, Slama-Schwok A. Endothelial nitric oxide synthase reduces nitrite anions to NO under anoxia. Biochem Biophys Res Commun. 2006;341:816–821. doi: 10.1016/j.bbrc.2006.01.031. [DOI] [PubMed] [Google Scholar]
- 15.Bolli R. Cardioprotective function of inducible nitric oxide synthase and role of nitric oxide in myocardial ischemia and preconditioning: an overview of a decade of research. J Mol Cell Cardiol. 2001;33:1897–1918. doi: 10.1006/jmcc.2001.1462. [DOI] [PubMed] [Google Scholar]
- 16.Bell RM, Maddock HL, Yellon DM. The cardioprotective and mitochondrial depolarising properties of exogenous nitric oxide in mouse heart. Cardiovasc Res. 2003;57:405–415. doi: 10.1016/s0008-6363(02)00675-2. [DOI] [PubMed] [Google Scholar]
- 17.Jones SP, Bolli R. The ubiquitous role of nitric oxide in cardioprotection. J Mol Cell Cardiol. 2006;40:16–23. doi: 10.1016/j.yjmcc.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 18.Gianetti J, Del Sarto P, Bevilacqua S, Vassalle C, De Filippis R, Kacila M, et al. Supplemental nitric oxide and its effect on myocardial injury and function in patients undergoing cardiac surgery with extracorporeal circulation. J Thorac Cardiovasc Surg. 2004;127:44–50. doi: 10.1016/j.jtcvs.2002.08.001. [DOI] [PubMed] [Google Scholar]
- 19.Jones SP, Bolli R. The ubiquitous role of nitric oxide in cardioprotection. Journal of Molecular and Cellular Cardiology. 2006;40:16–23. doi: 10.1016/j.yjmcc.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 20.Duranski MR, Greer JJ, Dejam A, Jaganmohan S, Hogg N, Langston W, et al. Cytoprotective effects of nitrite during in vivo ischemia-reperfusion of the heart and liver. J Clin Invest. 2005;115:1232–1240. doi: 10.1172/JCI22493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jung KH, Chu K, Ko SY, Lee ST, Sinn DI, Park DK, et al. Early intravenous infusion of sodium nitrite protects brain against in vivo ischemia-reperfusion injury. Stroke. 2006;37:2744–2750. doi: 10.1161/01.STR.0000245116.40163.1c. [DOI] [PubMed] [Google Scholar]
- 22.Lu P, Liu F, Yao Z, Wang CY, Chen DD, Tian Y, et al. Nitrite-derived nitric oxide by xanthine oxidoreductase protects the liver against ischemia-reperfusion injury. Hepatobiliary Pancreat Dis Int. 2005;4:350–355. [PubMed] [Google Scholar]
- 23.Webb A, Bond R, McLean P, Uppal R, Benjamin N, Ahluwalia A. Reduction of nitrite to nitric oxide during ischemia protects against myocardial ischemia-reperfusion damage. Proc Natl Acad Sci USA. 2004;101:13683–13688. doi: 10.1073/pnas.0402927101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gonzalez FM, Shiva S, Vincent PS, Ringwood LA, Hsu L-Y, Hon C, et al. Nitrite Provides Potent Cytoprotective and Anti-apoptotic Effects as Adjunctive Therapy to Reperfusion for Acute Myocardial Infarction. Manuscript in preparation. 2007 doi: 10.1161/CIRCULATIONAHA.107.748814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tripatara P, Patel NSA, Webb A, Rathod K, Lecomte FMJ, Mazzon E, et al. Nitrite-Derived Nitric Oxide Protects the Rat Kidney against Ischemia/Reperfusion Injury In Vivo: Role for Xanthine Oxidoreductase. J Am Soc Nephrol. 2007;18:570–580. doi: 10.1681/ASN.2006050450. [DOI] [PubMed] [Google Scholar]
- 26.Bryan NS, Fernandez BO, Bauer SM, Garcia-Saura MF, Milsom AB, Rassaf T, et al. Nitrite is a signaling molecule and regulator of gene expression in mammalian tissues. Nat Chem Biol. 2005;1:290–297. doi: 10.1038/nchembio734. [DOI] [PubMed] [Google Scholar]
- 27.Gladwin MT. Nitrite as an intrinsic signaling molecule. Nat Chem Biol. 2005;1:245–246. doi: 10.1038/nchembio1005-245. [DOI] [PubMed] [Google Scholar]
- 28.Kleinbongard P, Dejam A, Lauer T, Rassaf T, Schindler A, Picker O, et al. Plasma nitrite reflects constitutive nitric oxide synthase activity in mammals. Free Radic Biol Med. 2003;35:790–796. doi: 10.1016/s0891-5849(03)00406-4. [DOI] [PubMed] [Google Scholar]
- 29.Kleinbongard P, Dejam A, Lauer T, Jax T, Kerber S, Gharini P, et al. Plasma nitric concentrations reflect the degree of endothelial dysfunction in humans. Free Radic Biol Med. 2006;40:295–302. doi: 10.1016/j.freeradbiomed.2005.08.025. [DOI] [PubMed] [Google Scholar]
- 30.Dejam A, Hunter CJ, Pelletier MM, Hsu LL, Machado RF, Shiva S, et al. Erythrocytes are the major intravascular storage sites of nitrite in human blood. Blood. 2005;106:734–739. doi: 10.1182/blood-2005-02-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shingles R, Roh MH, McCarty RE. Direct measurement of nitrite transport across erythrocyte membrane vesicles using the fluorescent probe, 6-methoxy-N-(3-sulfopropyl) quinolinium. J Bioenerg Biomembr. 1997;29:611–616. doi: 10.1023/a:1022491220299. [DOI] [PubMed] [Google Scholar]
- 32.Jensen FB. Nitrite transport into pig erythrocytes and its potential biological role. Acta Physiol Scand. 2005;184:243–251. doi: 10.1111/j.1365-201X.2005.01448.x. [DOI] [PubMed] [Google Scholar]
- 33.May JM, Qu ZC, Xia L, Cobb CE. Nitrite uptake and metabolism and oxidant stress in human erythrocytes. Am J Physiol Cell Physiol. 2000;279:C1946–C1954. doi: 10.1152/ajpcell.2000.279.6.C1946. [DOI] [PubMed] [Google Scholar]
- 34.Spiegelhalder B, Eisenbrand G, Preussmann R. Influence of dietary nitrate on nitrite content of human saliva: possible relevance to in vivo formation of N-nitroso compounds. Food Cosmet Toxicol. 1976;14:545–548. doi: 10.1016/s0015-6264(76)80005-3. [DOI] [PubMed] [Google Scholar]
- 35.Shiva S, Wang X, Ringwood LA, Xu X, Yuditskaya S, Annavajjhala V, et al. Ceruloplasmin is a NO oxidase and nitrite synthase that determines endocrine NO homeostasis. Nat Chem Biol. 2006;2:486–493. doi: 10.1038/nchembio813. [DOI] [PubMed] [Google Scholar]
- 36.Pennington JAT. Dietary exposure models for nitrates and nitrites. Food Control. 1998;9:385–395. [Google Scholar]
- 37.White JW., Jr Relative significance of dietary sources of nitrate and nitrite. J Agric Food Chem. 1975;23:886–891. doi: 10.1021/jf60201a034. [DOI] [PubMed] [Google Scholar]
- 38.Fassett DW. Nitrates and Nitrites. Washington, DC: National Academy of Sciences; 1973. [Google Scholar]
- 39.Ysart G, Miller P, Barrett G, Farrington D, Lawrance P, Harrison N. Dietary exposures to nitrate in the UK. Food Addit Contam. 1999;16:521–532. doi: 10.1080/026520399283669. [DOI] [PubMed] [Google Scholar]
- 40.Dich J, Jarvinen R, Knekt P, Penttila PL. Dietary intakes of nitrate, nitrite and NDMA in the Finnish Mobile Clinic Health Examination Survey. Food Addit Contam. 1996;13:541–552. doi: 10.1080/02652039609374439. [DOI] [PubMed] [Google Scholar]
- 41.Bjorne HH, Petersson J, Phillipson M, Weitzberg E, Holm L, Lundberg JO. Nitrite in salvia increases gastric mucosal blood flow and mucus thickness. J Clin Invest. 2004;113:106–114. doi: 10.1172/JCI200419019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Samouilov A, Kuppusamy P, Zweier JL. Evaluation of the Magnitude and Rate of Nitric Oxide Production from Nitrite in Biological Systems. Archives of Biochemistry and Biophysics. 1998;357:1–7. doi: 10.1006/abbi.1998.0785. [DOI] [PubMed] [Google Scholar]
- 43.Mowat C, Carswell A, Wirz A, McColl KE. Omeprazole and dietary nitrate independently affect levels of vitamin C and nitrite in gastric juice. Gastroenterology. 1999;116:813–822. doi: 10.1016/s0016-5085(99)70064-8. [DOI] [PubMed] [Google Scholar]
- 44.Jansson EA, Petersson J, Reinders C, Sobko T, Bjorne H, Phillipson M, et al. Protection from nonsteroidal anti-inflammatory drug (NSAID)-induced gastric ulcers by dietary nitrate. Free Radic Biol Med. 2007;42:510–518. doi: 10.1016/j.freeradbiomed.2006.11.018. [DOI] [PubMed] [Google Scholar]
- 45.Bjorne HM, Govoni MM, Tornberg DCM, PhD, Lundberg JOM, PhD, Weitzberg EM, PhD. Intragastric nitric oxide is abolished in intubated patients and restored by nitrite*. Critical Care Medicine. 2005;33:1722–1727. doi: 10.1097/01.ccm.0000171204.59502.aa. [DOI] [PubMed] [Google Scholar]
- 46.Larsen FJ, Ekblom B, Sahlin K, Lundberg JM, Weitzberg E. Effects of Dietary Nitrate on Blood Pressure in Healthy Volunteers. N Engl J Med. 2006;355:2792–2793. doi: 10.1056/NEJMc062800. [DOI] [PubMed] [Google Scholar]
- 47.Lundberg JO, Feelisch M, Bjorne H, Jansson EA, Weitzberg E. Cardioprotective effects of vegetables. Is nitrate the answer? Nitric Oxide. 2006;15:359–362. doi: 10.1016/j.niox.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 48.Gladwin MT, Ognibene FP, Pannell LK, Nichols JS, Pease-Fye ME, Shelhamer JH, et al. Relative role of heme nitrosylation and beta-cysteine 93 nitrosation in the transport and metabolism of nitric oxide by hemoglobin in the human circulation. Proc Natl Acad Sci USA. 2000;97:9943–9948. doi: 10.1073/pnas.180155397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Joshi MS, Ferguson TB, Jr, Han TH, Hyduke DR, Liao JC, Rassaf T, et al. Nitric oxide is consumed, rather than conserved, by reaction with oxyhemoglobin under physiological conditions. Proc Natl Acad Sci USA. 2002;99:10341–10346. doi: 10.1073/pnas.152149699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu X, Miller MJ, Joshi MS, Sadowska-Krowicka H, Clark DA, Lancaster JR., Jr Diffusion-limited reaction of free nitric oxide with erythrocytes. J Biol Chem. 1998;273:18709–18713. doi: 10.1074/jbc.273.30.18709. [DOI] [PubMed] [Google Scholar]
- 51.Martin F, Linden T, Katschinski DM, Oehme F, Flamme I, Mukhopadhyay CK, et al. Copper-dependent activation of hypoxia-inducible factor (HF)-1: implications for ceruloplasmin regulation. Blood. 2005;105:4613–4619. doi: 10.1182/blood-2004-10-3980. [DOI] [PubMed] [Google Scholar]
- 52.Mukhopadhyay CK, Mazumder B, Fox PL. Role of hypoxia-inducible factor-1 in transcriptional activation of ceruloplasmin by iron deficiency. J Biol Chem. 2000;275:21048–21054. doi: 10.1074/jbc.M000636200. [DOI] [PubMed] [Google Scholar]
- 53.Zweier JL, Wang P, Samouilov A, Kuppusamy P. Enzyme-independent formation of nitric oxide in biological tissues. Nat Med. 1995;1:804–809. doi: 10.1038/nm0895-804. [DOI] [PubMed] [Google Scholar]
- 54.Lundberg JO, Weitzberg E, Lundberg JM, Alving K. Intragastric nitric oxide production in humans: measurements in expelled air. Gut. 1994;35:1543–1546. doi: 10.1136/gut.35.11.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cannon RO, 3rd, Schechter AN, Panza JA, Ognibene FP, Pease-Fye ME, Waclawiw MA, et al. Effects of inhaled nitric oxide on regional blood flow are consistent with intravascular nitric oxide delivery. J Clin Invest. 2001;108:279–287. doi: 10.1172/JCI12761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Crawford JH, Isbell TS, Huang Z, Shiva S, Chacko BK, Schechter AN, et al. Hypoxia, red blood cells, and nitrite regulate NO-dependent hypoxic vasodilation. Blood. 2006;107:566–74. doi: 10.1182/blood-2005-07-2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brooks J. The action of nitrite on haemoglobin in the absence of oxygen. Proc R Soc Med. 1937;123:368–382. [Google Scholar]
- 58.Furchgott RF, Bhadrakom S. Reactions of strips of rabbit aorta to epinephrine, isopropylarterenol, sodium nitrite and other drugs. J Pharmacol Exp Ther. 1953;108:129–143. [PubMed] [Google Scholar]
- 59.Basireddy M, Isbell TS, Teng X, Patel RP, Agarwal A. Effects of sodium nitrite on ischemia-reperfusion injury in the rat kidney. Am J Physiol Renal Physiol. 2006;290:F779–F786. doi: 10.1152/ajprenal.00334.2005. [DOI] [PubMed] [Google Scholar]
- 60.Okamoto M, Tsuchiya K, Kanematsu Y, Izawa Y, Yoshizumi M, Kagawa S, et al. Nitrite-derived nitric oxide formation following ischemia-reperfusion injury in kidney. Am J Physiol Renal Physiol. 2005;288:F182–F187. doi: 10.1152/ajprenal.00036.2004. [DOI] [PubMed] [Google Scholar]
- 61.Tiravanti E, Samouilov A, Zweier JL. Nitrosyl-heme complexes are formed in the ischemic heart: evidence of nitrite-derived nitric oxide formation, storage, and signaling in post-ischemic tissues. J Biol Chem. 2004;279:11065–11073. doi: 10.1074/jbc.M311908200. [DOI] [PubMed] [Google Scholar]
- 62.Lundberg JO, Weitzberg E. NO generation from nitrite and its role in vascular control. Arterioscler Thromb Vasc Biol. 2005;25:915–922. doi: 10.1161/01.ATV.0000161048.72004.c2. [DOI] [PubMed] [Google Scholar]
- 63.Roche CJ, Dantsker D, Samuni U, Friedman JM. Nitrite reductase activity of sol-gel-encapsulated deoxyhemoglobin. Influence of quaternary and tertiary structure. J Biol Chem. 2006;281:36874–36882. doi: 10.1074/jbc.M603914200. [DOI] [PubMed] [Google Scholar]
- 64.Gladwin MT. Hemoglobin as a nitrite reductase regulating red cell-dependent hypoxic vasodilation. Am J Respir Cell Mol Biol. 2005;32:363–366. doi: 10.1165/rcmb.f294. [DOI] [PubMed] [Google Scholar]
- 65.Castello PR, David PS, McClure T, Crook Z, Poyton RO. Mitochondrial cytochrome oxidase produces nitric oxide under hypoxic conditions: implications for oxygen sensing and hypoxic signaling in eukaryotes. Cell Metab. 2006;3:277–287. doi: 10.1016/j.cmet.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 66.Schulz R, Kelm M, Heusch G. Nitric oxide in myocardial ischemia/reperfusion injury. Cardiovasc Res. 2004;61:402–413. doi: 10.1016/j.cardiores.2003.09.019. [DOI] [PubMed] [Google Scholar]
- 67.Hanley PJ, Daut J. KATP channels and preconditioning: A re-examination of the role of mitochondrial KATP channels and an overview of alternative mechanisms. Journal of Molecular and Cellular Cardiology. 2005;39:17–50. doi: 10.1016/j.yjmcc.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 68.Sasaki N, Sato T, Ohler A, O'Rourke B, Marban E. Activation of mitochondrial ATP-dependent potassium channels by nitric oxide. Circulation. 2000;101:439–445. doi: 10.1161/01.cir.101.4.439. [DOI] [PubMed] [Google Scholar]
- 69.Forbes RA, Steenbergen C, Murphy E. Diazoxide-induced cardioprotection requires signaling through a redox-sensitive mechanism. Circ Res. 2001;88:802–809. doi: 10.1161/hh0801.089342. [DOI] [PubMed] [Google Scholar]
- 70.Pain T, Yang XM, Critz SD, Yue Y, Nakano A, Liu GS, et al. Opening of mitochondrial K(ATP) channels triggers the preconditioned state by generating free radicals. Circ Res. 2000;87:460–466. doi: 10.1161/01.res.87.6.460. [DOI] [PubMed] [Google Scholar]
- 71.Rakhit RD, Mojet MH, Marber MS, Duchen MR. Mitochondria as targets for nitric oxide-induced protection during simulated ischemia and reoxygenation in isolated neonatal cardiomyocytes. Circulation. 2001;103:2617–2623. doi: 10.1161/01.cir.103.21.2617. [DOI] [PubMed] [Google Scholar]
- 72.Borutaite V, Morkuniene R, Brown GC. Release of cytochrome c from heart mitochondria is induced by high Ca2+ and peroxynitrite and is responsible for Ca2+-induced inhibition of substrate oxidation. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 1999;1453:41–48. doi: 10.1016/s0925-4439(98)00082-9. [DOI] [PubMed] [Google Scholar]
- 73.Di Lisa F, Menabo R, Canton M, Barile M, Bernardi P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J Biol Chem. 2001;276:2571–2575. doi: 10.1074/jbc.M006825200. [DOI] [PubMed] [Google Scholar]
- 74.Korge P, Honda HM, Weiss JN. Protection of cardiac mitochondria by diazoxide and protein kinase C: implications for ischemic preconditioning. Proc Natl Acad Sci USA. 2002;99:3312–3317. doi: 10.1073/pnas.052713199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cleeter MW, Cooper JM, Darley-Usmar VM, Moncada S, Schapira AH. Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett. 1994;345:50–54. doi: 10.1016/0014-5793(94)00424-2. [DOI] [PubMed] [Google Scholar]
- 76.Shiva S, Brookes PS, Patel RP, Anderson PG, Darley-Usmar VM. Nitric oxide partitioning into mitochondrial membranes and the control of respiration at cytochrome c oxidase. Proc Natl Acad Sci USA. 2001;98:7212–7217. doi: 10.1073/pnas.131128898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brunori M, Giuffre A, Forte E, Mastronicola D, Barone MC, Sarti P. Control of cytochrome c oxidase activity by nitric oxide. Biochim Biophys Acta. 2004;1655:365–371. doi: 10.1016/j.bbabio.2003.06.008. [DOI] [PubMed] [Google Scholar]
- 78.Sarti P, Arese M, Bacchi A, Barone MC, Forte E, Mastronicola D, et al. Nitric oxide and mitochondrial complex IV. IUBMB Life. 2003;55:605–611. doi: 10.1080/15216540310001628726. [DOI] [PubMed] [Google Scholar]
- 79.Brown GC, Cooper CE. Nanomolar concentrations of nitric oxide reversibly inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS Lett. 1994;356:295–298. doi: 10.1016/0014-5793(94)01290-3. [DOI] [PubMed] [Google Scholar]
- 80.Brookes P, Darley-Usmar VM. Hypothesis: the mitochondrial NO(*) signaling pathway, and the transduction of nitrosative to oxidtive cell signals: an alternative function for cytochrome C oxidase. Free Radic Biol Med. 2002;32:370–374. doi: 10.1016/s0891-5849(01)00805-x. [DOI] [PubMed] [Google Scholar]
- 81.Poderoso JJ, Carreras MC, Lisdero C, Riobo N, Schopfer F, Boveris A. Nitric oxide inhibits electron transfer and increases superoxide radical production in rat heart mitochondria and submitochondrial particles. Arch Biochem Biophys. 1996;328:85–92. doi: 10.1006/abbi.1996.0146. [DOI] [PubMed] [Google Scholar]
- 82.Thomas DD, Liu X, Kantrow SP, Lancaster JR., Jr The biological lifetime of nitric oxide: implications for the perivascular dynamics of NO and O2. Proc Natl Acad Sci USA. 2001;98:355–360. doi: 10.1073/pnas.011379598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Anderson TC, Li CQ, Shao ZH, Hoang T, Chan KC, Hamann KJ, et al. Transient and partial mitochondrial inhibition for the treatment of postresuscitation injury: Getting it just right. Crit Care Med. 2006;34:S474–S482. doi: 10.1097/01.CCM.0000246014.19486.A1. [DOI] [PubMed] [Google Scholar]
- 84.Nadtochiy SM, Burwell LS, Brookes PS. Cardioprotection and mitochondrial S-nitrosation: Effects of S-nitroso-2-mercaptopropionyl glycine (SNO-MPG) in cardiac ischemia-reperfusion injury. Journal of Molecular and Cellular Cardiology. doi: 10.1016/j.yjmcc.2007.01.010. In Press, Corrected Proof. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gryglewski RJ, Palmer RMJ, Moncada S. Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature. 1986;320:454–456. doi: 10.1038/320454a0. [DOI] [PubMed] [Google Scholar]
- 86.Ferdinandy P. Peroxynitrite: just an oxidative/nitrosative stressor or a physiological regulator as well? Br J Pharmacol. 2006;148:1–3. doi: 10.1038/sj.bjp.0706693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ferdinandy P, Schulz R. Peroxynitrite: Toxic or Protective in the Heart? Circ Res. 2001;88:12e–13e. doi: 10.1161/01.res.88.2.e12. [DOI] [PubMed] [Google Scholar]
- 88.Ferdinandy P, Schulz R. Nitric oxide, superoxide, and peroxynitrite in myocardial ischaemia-reperfusion injury and preconditioning. Br J Pharmacol. 2003;138:532–543. doi: 10.1038/sj.bjp.0705080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hataishi R, Rodrigues AC, Neilan TG, Morgan JG, Buys E, Sruti S, et al. Inhaled Nitric Oxide Decreases Infarction Size and Improves Left Ventricular Function in a Murine Model of Myocardial Ischemia-Reperfusion Injury. Am J Physiol Heart Circ Physiol. 2006 doi: 10.1152/ajpheart.01172.2005. [DOI] [PubMed] [Google Scholar]
- 90.Lefer DJ, Nakanishi K, Johnston WE, Vinten-Johansen J. Antineutrophil and myocardial protecting actions of a novel nitric oxide donor after acute myocardial ischemia and reperfusion of dogs. Circulation. 1993;88:2337–2350. doi: 10.1161/01.cir.88.5.2337. [DOI] [PubMed] [Google Scholar]
- 91.Pabla R, Buda AJ, Flynn DM, Blesse SA, Shin AM, Curtis MJ, et al. Nitric oxide attenuates neutrophil-mediated myocardial contractile dysfunction after ischemia and reperfusion. Circ Res. 1996;78:65–72. doi: 10.1161/01.res.78.1.65. [DOI] [PubMed] [Google Scholar]
- 92.Bryan NS, Rassaf T, Maloney RE, Rodriguez CM, Saijo F, Rodriguez JR, et al. Cellular targets and mechanisms of nitros(yl)ation: an insight into their nature and kinetics in vivo. Proc Natl Acad Sci USA. 2004;101:4308–4313. doi: 10.1073/pnas.0306706101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Janero DR, Bryan NS, Saijo F, Dhawan V, Schwalb DJ, Warren MC, et al. Differential nitros(yl)ation of blood and tissue constituents during glyceryl trinitrate biotransformation in vivo. PNAS. 2004;101:16958–16963. doi: 10.1073/pnas.0406075101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 95.Mannick JB, Hausladen A, Liu L, Hess DT, Zeng M, Miao QX, et al. Fas-induced caspase denitrosylation. Science. 1999;284:651–654. doi: 10.1126/science.284.5414.651. [DOI] [PubMed] [Google Scholar]
- 96.Maejima Y, Adachi S, Morikawa K, Ito H, Isobe M. Nitric oxide inhibits myocardial apoptosis by preventing caspase-3 activity via S-nitrosylation. J Mol Cell Cardiol. 2005;38:163–174. doi: 10.1016/j.yjmcc.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 97.Sun J, Picht E, Ginsburg KS, Bers DM, Steenbergen C, Murphy E. Hypercontractile female hearts exhibit increased S-nitrosylation of the L-type Ca2+ channel alphal subunit and reduced ischemia/reperfusion injury. Circ Res. 2006;98:403–411. doi: 10.1161/01.RES.0000202707.79018.0a. [DOI] [PubMed] [Google Scholar]
- 98.Oldenburg O, Cohen MV, Downey JM, Mitochondrial K. (ATP) channels in preconditioning. J Mol Cell Cardiol. 2003;35:569–575. doi: 10.1016/s0022-2828(03)00115-9. [DOI] [PubMed] [Google Scholar]
- 99.Oldenburg O, Qin Q, Krieg T, Yang XM, Philipp S, Critz SD, et al. Bradykinin induces mitochondrial ROS generation via NO, cGMP, PKG, and mitoKATP channel opening and leads to cardioprotection. Am J Physiol Heart Circ Physiol. 2004;286:H468–H476. doi: 10.1152/ajpheart.00360.2003. [DOI] [PubMed] [Google Scholar]
- 100.Xu Z, Ji X, Boysen PG. Exogenous nitric oxide generates ROS and induces cardioprotection: involvement of PKG, mitochondrial KATP channels, and ERK. Am J Physiol Heart Circ Physiol. 2004;286:H1433–H1440. doi: 10.1152/ajpheart.00882.2003. [DOI] [PubMed] [Google Scholar]
- 101.Zweier JL, Flaherty JT, Weisfeldt ML. Direct measurement of free radical generation following reperfusion of ischemic myocardium. Proc Natl Acad Sci USA. 1987;84:1404–1407. doi: 10.1073/pnas.84.5.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Das DK. Cellular, biochemical, and molecular aspects of reperfusion injury. Introduction. Ann NY Acad Sci. 1994;723:xiii–xvi. [PubMed] [Google Scholar]
- 103.Burwell LS, Nadtochiy SM, Tompkins AJ, Young S, Brookes PS. Direct evidence for S-nitrosation of mitochondrial complex I. Biochem J. 2006;394:627–634. doi: 10.1042/BJ20051435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dahm CC, Moore K, Murphy MP. Persistent S-nitrosation of complex I and other mitochondrial membrane proteins by S-nitrosothiols but not nitric oxide or peroxynitrite: implications for the interaction of nitric oxide with mitochondria. J Biol Chem. 2006;281:10056–10065. doi: 10.1074/jbc.M512203200. [DOI] [PubMed] [Google Scholar]
- 105.Lesnefsky EJ, Chen Q, Moghaddas S, Hassan MO, Tandler B, Hoppel CL. Blockade of Electron Transport during Ischemia Protects Cardiac Mitochondria. J Biol Chem. 2004;279:47961–47967. doi: 10.1074/jbc.M409720200. [DOI] [PubMed] [Google Scholar]
- 106.Chen Q, Hoppel CL, Lesnefsky EJ. Blockade of Electron Transport before Cardiac Ischemia with the Reversible Inhibitor Amobarbital Protects Rat Heart Mitrochondria. J Pharmacol Exp Ther. 2006;316:200–207. doi: 10.1124/jpet.105.091702. [DOI] [PubMed] [Google Scholar]
- 107.Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ. Reversible Blockade of Electron Transport during Ischemia Protects Mitochondria and Decreases Myocardial Injury following Reperfusion. J Pharmacol Exp Ther. 2006;319:1405–1412. doi: 10.1124/jpet.106.110262. [DOI] [PubMed] [Google Scholar]
- 108.Fontaine E, Eriksson O, Ichas F, Bernardi P. Regulation of the permeability transition pore in skeletal muscle mitochondria. Modulation By electron flow through the respiratory chain complex i. J Biol Chem. 1998;273:12662–12668. doi: 10.1074/jbc.273.20.12662. [DOI] [PubMed] [Google Scholar]
- 109.Bolli R, Becker L, Gross G, Mentzer R, Jr, Balshaw D, Lathrop DA. Myocardial Protection at a Crossroads: The Need for Translation Into Clinical Therapy. Circ Res. 2004;95:125–134. doi: 10.1161/01.RES.0000137171.97172.d7. [DOI] [PubMed] [Google Scholar]
- 110.Yasmin W, Strynadka KD, Schulz R. Generation of peroxynitrite contributes to ischemia-reperfusion injury in isolated rat hearts. Cardiovascular Research. 1997;33:422–432. doi: 10.1016/s0008-6363(96)00254-4. [DOI] [PubMed] [Google Scholar]
- 111.Elrod JW, Duranski MR, Langston W, Greer JJM, Tao L, Dugas TR, et al. eNOS Gene Therapy Exacerbates Hepatic Ischemia-Reperfusion Injury in Diabetes: A Role for eNOS Uncoupling. Circ Res. 2006;99:78–85. doi: 10.1161/01.RES.0000231306.03510.77. [DOI] [PubMed] [Google Scholar]
- 112.Takimoto E, Champion HC, Li M, Ren S, Rodriguez ER, Tavazzi B, et al. Oxidant stress from nitric oxide synthase-3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J Clin Invest. 2005;115:1221–1231. doi: 10.1172/JCI21968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Duranski MR, Greer JJ, Dejam A, Sathya J, Hogg N, Langston W, et al. Cytoprotective effects of nitrite during in vivo ischemia-reperfusion of the heart and liver. J Clin Invest. 2005 doi: 10.1172/JCI22493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lefer DJ. Do neutrophils contribute to myocardial reperfusion injury? Basic Res Cardiol. 2002;97:263–267. doi: 10.1007/s00395-002-0363-x. [DOI] [PubMed] [Google Scholar]
- 115.Sogo N, Magid KS, Shaw CA, Webb DJ, Megson IL. Inhibition of human platelet aggregation by nitric oxide donor drugs: relative contribution of cGMP-independent mechanisms. Biochem Biophys Res Commun. 2000;279:412–419. doi: 10.1006/bbrc.2000.3976. [DOI] [PubMed] [Google Scholar]
- 116.Palatianos GM, Paziouros K, Vassili MI, Stratigi P, Kaklamanis IL, Prapas S, et al. Effect of exogenous nitric oxide during cardiopulmonary bypass on lung postperfusion histology. Asaio J. 2005;51:398–403. doi: 10.1097/01.mat.0000169274.42302.14. [DOI] [PubMed] [Google Scholar]