

Abstract
We report that the novel anthracycline analog, 13-deoxy, 5-iminodoxorubicin (DIDOX), represents a potentially new class of immunosuppressive agents. DIDOX has been structurally modified from the parent compound, doxorubicin, to remove the carbonyl group at carbon-13 and the quinone moiety at carbon-5 since these structures likely mediate the cardiotoxic side effects of this family of chemotherapeutic drugs. Our studies demonstrate that DIDOX inhibits T cell proliferation and the expression of the T cell activation molecules, CD25 and CD40L. DIDOX also inhibits the production of the proinflammatory cytokine, TNF-α and IL-2. Studies using animal models demonstrate that DIDOX inhibits the inflammation accompanying contact hypersensitivity reactions and possesses reduced cardiotoxicity compared to doxorubicin. These findings indicate that DIDOX has important immunosuppressive activities that may warrant the development of this new and improved anthracycline for the treatment of T cell-mediated inflammatory diseases.
1. Introduction
The identification and characterization of novel T cell inhibitory compounds is important for developing new strategies for the prevention and treatment of autoimmune and allergic disorders. In particular, treatments that are safe, well-tolerated, and capable of suppressing T cell activation processes or modulating the balance of Th1/Th2 subsets are especially promising. Although anthracyclines, including doxorubicin (DOX), are a well recognized class of anti-mitogenic compounds that are commonly used in the treatment of many cancer types including acute leukemias, lymphomas, soft tissue sarcomas, breast, and lung cancers [1], their clinical utilization is compromised by a cumulative dose-dependent, life-threatening cardiomyopathy [1,2]. Early studies revealed that 10% of patients receiving more than 550 mg/m2 developed congestive heart failure [3] which was lethal in 60% of the cases [4]. Thus, cardiotoxicity has limited the clinical use of anthracyclines in the treatment of cancer and prevented their use for other serious, albeit, non-life threatening diseases. Structure-based drug design has led to the discovery of new anthracycline analogs with anti-mitogenic properties and reduced cardiotoxicity. These structural modifications may expand the use of novel anthracyclines for the treatment of immune disorders.
In spite of the wide use of anthracyclines for the treatment of malignancies, the molecular effect of these drugs on T lymphocyte function is not well characterized. The majority of available data was obtained using transformed leukemic T cell lines in which cytotoxicity appears to be a major mechanism of action. Some evidence indicates that DOX increases Jurkat T cell apoptosis via activation of the NF-κB pathway [5], although other data indicates necrosis is involved since loss of membrane integrity is an early event and precedes the induction of limited traits of programmed cell death [6]. Interestingly, a slight structural modification of DOX to generate the anthracycline analog, aclarubicin, induces apoptosis rather than necrosis in Jurkat T cells [6] indicating that relatively minor changes in chemical structure can produce fundamental differences in biological action.
In nontransformed T cells, only limited information is available regarding the effects of anthracyclines. In some studies, DOX prevented allogeneic mixed lymphocyte reactions and prolonged allotransplantation survival indicating an overall inhibition of antigen presentation or T cell activation processes [7]. However, other data indicate DOX can exert opposing roles on T cells that is related to differences between in vitro versus in vivo microenvironments. In these studies, in vitro drug administration suppressed T cell activation while in vivo drug administration increased T cell activation [8]. Thus, the effects of anthracyclines on normal T cells warrants further clarification, especially with respect to new anthracycline analogs.
The rationale for developing new anthracycline analogs has been to eliminate the molecular components of the parent compound giving rise to negative side effects while retaining the desired biological activity. The mechanism of anthracycline-induced cardiotoxicity is believed to result from free radical injury which is dependent on two distinct structures of the DOX molecule. These structures result in the generation of reactive oxygen species via redox-cycling of the semiquinone radical intermediate [9] and the generation of a cardiotoxic alcohol metabolite, doxorubicinol (DOXol) [10]. In this study we investigate the immunosuppressive properties of a novel DOX analog, 13-deoxy, 5-iminodoxorubicin (DIDOX), which has been modified to remove both of the sites implicated in cardiotoxicity. The C-13 carbonyl moiety has been reduced to a methylene preventing formation of DOXol and the quinone at C-5 has been changed to an imino group to prevent redox cycling and free radical generation. Our findings indicate that DIDOX possesses important immunosuppressive activities on T cells that may warrant its development for the treatment of inflammatory disease.
2. Materials and Methods
2.1 Primary T cell isolation and T cell lines
For the isolation of human primary CD4+ T cells, peripheral blood mononuclear cells (PBMC) were obtained by Ficoll-Hypaque (Histopaque-1077, Sigma, St. Louis, MO) gradient centrifugation using heparinized phlebotomy samples [11]. Cells were washed 3 times with Hank’s buffer (Sigma) and incubated at 1 × 106 cells/ml in RPMI-1640 (Sigma) containing 10% fetal bovine serum. CD4+ cells were obtained by negative immunomagnetic selection using a cocktail of antibodies against CD45RO, CD8, CD19, CD14, CD16, CD56, CD8, and glycophorin A (StemCell Technologies, Vancouver, Canada) with collection of unlabeled T cells (typically >96% CD4+ as assessed by flow cytometry). Purified T cells were cultured in RPMI-1640 supplemented with 10% FCS, 100 U/ml penicillin, and 100 μg/ml streptomycin sulfate and grown at 37°C and 5% CO2. Written, informed consent was obtained from all blood donors and the University Institutional Review Board approved this study.
2.2 T cell activation and culture condition
Purified CD4+ cells were cultured in RPMI/10% FCS at 1 × 106 cells/ml and activated with immobilized CD3 (0.3 μg/well of clone OKT3, ATCC, Rockville, MD) +/− CD28 antibodies (Abs) (0.08 μg/well of clone CD28.2, PharMingen, San Diego, CA), +/− 100 U/ml human recombinant IL-2 (Sigma) on 96 well plates in 200 μl total volume. Cultures were concurrently treated with various concentrations of DIDOX (Gem Pharmaceuticals LLC, Birmingham, AL) or vehicle control for 6h prior to analysis. Alternatively, purified T cells were pretreated with DIDOX or vehicle control for 18h prior to CD3/CD28 activation for an additional 6h. Cells were subsequently harvested and stained for markers of T cell activation and analyzed by flow cytometry.
2.3 Flow cytometry and mAbs
Methods of immunofluorescent staining and flow cytometry were performed as previously described [11]. Briefly, cells were stained with fluorescently labeled antibodies for 30 minutes at 4°C, washed two times, fixed and analyzed on a 3-color Epics flow cytometer (Beckman Coulter, Miami, FL). Ten thousand events gated on size and side scatter were analyzed and the expression of the percent positively staining cells or the mean fluorescence intensity (MFI) determined by comparisons to appropriate isotype control Ab. FITC-conjugated anti-CD40L, anti-CD25, or isotype control Abs were purchased from Beckman Coulter. Appropriate concentrations of each Ab were determined by titration for optimal staining prior to experimental use.
2.4 Cell proliferation
Negatively selected peripheral blood CD4+ T cells were plated into 96-well plates coated with immobilized CD3 (0.16 μg) and CD28 (0.03 μg) Abs at a concentration of 2 × 105 cells/well in 180 μl. Serial dilutions of DIDOX, DOX, or vehicle control were added to wells in quadruplicate as 2 μl aliquots. After 24h of culture, 1 μCi of 3[H] thymidine was added to each well for and additional 24h of culture. Cells were harvested on a Brandel cell harvester (Gaithersburg, MD) using glass fiber filters and extensive filter washing to remove unincorporated thymidine. Filters were air dried and the disintegrations/min determined using scintillation counting. Cell viability was determined by trypan blue staining.
2.5 ELISA
Expression of TNF-α and IL-2 were assessed by ELISA. For TNF-α determinations, primary CD4+ T cells at 1 × 106 cells were activated for 48h with immobilized anti-CD3/CD28 Abs (0.3 μg/well of clone OKT3, ATCC, Rockville, MD)/CD28 (0.08 μg/well of clone CD28.2, PharMingen, San Diego, CA) with the inclusion of 40 U/ml IL-2, and treated with various concentrations of DIDOX in a total volume of 230 μl. Cell-free conditioned media was harvested via successive centrifugations and analyzed by ELISA performed by the Cytokine Core Laboratory (Baltimore, MD). Expression of IL-2 was assessed as described above except that exogenous IL-2 was not added to cultures.
2.6 Contact hypersensitivity assay
BALB/c mice (Harlan, Indianapolis, IN) were sensitized by application of 15 μl of 0.5% 2,4-dinitrofluorobenzene (DNFB, Sigma) in acetone/olive oil (4:1) on a 2 cm2 area of the shaved abdomen [12]. Mice were randomized into groups of 10 mice each and received either an i.p. injection of DIDOX (3.2 mg/kg) or saline control on days 0–5. On day 6, sensitized and unsensitized animals received 20 μl of 0.3% DNFB on the left ear. Ear thickness was measured before challenge and 24h after DNFB challenge using a micrometer. Data are expressed as the change (from pre-challenge levels) in ear thickness (mm) ± S.E. Animal studies were approved by the local IACUC committee.
2.7 Assessment of chronic cardiotoxicity of DIDOX in a rabbit model
Since the potency of DIDOX was likely to differ from DOX, it was necessary to compare cardiotoxicity at equivalent DOX and DIDOX doses. Because the utilization of DOX in chemotherapy applications is well recognized to cause myelosuppression, a high level DIDOX dosing regime was used to generate myelosuppression to an equivalent degree of DOX. White blood cell (WBC) counts were used as the dosing index and three groups of six New Zealand white rabbits per group were chronically treated with DOX, DIDOX, or vehicle (0.9% NaCl). Doses of DOX (1.25 mg/kg twice per week for 7 weeks) and DIDOX (5.0 mg/kg twice a week for 12 weeks)(cumulative doses of DOX and DIDOX were 17.5 mg/kg and 120 mg/kg, respectively) or vehicle were administered i.v. into an ear vein and blood samples obtained at weekly intervals. This dose of DIDOX was chosen since it produced a similar decrease in WBC counts as the cardiotoxic dose of DOX thus allowing comparison of cardiotoxicity of both drugs. Food consumption was measured daily and body weight was measured weekly. Control rabbits were fed the same amount of food as DOX treated rabbits (pair-fed). At sacrifice, left atria were removed and function analyzed to assess cardiotoxicity. Rabbits were sacrificed if they exhibited life threatening or debilitating toxicities (i.e., severe anemia or mucositis), or 13 weeks after beginning the study.
When New Zealand White rabbits were euthanized, the hearts were rapidly removed. Left atria were carefully excised, cut in half and placed in a muscle bath (30° C) containing Krebs-bicarbonate buffer (pH 7.4) of the following composition: 127 mM NaCl, 2.5 mM CaCl2, 2.3 mM KCl, 25 mM NaHCO3, 1.3 mM KH2PO4, 0.6 mM MgSO4 and 5.6 mM glucose. The buffer was continuously bubbled with a mixture of 95% 02 and 5% CO2. Each atrial strip was affixed to a force transducer (Kulite BG 25) and electrically stimulated (Grass Instruments) to contract isometrically with square wave pulses (3 msec duration) 10% higher than threshold voltage. Left atrial strips were initially stabilized at a rate of contraction of 1 Hz and a resting force of 0.5 gm. Force tracing were recorded at 100 mm/sec (Gould Instruments) and analyzed with a Buxco Pulsatile analyzer to obtain contractile function (developed force (DF)). Isolated atrial preparations obtained from rabbits chronically treated with anthracyclines or vehicle were stimulated to contract at 3 contraction rates, 1, 2 and 3 contractions/sec (Hz). High speed oscillographic tracing and Buxco Pulsatile analysis were obtained at each contraction rate after the contractions were stable.
2.8 Echocardiography
Two groups of six rabbits per group were used to assess effects of DIDOX on cardiac function measured by M-mode echocardiography. One group of rabbits received 2 infusions of DIDOX (5 mg/kg/infusion, i.v.) per week for two weeks. The other group received only the vehicle (0.9% NaCl). Before drug or vehicle infusion (baseline) and two weeks after beginning drug or vehicle infusion, echocardiography was performed on each rabbit to obtain left ventricular fractional shortening (LVFS). Briefly, each rabbit was held supine by an assistant and the left ventricle was imaged by 2-D echocardiography and the left ventricular dimensions from the left ventricular free wall to the septal endocardium were measured in M-mode during systole and diastole to obtain LVFS. Cardiotoxicity is defined as LVFS<30% as previously reported [13].
2.9 Determinations of plasma concentrations of DIDOX
Blood samples for analysis of DIDOX concentrations were obtained from 2 rabbits 5, 15 and 30 minutes after receiving i.v. infusions of DIDOX (5 mg/kg). Each sample (~2 ml) was centrifuged to obtain plasma and stored at −80 °C. On the day of analysis, samples were thawed at 37°C and 500 μl of plasma added to a vial containing 430 μl ultrapure H2O and 70 μl daunorubicin (10 μg/ml; Sigma) as an internal standard. Each vial was vortexed and extracted via an Oasis HLB cartridge (Waters and Associates, Milford, MA). The standard curve was prepared by adding DIDOX to vials containing human plasma and water (1:1) to yield concentrations of 10.0, 7.0, 3.0, 1.0, 0.7, 0.3, 0.1, 0.07, 0.03, and 0.01 μg/ml and daunorubicin added as an internal standard at 0.7 μg/ml.
For anthracycline extraction, Oasis HLB cartridges were activated with low vacuum using 1 ml 100% MeOH, followed by 1ml ultrapure H2O being careful not to allow air to enter the cartridge. Samples were loaded onto cartridges and pulled through using vaccum and then washed twice with 1 ml of Oasis wash buffer (40 ml of 20% methanol, 4 ml 2% NH4OH and 156 ml H2O). Cartridges were dried and gravity used to elute the samples using 1.5ml 100% MeOH. The MeOH was evaporated and samples resuspended in 1 ml initial condition mobile phase for the 10, 7, and 3 μg/ml standard curve points; 500 μl initial condition mobile phase for the 1, 0.7, 0.3 and 0.1 μg/ml standards c; and 200 μl initial condition mobile phase for the 0.07, 0.03, and 0.01 μg/ml standards, and all plasma samples. The samples were injected onto a Agilent 1100 series HPLC (Agilent Technoligies, Wilmington, DE) using a Phenomenex Luna hexa/phenyl/5 micron (Phenomenex, Torrance, CA) column and fluorescence detector (daunorubicin - 470 nm excitation/550 emission; DIDOX - 545 nm excitation/630 nm emission). An HPLC gradient was used going from initial condition mobile phase of 76:24 ammonium formate buffer (AFB)/acetonitrile to final 10% AFB; 90% acentonitrile by 18 min. AFB contains 1 g ammonium formate in 1 liter H2O (pH 4 via formic acid). The chromatographic area under the curves for DIDOX and daunorubicin were used for the determinations of plasma concentrations of DIDOX using values obtained from the standard curve with the limit of detection for DIDOX and daunorubicin being 0.1 ng.
2.10 Statistics
Dose-response means were compared to the control baseline value using one-way ANOVA for repeated measures and Dunnett’s test. All pair-wise treatment means were made using one-way ANOVA for repeated measures and Student-Neuman-Keuls test for multiple mean comparisons. Significance was assessed as P<0.05 and is indicated by an asterisk.
3. Results
The mechanism of anthracycline cardiotoxicity is believed to result from free radical injury which is dependent on two distinct structures of the doxorubicin (DOX) molecule (Figure 1). DIDOX is the DOX molecule modified at both sites. The C-13 carbonyl moiety has been reduced to a methylene preventing formation of the cardiotoxic alcohol metabolite doxorubicinol (DOXol) and the quinone moiety at C-5 has been changed to an imino moiety to prevent redox cycling and free radical generation. We postulated that these changes allow for a reduced toxicity profile while retaining the anti-mitogenic properties of anthracyclines, thereby making DIDOX a potential candidate for use in inflammatory disease.
Figure 1.
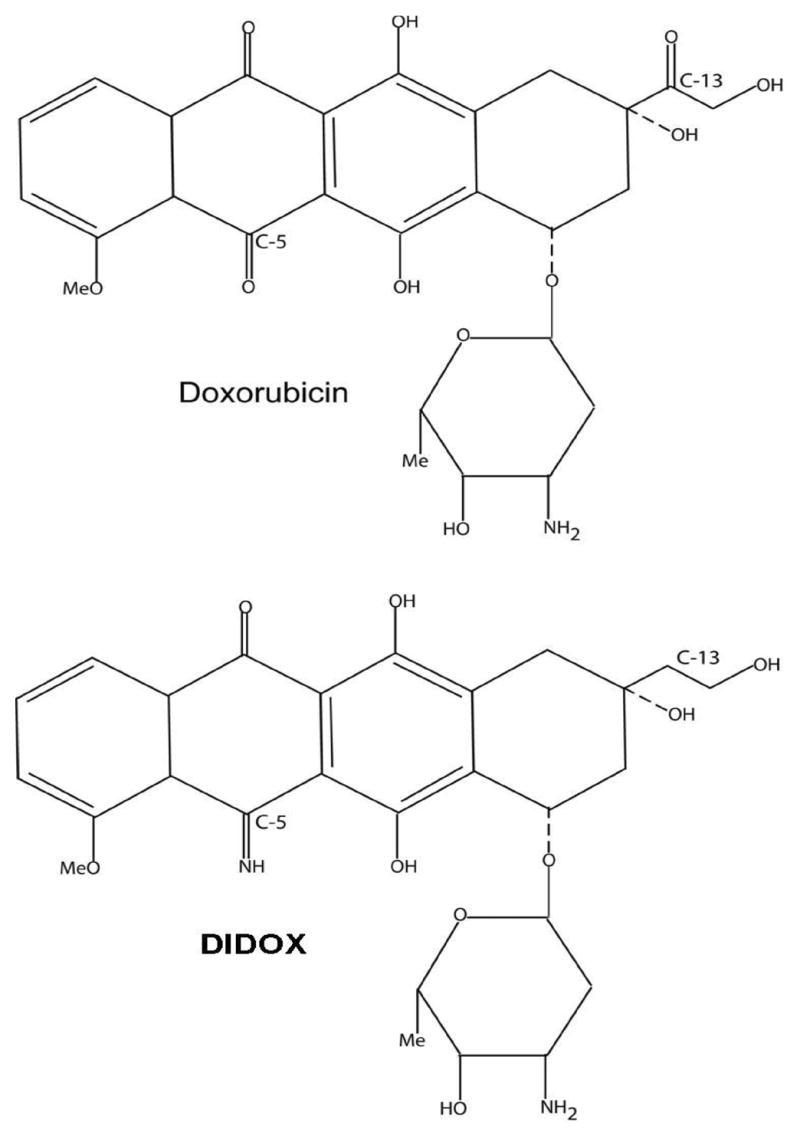
Structural formulae of the anthracyclines doxorubicin and DIDOX.
We first investigated the effect of DIDOX on T cell proliferation. Human primary CD4+ T cells were treated with various concentrations of DIDOX, DOX, or vehicle control and the effect on proliferation determined using a 3[H] thymidine incorporation assay. DIDOX inhibited T cell proliferation with an IC50 of 1.25 μM while an IC50 of 0.08 μM was observed for DOX (Figure 2). Importantly, both drugs were capable of inhibiting cell proliferation by ~90% or more at concentrations of 5 μM.
Figure 2.
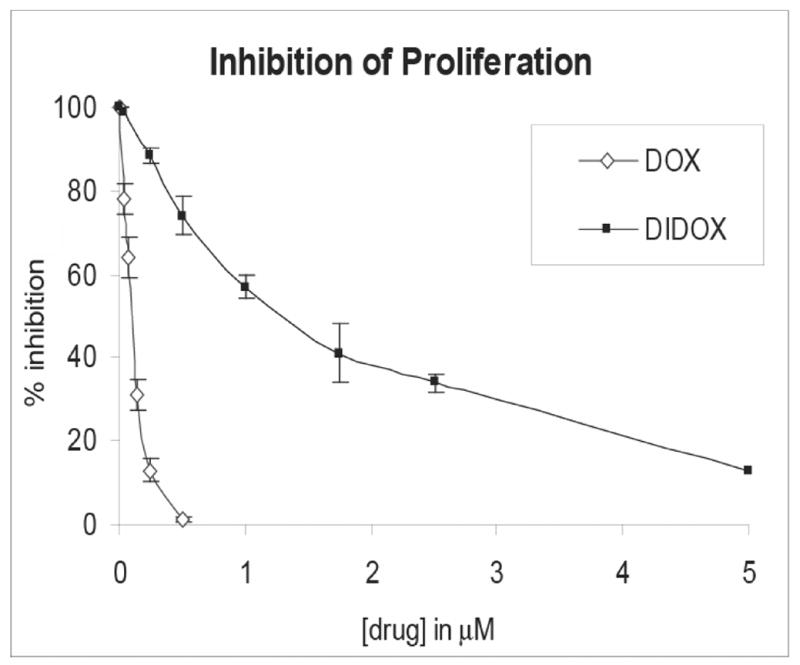
DIDOX inhibits the proliferation of human primary T cells. CD4+ T cells were isolated by negative selection (>97% purity), activated with immobilized CD3/CD28 Abs, and concurrently treated with the indicated concentrations of DIDOX, DOX, or vehicle control. After 24h of culture, 1 μCi of 3[H] thymidine was added to each well for an additional 24h of culture and proliferation measured using a thymidine incorporation assay. Data are normalized relative to values obtained in cultures treated with vehicle control. Data from three independent experiments are presented with error bars depicting S.E.
To further characterize the inhibitory effects of DIDOX on T cells, changes in the surface expression of T cell activation molecules were measured. CD40L (CD154) and CD25 (IL-2 receptor) were evaluated since they represent early markers of T cell activation and play central roles in the initiation and maintenance of T cell responses. In control cultures (Figure 3A & B), a robust induction of CD40L was observed on purified primary CD4+ T cells following T cell receptor (TCR) activation using CD3/CD28 antibodies (62 ± 4 % percent positive cells, n=4). The inclusion of DIDOX, however, produced a significant decrease in the percentage of CD40L positive cells at concentrations as low at 5 μM. 50 μM DIDOX inhibited the percent CD40L positive cells by 37% and the mean fluorescence intensity (MFI) was inhibited by >50%. Interestingly, the pretreatment of T cells with DIDOX prior to TCR activation produced a more striking inhibition of CD40L expression (Figure 3C & D). No appreciable decrease in cell viability (≤5%) was detected in these culture conditions.
Figure 3.
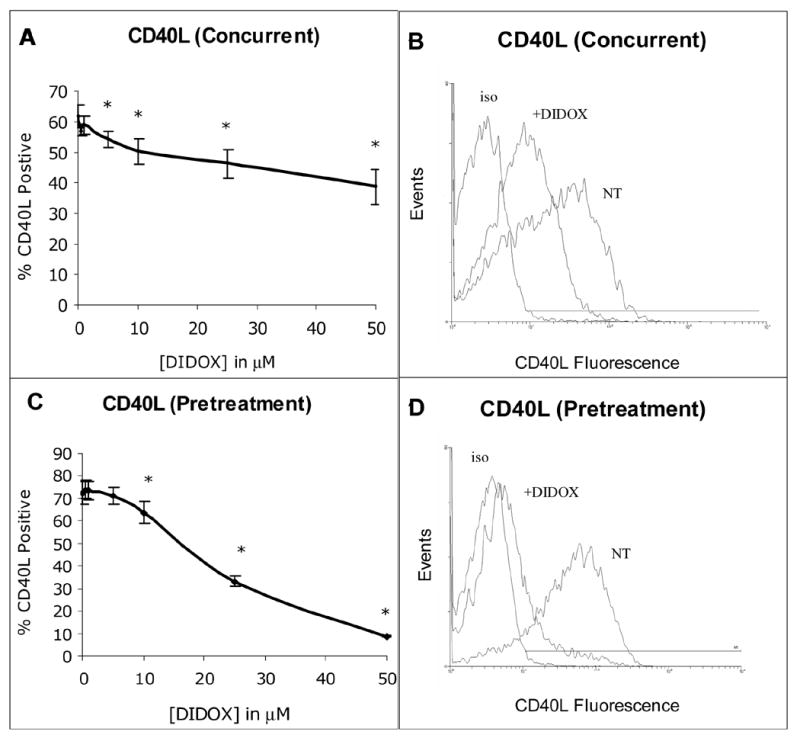
DIDOX inhibits T cell CD40L expression. Primary human CD4+ T cells were isolated by negative immunomagnetic selection (~98% purity). Cells were activated with immobilized CD3/CD28 Abs either concurrent to treatment with GPX -150 for 6h (A & B) or following an 18h DIDOX pretreatment followed by 6h CD3/CD28 activation (C & D). After activation, cells were harvested, stained with a FITC-conjugated CD40L Ab and analyzed by flow cytometry. A) Concurrent DIDOX treatment and TCR activation (6h) inhibits T cell CD40L expression, n=4. B) A representative histogram is depicted with control cultures staining 57% positive for CD40L and 50 μM DIDOX treated cultures staining 32.6% positive, iso = isotype, NT = no treatment. C) DIDOX pretreatment (18h) prior to TCR activation (6h) inhibits CD40L expression, n=5. D) A representative histogram is depicted with control cultures staining 74.6% CD40L positive and 50 μM DIDOX treated cultures staining 12.0% positive. Error bars depict S.E and asterisks indicate statistical significance from control.
As shown in Figure 4, CD25 expression was also suppressed by DIDOX. An 18h pretreatment with DIDOX prior to TCR activation inhibited the percentage of CD25 positive cells (IC50 of 18.5 μM) similar to the effects observed with CD40L (IC50 of 22 μM) with significant effects observed at concentrations as low as 5 μM. The CD25 MFI was also dose-dependently inhibited (~26% at 5 μM, 51% at 10 μM DIDOX). Interestingly, in parallel experiments, the addition of exogenous IL-2 failed to prevent the inhibitory effects of DIDOX on either CD25 or CD40L expression (data not shown).
Figure 4.
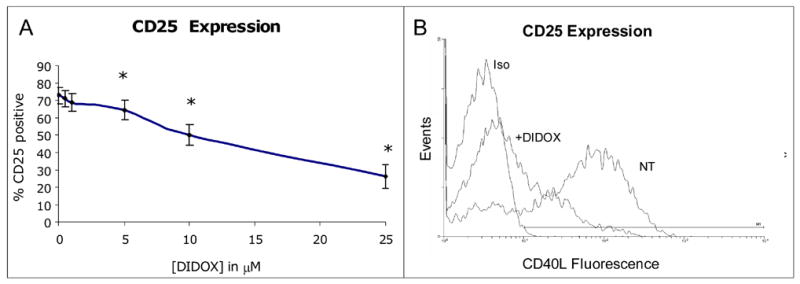
DIDOX inhibits T cell CD25 expression. Primary CD4+ T cells were isolated by negative selection (~95% purity) and pretreated with various concentrations of DIDOX or vehicle for 18h. Cells were subsequently activated with CD3/CD28 antibodies for an additional 24h, stained with a FITC-conjugated CD25 Ab, and analyzed by flow cytometry. A) Data are normalized relative to values obtained in TCR activated cultures treated with vehicle alone, n=6. Error bars depict S.E and asterisks indicate significance from baseline control. B) A representative histogram depicts CD25 staining in control cultures (NT; 73.4% CD25 positive staining) and cultures treated with 50 μM DIDOX (20.1% CD25 positive) relative to staining with an isotype control antibody (iso).
To further investigate the effects of DIDOX on T cell activation, changes in the expression of the key proinflammatory cytokine, TNF-α, was measured. Cultures of purified primary T cells were activated with CD3/CD28 antibodies, DIDOX or vehicle control, and supernatants evaluated by ELISA. Treatment with DIDOX inhibited the production of TNF-α by 50% at 10 μM (Figure 5). A more modest, albeit, significant reduction was also observed at 2 μM DIDOX (Figure 5). No decrease in cell viability was observed in T cells treated with 2 μM DIDOX and only a slight loss of viability (7%) was observed in cultures treated with 10 μM by the end of the culture period.
Figure 5.
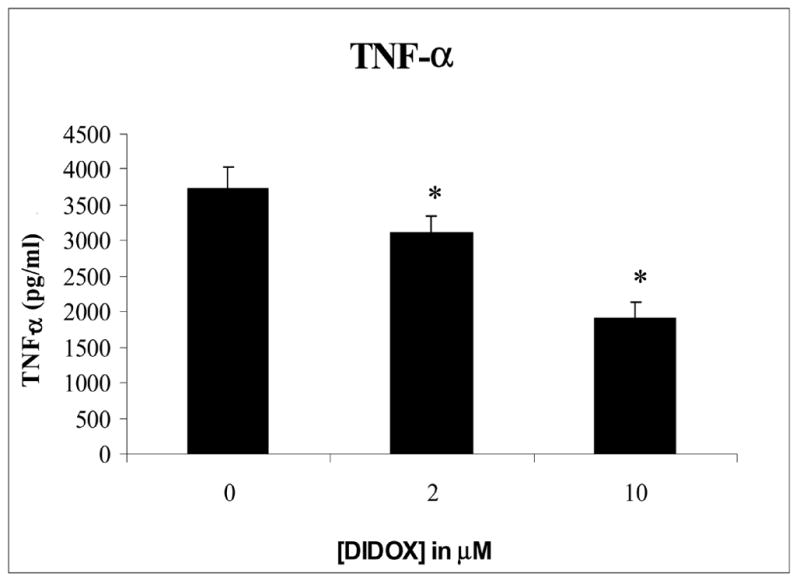
DIDOX inhibits T cell production of TNF-α Primary CD4+ T cells were isolated by negative immunomagnetic selection (>98% purity). Cells were activated for 48h with immobilized CD3/CD28 antibodies, 40 U/ml IL-2, and treated with various concentrations of DIDOX. Cell-free conditioned media was harvested via successive centrifugations and analyzed for TNF-α by ELISA. Data from 5 representative experiments is presented. Error bars depict S.E. and asterisks indicates statistical significance from baseline control.
Similar studies were performed to determine the effects of DIDOX on IL-2 production. Primary T cells were activated with CD3/CD28 antibodies and treated with drug and supernatants assayed by ELISA. As shown in figure 6, DIDOX also inhibited the production of IL-2 with a ≥ 50% reduction observed at 5 μM.
Figure 6.
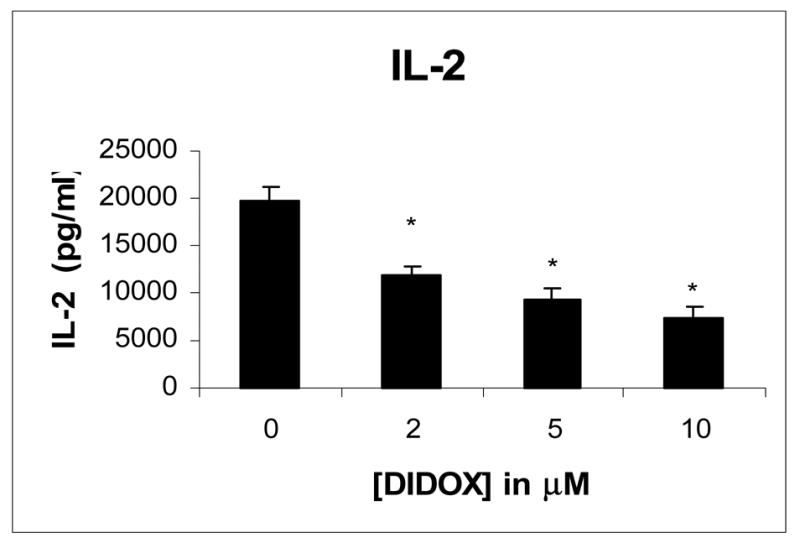
DIDOX inhibits T cell production of IL-2. Primary CD4+ T cells were isolated by negative immunomagnetic selection (>98% purity). Cells were activated for 48h with immobilized CD3/CD28 antibodies and treated with various concentrations of DIDOX or vehicle control. Cell-free conditioned media was harvested via successive centrifugations and analyzed by ELISA. Data from 3 representative experiments is presented. Error bars depict S.E. and asterisks indicates statistical significance from baseline control.
A murine model of contact hypersensitivity was used to evaluate the anti-inflammatory properties of DIDOX (Figure 7). BALB/C mice were sensitized to dinitrofluorobenzene (DNFB) on day 0 and randomized into two groups of 10 mice per group. Mice received either an i.p. injection of DIDOX or saline control on days 1–5 and ear thickness was measured on day 5. After baseline measurement, allergic inflammation was elicted by applying DNFB to the ear. Twenty four hours later the ear thickness was recorded and the difference ascribed to the degree of edema and inflammation. As shown in Figure 7, a 33% suppression in inflammation was observed in animals treated with DIDOX compared to saline controls.
Figure 7.
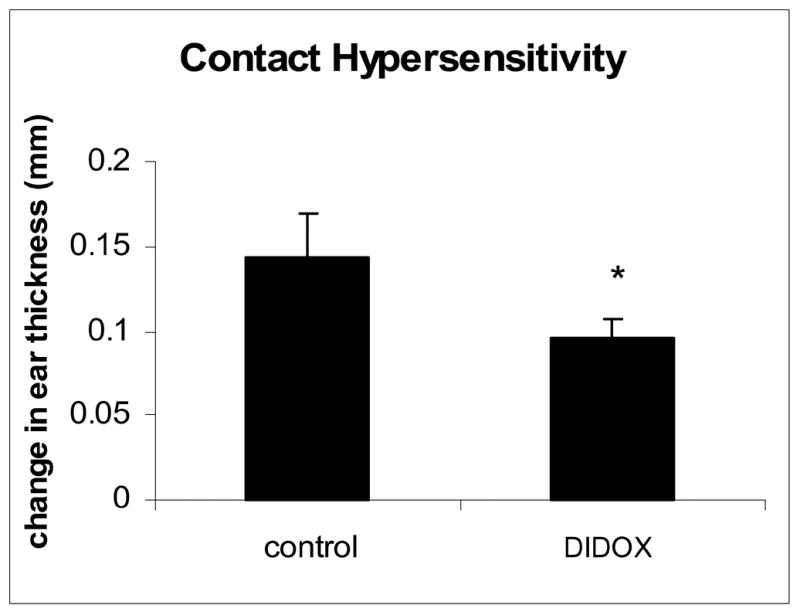
DIDOX inhibits inflammation in an allergic model of contact hypersensitivity. BALB/C mice were sensitized to DNFB on day 0 by applying 15 μl of a 0.5% DNFB acetone/olive oil solution to their shaved abdomens. Mice were randomized into groups of 10 mice and received an i.p. injection of DIDOX (3.2 mg/kg) or saline control on days 0–5. On day 6, the right ear thickness was measured with a micrometer and the ear painted with 20 μl 0.3% DNFB. Twenty four hours later the ear thickness was recorded. Data is presented as the change in ear thickness (mm) after allergen treatment with error bars depicting S.E. An asterisk indicates statistical significance with P<0.05.
Studies were performed to compare the cardiotoxicity of DOX to DIDOX in a chronic rabbit model. In order to perform this study, it was necessary to compare cardiotoxicity at equivalent DOX and DIDOX doses since the potency of the two drugs likely differed. Because myelosuppression is commonly observed following chemotherapy treatment with DOX, white blood cell (WBC) counts were used as the dosing index. Doses of drug (DIDOX, 5 mg/kg; DOX, 1.25 mg/kg) or vehicle were administered i.v. into an ear vein two times per week and blood samples obtained at weekly intervals. At seven weeks of anthracycline administration, WBC counts decreased to the same level in both anthracycline treated groups (Figure 8) with DIDOX and DOX treatment causing a 29% inhibition in WBC counts relative to control animals. Importantly, a more aggressive DIDOX dosing regimen was required (4-fold higher) to achieve similar myelosuppression to DOX. In addition, seven weeks after beginning drug administration, the DOX treated rabbits had to be sacrificed because of toxicities and DIDOX rabbits were able to tolerate treatment for at least another 5 weeks without clinical evidence of toxicity.
Figure 8.
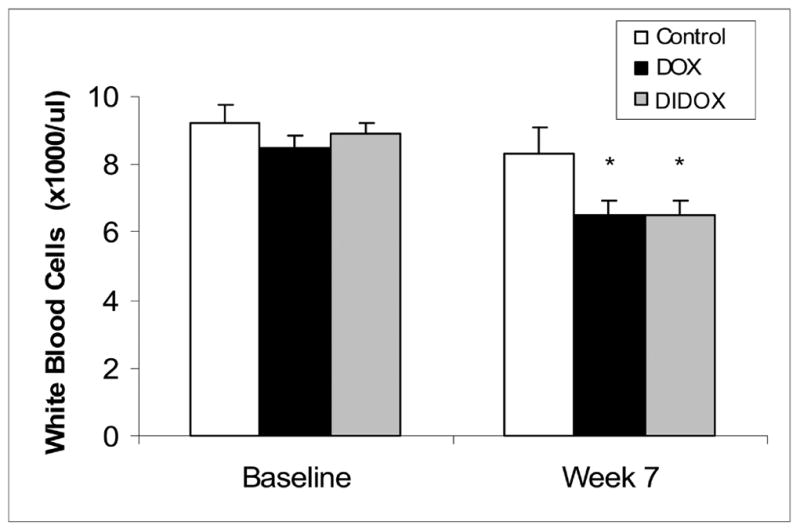
White blood cell counts were performed on rabbits chronically treated with DOX (1.25 mg/kg twice/week for 7 weeks), DIDOX (5.0 mg/kg twice/week for 7 weeks) and 0.9% NaCl (controls) at time 0 and 7 weeks after beginning anthracycline administration. Values are mean ± SE (n=6/group) and asterisks indicate treatments are statistically different (P<0.05) from controls as determined by one-way ANOVA and Tukey’s post-hoc test.
By using comparable myelosuppresive dosing regimens, it was possible to assess the cardiotoxicity of DOX and DIDOX. Cardiac contractile function was assessed by determining developed force (DF, measured in grams). Developed force is the amount of force generated during an individual contraction and is a measure of muscle strength in isolated cardiac muscle preparations in vitro [14]. As shown in Figure 9, atria from DOX treated rabbits displayed impaired developed force over the entire force-frequency range (1, 2, and 3 Hz). Atria from rabbits treated with DIDOX did not exhibit impaired developed force compared to controls.
Figure 9.
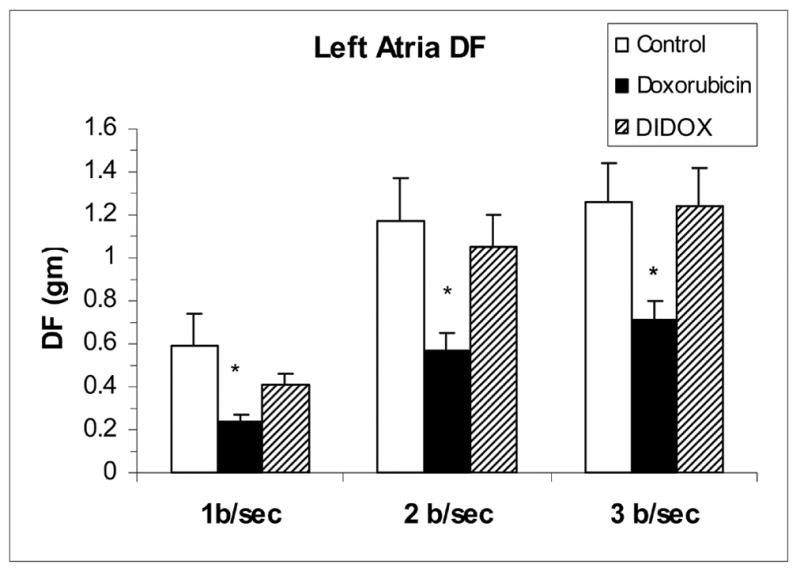
Cardiac contractile function (developed force (DF), expressed in grams (gm)) at 3 frequencies of contraction (1, 2 and 3 Hz (beat/sec)) of atria isolated from rabbits at sacrifice. Rabbits were chronically treated with DOX (1.25 mg/kg twice/week for 7 weeks), DIDOX (5.0 mg/kg twice/week for 12 weeks) and 0.9% NaCl (controls). Values are mean ± SE (n=6/group) and asterisks indicate statistical difference (P<0.05) from control at each frequency as determined by one-way ANOVA and Tukey’s post-hoc test.
To verify that concentrations of DIDOX affecting T cell function can be achieved in vivo and have acceptable cardiotoxic effects, the following studies were performed. Six rabbits were treated with a dose of DIDOX of 5 mg/kg twice a week for two weeks since this dosing regimen had no measurable effect on cardiac contractile function at animal sacrifice (Figure 9). In drug treated rabbits, echocardiography was performed to assess left ventricular fractional shortening (LVFS) as an additional measure of cardiotoxicity. After two weeks of DIDOX administration, LFVS was not significantly different than baseline (pre-DIDOX values were 37.3% ± 1.1% versus 36.6% ± 0.4% in DIDOX treated animals, mean ± S.E.). In addition, the values from DIDOX treated rabbits were not different than control rabbits receiving only the DIDOX vehicle (37.5% ± 0.3% baseline, 39.4% ± 0.7% at 2 weeks). These results indicate that 5 mg/kg doses of DIDOX are not cardiotoxic as assessed by echocardiography while similar doses of DOX have been previously shown to have cardiotoxic effects [13].
Subsequent experiments were performed to determine the achievable plasma concentrations of DIDOX in rabbits receiving noncardiotoxic dosing. A single i.v. dose of 5 mg/kg DIDOX in rabbits was found to produce plasma concentrations of 5.0 ± 0.2 μM at 5 minutes, 1.8 ± 0.02 μM at 15 minutes, and 1.4 ± 0.2 μM at 30 minutes following DIDOX administration (mean ± S.E., n=2). Thus, the plasma level of DIDOX obtained in rabbits following a single 5 mg/kg dose is within the range of concentrations capable of producing significant effects on T cell proliferation, activation, and cytokine production in vitro.
4. Discussion
The results of this study demonstrate that the novel anthracycline analog, DIDOX, displays reduced cardiotoxicity yet inhibits T cell function and inflammation. Because T cell proliferation and activation are involved in the initiation and maintenance of autoimmune inflammatory processes and allergic disease, DIDOX may prove clinically useful in the treatment of these disorders.
In this report we show that DIDOX potently inhibits T cell proliferation. At concentrations as low as 5 μM DIDOX, nearly 90% of cell growth was blocked in primary CD4+ T cells (Figure 2). Cell viability assessments were also performed, and at concentrations of DIDOX capable of effectively reducing T cell proliferation (2–5 μM; 24 h), no appreciable effects on viability were observed. However at longer periods of exposure, some loss of viability was detected (~6% at 10 μM, 26% at 50 μM; 48h).
DIDOX also inhibited the expression of T cell markers of activation. The expression of CD25 in primary T cells was inhibited with an IC50 of 18.5 μM. Significant inhibitory effects were also observed at 5 μM DIDOX (Figure 4). The CD25 protein is an important measure of immune function since the CD25 subunit of the IL-2 receptor is required for T cell growth and IL-2 receptor signaling regulates the growth and differentiation of B cells, NK cells, and macrophages [15].
The effects of DIDOX on T cell CD40L expression were also studied due to the role of this protein in the activation of B cells, CD8+ T cells, dendritic cells, and macrophages [16] and its rate limiting function in the regulation of humoral and cell mediated immune responses [17,18]. It was observed that concurrent T cell activation and DIDOX treatment produced a 40% inhibition in the percentage of CD40L positive cells and a >50% inhibition in MFI (Figure 3). Interestingly, an 18h pretreatment with DIDOX prior to T cell activation was even more effective at reducing CD40L (88% inhibition) indicating that DIDOX may be particularly effective at preventing an immune response if administered prior to antigen encounter. Because elevated or constitutive expression of CD40L is frequently observed during autoimmune attacks and the blockade of CD40L/CD40 interactions is recognized as a beneficial treatment for autoimmune disorders [19], actions of DIDOX to inhibit CD40L support a potential role of this compound in the treatment of inflammatory disease.
The effects of DIDOX on the T cell production of the pro-inflammatory cytokine, TNF-α, and IL-2 were also studied. TNF-α is expressed by activated T cells and has a significant role in a variety of pathophysiological processes involving inflammation and apoptosis. It is among the most potent of pro-inflammatory molecules and promotes inflammatory cell infiltration and phagocytosis [20]. Anti-TNF therapy is currently used to treat a number of inflammatory diseases including psoriasis, rheumatoid arthritis, and Crohn’s disease and is implicated in the inflammation associated with asthma, septic shock, and lupus [21]. DIDOX inhibited TNF-α expression in a concentration dependent manner. 10 μM DIDOX reduced cytokine levels to 50% of control and a significant inhibitory effect was also observed at 2 μM (Figure 5). Because reduction of TNF-α is a common goal of anti-inflammatory therapies, actions of DIDOX to inhibit TNF-α is consistent with its potential use in immunosuppressive therapy. A similar inhibition of the T cell growth factor, IL-2 was also observed (Figure 6) and is consistent with a potential role of DIDOX in the treatment of T cell mediated inflammatory diseases.
To assess in vivo actions of DIDOX to inhibit inflammation, an animal model of contact hypersensitivity was used. DIDOX significantly suppressed inflammation (33%) in this model (Figure 7). Because T cells are recognized as a primary mediator of this in vivo response [22,23], this data further supports a potential use for DIDOX in the treatment of T cell-mediated inflammatory diseases. The investigation of DIDOX on additional animal models of inflammatory disease, including Th-2 mediated models, is a current area of research in our laboratory.
DIDOX was designed to remove the two molecular moieties on the parent drug implicated in anthracycline-induced cardiotoxicity. The mechanism of cardiotoxicity likely involves free radical injury which is dependent on two distinct structures of the DOX molecule which result in the generation of reactive oxygen species [9] and the generation of a cardiotoxic alcohol metabolite, DOXol [10]. The first modification involves the quinone structure on the “C” ring of the parent molecule, DOX (Figure 1). This structure has been removed from DIDOX since the quinone group participates in single electron reduction reactions, yielding semiquinone free radicals that reduce molecular oxygen and generate superoxide anions [9]. The second structural modification occurs at the C-13 carbonyl group. The removal of this group prevents the reduction to a hydroxyl via carbonyl reductases present in the heart, to form the primary alcohol metabolite, DOXol. DOXol is believed to inhibit the ability of myocytes to regulate Fe levels causing Fe catalyzed free radical reactions and reactive oxygen-induced myocardial injury [10].
To test whether the structural modifications of DIDOX result in reduced cardiotoxicity, studies were performed to compare the cardiotoxicity of DOX to DIDOX at equivalent drug doses in a chronic rabbit model. Cardiotoxicity was assessed at sacrifice by evaluating contractile function of left atrial preparations isolated from control, DOX, and DIDOX treated rabbits. The left atrial preparations were studied in a tissue bath where afterload and preload remained constant allowing more control over contractile function. Contractile function was assessed at varying frequencies of contraction (force-frequency relation) to unmask cardiotoxicity that might not be apparent at a single slow contraction frequency (steady-state contractions). Utilizing this model of assessing cardiac function, atria from DOX-treated rabbits displayed impaired maximal developed force over the entire force-frequency range (1, 2, and 3 Hz) when compare to atria from control rabbits (Figure 9). Atria from rabbits treated with DIDOX did not exhibit impaired developed force compared to control atria. Thus, DIDOX appears significantly less cardiotoxic than DOX in this chronic model of anthracycline cardiotoxicity.
The cardiotoxicity studies were assessed using equivalent doses of DOX and DIDOX since the two drugs likely differed in potency. To identify equivalent doses, white blood cell counts were used as the dosing index since myelosuppresssion is commonly observed following anthracycline treatment in cancer patients. Importantly, a more aggressive DIDOX dosing regime (4-fold higher) was required to decrease WBC counts similar to the level occurring with DOX (Figure 8). Thus, the dosing regime of DIDOX required to cause myelosuppression used both large amounts of drug and repeated systemic administration for an extended period of time (7 weeks). The myelosuppressive properties of DIDOX may be of little to no consequence in the topical treatment of inflammatory diseases such as psoriasis or for the treatment of inflammatory diseases responding to lower doses or less frequent administration than doses required to induce myelosuppression for cardiotoxicity assessements.
Lastly, studies were performed to verify that concentrations of DIDOX having clear and substantial effects on T cell function could be achieved in vivo. A single i.v. injection of 5 mg/kg was shown to produce plasma concentrations of DIDOX of 5 μM. This same concentration was capable of inhibiting T cell proliferation by ~90% and significantly inhibiting parameters of T cell activation and cytokine production in in vitro studies. Importantly, a twice weekly dose of DIDOX (5 mg/kg) given over two week period did not cause measurable cardiotocity as assessed by echocardiographic measurements of left ventricular fractional shortening while similar doses of DOX have cardiotoxic effects [13]. No loss of cardiac contractile function was observed at animal sacrifice using this same dosing regime for twelve weeks (Figure 9).
In conclusion, the results of this study indicate that DIDOX is a potent inhibitor of T cell function both in vitro and in vivo. DIDOX inhibits the expression of T cell markers of activation, cell proliferation, and TNF-α and IL-2 production at physiologically attainable concentrations. These concentrations do not appear to be associated with cardiotoxicity. DIDOX also significantly reduces inflammation in a T cell mediated in vivo model of contact hypersensitivity. Collectively, these immunosuppressive actions suggest that DIDOX may represent a new candidate for the clinical treatment of immune disorders. If this drug is to be clinically exploited, further studies are needed to fully elucidate mechanisms of action and identify appropriate methodologies of administration and dosing regimes.
Abbreviations
- Ab
antibody
- APC
antigen presenting cell
- DIDOX
13-deoxy, 5-iminodoxorubicin
- DOX
doxorubicin
- DOXol
doxorubicinol
- DNFB
dinitrofluorobenzene
- ELISA
enzyme-linked immunosorbent assay
- HRP
horse radish peroxidase
- i.p
intraperitoneal
- IRP-1
iron regulatory protein-1
- MFI
mean fluorescence intensity
- PBMC
peripheral blood mononuclear cell
- PKC
protein kinase C
- PMA
phorbol 12-myristate 13-acetate
- S.E
standard error
- TCR
T cell receptor
- WBC
white blood cell
Footnotes
This work was supported by a grant from the National Institutes of Health (R41 AR052955-01)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Young RC, Ozols RF, Myers CE. The anthracycline antineoplastic drugs. N Engl J Med. 1981;305:139–153. doi: 10.1056/NEJM198107163050305. [DOI] [PubMed] [Google Scholar]
- 2.Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. N Engl J Med. 1998;339:900–905. doi: 10.1056/NEJM199809243391307. [DOI] [PubMed] [Google Scholar]
- 3.Gottdiener JS, Mathisen DJ, Borer JS, Bonow RO, Myers CE, Barr LH, Schwartz DE, Bacharach SL, Green MV, Rosenberg SA. Doxorubicin cardiotoxicity: assessment of late left ventricular dysfunction by radionuclide cineangiography. Ann Intern Med. 1981;94:430–435. doi: 10.7326/0003-4819-94-4-430. [DOI] [PubMed] [Google Scholar]
- 4.Von Hoff DD, Layard MW, Basa P, Davis HL, Jr, Von Hoff AL, Rozencweig M, Muggia FM. Risk factors for doxorubicin-induced congestive heart failure. Ann Intern Med. 1979;91:710–717. doi: 10.7326/0003-4819-91-5-710. [DOI] [PubMed] [Google Scholar]
- 5.Ashikawa K, Shishodia S, Fokt I, Priebe W, Aggarwal BB. Evidence that activation of nuclear factor-kappaB is essential for the cytotoxic effects of doxorubicin and its analogues. Biochem Pharmacol. 2004;67:353–364. doi: 10.1016/j.bcp.2003.08.039. [DOI] [PubMed] [Google Scholar]
- 6.Dartsch DC, Schaefer A, Boldt S, Kolch W, Marquardt H. Comparison of anthracycline-induced death of human leukemia cells: programmed cell death versus necrosis. Apoptosis. 2002;7:537–548. doi: 10.1023/a:1020647211557. [DOI] [PubMed] [Google Scholar]
- 7.Eckert R, Gruner S, Volk HD, Giese C, von Baehr R. Studies on the immunomodulatory effects of anthracycline antibiotics in mice: effects on immune responses and graft immunogenicity. Immunobiology. 1989;179:445–455. doi: 10.1016/S0171-2985(89)80048-8. [DOI] [PubMed] [Google Scholar]
- 8.Orsini FR, Henderson ES. Doxorubicin and daunorubicin modulation of macrophages, T and B lymphocytes (anthracycline lymphocyte modulation) Immunopharmacology. 1980;2:375–384. doi: 10.1016/0162-3109(80)90021-1. [DOI] [PubMed] [Google Scholar]
- 9.Kalyanaraman B, Morehouse KM, Mason RP. An electron paramagnetic resonance study of the interactions between the adriamycin semiquinone, hydrogen peroxide, iron-chelators, and radical scavengers. Arch Biochem Biophys. 1991;286:164–170. doi: 10.1016/0003-9861(91)90023-c. [DOI] [PubMed] [Google Scholar]
- 10.Minotti G, Cavaliere AF, Mordente A, Rossi M, Schiavello R, Zamparelli R, Possati G. Secondary alcohol metabolites mediate iron delocalization in cytosolic fractions of myocardial biopsies exposed to anticancer anthracyclines. Novel linkage between anthracycline metabolism and iron-induced cardiotoxicity. J Clin Invest. 1995;95:1595–1605. doi: 10.1172/JCI117833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coligan JE. Current Protocals in Immunology. Greene Publishing Associates and Wiley-Interscience; New York: 1995. [Google Scholar]
- 12.Takanami-Ohnishi Y, Amano S, Kimura S, Asada S, Utani A, Maruyama M, Osada H, Tsunoda H, Irukayama-Tomobe Y, Goto K, Karin M, Sudo T, Kasuya Y. Essential role of p38 mitogen-activated protein kinase in contact hypersensitivity. J Biol Chem. 2002;277:37896–37903. doi: 10.1074/jbc.M207326200. [DOI] [PubMed] [Google Scholar]
- 13.Olson RD, Gambliel HA, Vestal RE, Shadle SE, Charlier HA, Jr, Cusack BJ. Doxorubicin cardiac dysfunction: effects on calcium regulatory proteins, sarcoplasmic reticulum, and triiodothyronine. Cardiovasc Toxicol. 2005;5:269–283. doi: 10.1385/ct:5:3:269. [DOI] [PubMed] [Google Scholar]
- 14.Olson RD, Mushlin PS. Doxorubicin cardiotoxicity: analysis of prevailing hypotheses. FASEB J. 1990;4:3076–3086. [PubMed] [Google Scholar]
- 15.Waldmann TA. The interleukin-2 receptor. J Biol Chem. 1991;266:2681–2684. [PubMed] [Google Scholar]
- 16.Banchereau J, Bazan F, Blanchard D, Briere F, Galizzi JP, Van Kooten C, Liu YJ, Rousset F, Saeland S. The CD40 antigen and its ligand. Annu Rev Immunol. 1994;12:881–922. doi: 10.1146/annurev.iy.12.040194.004313. [DOI] [PubMed] [Google Scholar]
- 17.Wingett D, Nielson CP. Divergence in NK cell and cyclic AMP regulation of T cell CD40L expression in asthmatic subjects. J Leukoc Biol. 2003;74:531–541. doi: 10.1189/jlb.0303103. [DOI] [PubMed] [Google Scholar]
- 18.Perez-Melgosa M, Hollenbaugh D, Wilson CB. Cutting edge: CD40 ligand is a limiting factor in the humoral response to T cell-dependent antigens. J Immunol. 1999;163:1123–1127. [PubMed] [Google Scholar]
- 19.Daikh DI, Wofsy D. Treatment of autoimmunity by inhibition of T cell costimulation. Adv Exp Med Biol. 2001;490:113–117. doi: 10.1007/978-1-4615-1243-1_12. [DOI] [PubMed] [Google Scholar]
- 20.Hehlgans T, Mannel DN. The TNF-TNF receptor system. Biol Chem. 2002;383:1581–1585. doi: 10.1515/BC.2002.178. [DOI] [PubMed] [Google Scholar]
- 21.MacEwan DJ. TNF ligands and receptors--a matter of life and death. Br J Pharmacol. 2002;135:855–875. doi: 10.1038/sj.bjp.0704549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kish DD, Gorbachev AV, Fairchild RL. CD8+ T cells produce IL-2, which is required for CD4+CD25+ T cell regulation of effector CD8+ T cell development for contact hypersensitivity responses. J Leukoc Biol. 2005 doi: 10.1189/jlb.0205069. [DOI] [PubMed] [Google Scholar]
- 23.Wang LF, Sun CC, Wu JT, Lin RH. Epicutaneous administration of hapten through patch application augments TH2 responses which can downregulate the elicitation of murine contact hypersensitivity. Clin Exp Allergy. 1999;29:271–279. doi: 10.1046/j.1365-2222.1999.00498.x. [DOI] [PubMed] [Google Scholar]