

Abstract
A system for addressing in the construction of macromolecular assemblies can be based on the biospecificity of DNA (cytosine-5) methyltransferases and the capacity of these enzymes to form abortive covalent complexes at targeted 5-fluorocytosine residues in DNA. Using this system, macromolecular assemblies have been created using two representative methyltransferases: M·HhaI and M·MspI. When 5-fluorocytosine (F) is placed at the targeted cytosine in each recognition sequence in a synthetic oligodeoxynucleotide (GFGC for M·HhaI or FCGG for M·MspI), we show that the first recognition sequence becomes an address for M·HhaI, while the second sequence becomes an address for M·MspI. A chimeric enzyme containing a dodecapeptide antigen linked to the C terminus of M·HhaI retained its recognition specificity. That specificity served to address the linked peptide to the GFGC recognition site in DNA. With this assembly system components can be placed in a preselected order on the DNA helix. Axial spacing for adjacent addresses can be guided by the observed kinetic footprint of each methyltransferase. Axial rotation of the addressable protein can be guided by the screw axis of the DNA helix. The system has significant potential in the general construction of macromolecular assemblies. We anticipate that these assemblies will be useful in the construction of regular protein arrays for structural analysis, in the construction of protein–DNA systems as models of chromatin and the synaptonemal complex, and in the construction of macromolecular devices.
Macromolecular assembly is easily approached with DNA. Branching through the formation of Watson–Crick paired duplexes in the shape of a Y or an X is now well known (1–4), and the feasibility of assembling 2-dimensional quadrilaterals and 3-dimensional cubes on which more extended structures can be based has been demonstrated (5, 6). However, the stable, site-directed attachment of labile enzymes and proteins to a DNA scaffold presents a formidable challenge in macromolecular fabrication. Candidate procedures in which the Watson–Crick base-pairing homology or triple-helix base-pairing homology of an oligodeoxynucleotide is used to direct a tethered moiety to a preselected site in DNA (3, 4, 7–9) involve extremes of pH or temperature that can destroy the native structure of these proteins. Attachment systems based on antibodies directed against DNA are likely to lack specificity. On the other hand, antibodies to a hapten could be used to decorate a matrix depending on the pattern of haptens laid down during synthesis. The disadvantage here is that all hapten moieties are equivalent, and thus selective addressing would not be possible unless a series of haptens and antibodies directed to them could be developed. While a system of distinct haptens and antibodies is possible (3), it would be necessary to develop a set of hapten–phosphoramidites and the corresponding series of bifunctional antibodies to utilize this approach. Moreover, the use of noncovalent linkages sacrifices stability.
The DNA (cytosine-5)methyltransferases provide a key advance in addressable linking (10). They have well characterized DNA sequence specificities (11) and they form abortive covalent complexes (Fig. 1) between an active-site cysteine and 5-fluorocytosine in their DNA recognition sequences (12–15). In this report, we demonstrate this approach to macromolecular assembly using two representative methyltransferases: M·HhaI and M·MspI. When 5-fluorocytosine (F) is placed at the targeted cytosine in each recognition sequence (GFGC for M·HhaI and FCGG for M·MspI), we show that the first recognition site becomes a unique address for M·HhaI and the second recognition site becomes a unique address for M·MspI. Moreover, when a dodecapeptide (dod) was linked to the C terminus of M·HhaI by molecular cloning techniques, we found that the chimeric enzyme retained the recognition specificity of M·HhaI and served to address the linked peptide to the M·HhaI recognition site. With this approach, it is now possible to order functional proteins in a specified manner along DNA.
Figure 1.
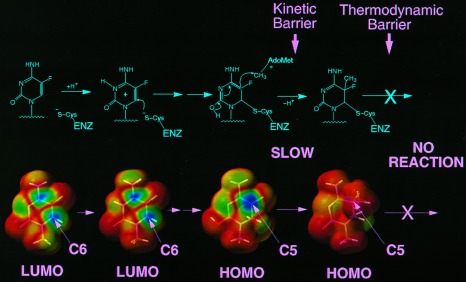
Attack of 5-fluorodeoxycytosine by DNA (cytosine-5)methyltransferases. (Upper) Chemical depiction of intermediates. Protonation of cytosine-N3 by a group at the active site of the enzyme activates the ring for nucleophilic attack by a cysteine residue also at the active site. Once nucleophilic attack occurs, a 5,6 saturated enol can attack the methyl group on S-adenosylmethionine to generate a dihydrocytosine intermediate. In the normal reaction this intermediate would undergo β-elimination to generate a 5-methylcytosine product in DNA and active enzyme. 5-Fluorodeoxycytosine blocks β-elimination because the fluorine at C5 cannot be abstracted. (Lower) Quantum chemical depiction of models of the intermediates. Ab initio geometries were calculated at the Hartree–Fock level of theory using the STO-3G basis set with spartan 4.0 (Wavefunction, Irvine, CA) running on a network of Silicon Graphics workstations. Single-point ab initio orbital energies calculated with the 6-31G* basis set were used to construct electron density surfaces with color maps of frontier orbital values. Blue indicates a high value for the orbital, and red indicates a low value. High values for the lowest unoccupied molecular orbital (LUMO) are seen over C6 and C4 for both 1-methylcytosine and N3 protonated 1-methylcytosine, indicating that these are potential sites for nucleophilic attack. The 1-methyl-6-sulfhydryl-enol derivative of cytosine was used to model the intermediate formed by the enzyme after nucleophilic attack at C6. High values of the highest occupied molecular orbital (HOMO) are confined to C5. This highly reactive orbital is then poised for attack on the methyl of S-adenosylmethionine. Once methyl transfer takes place, the high values of the HOMO remain at C5 but are on the side of the ring (hidden from view) opposite the C5 methyl near the fluorine atom in the model compound (1-methyl-6-sulfhydryl-5-methyldihydrocytosine).
MATERIALS AND METHODS
Molecular Cloning of the M·HhaI-dod Fusion Protein.
A HindIII fragment containing the complete hsdM gene was subcloned from plasmid pJM139RM4.2 (ATCC 40872) into the HindIII site of pSP72 (Promega). The orientation of the insert was such that the EcoRI site on the vector was on the 3′ side of hsdM. The resulting plasmid, pSP72.HhaI, was transformed into the RRI strain of Escherichia coli for production of unmodified HhaI methyltransferase.
A dod coding sequence was coupled in-frame to the 3′ end of hsdM at a multicloning site (SpeI-stop-AgeI-EcoRI) that had been inserted immediately downstream of the M·HhaI coding sequence using the PCR and methods described by Higuchi (16). The inserted amino acids Thr-Ser-Met-Arg-Gly-Ser-His6 contain an epitope recognized by the anti-MRGShis6-antibody (Qiagen, Chatsworth, CA; ref. 17). The sequence of the entire PCR-amplified region in the final modified version of hsdM (pSP72.Hha-dod), from nucleotide 1243 to the end of the dodecamer insert, was confirmed by automated dye terminator sequencing using an Applied Biosystems model 373A sequencer.
Enzymatic Activity.
Activity was determined using trichloroacetic acid-precipitable counts retained on glass fiber filters as described (14). M·HhaI and M·HhaI-dod were assayed in a buffer containing 50 mM Tris·HCl (pH 7.5), 10 mM EDTA, 5 mM 2-mercaptoethanol, 6 μM S-adenosyl-l-[3H-methyl] methionine (Amersham) and 20 μM oligodeoxynucleotide duplex 30-mer as described below. M·MspI was assayed in the same reaction buffer containing 100 mM NaCl.
Preparation of M·HhaI and M·HhaI-dod.
For the purification of M·HhaI and M·HhaI-dod, E. coli RRI cells containing pSP72.HhaI or pSP72.HhaI-dod were cultured in Luria–Bertani medium with 50 μg/ml ampicillin, and the enzyme was purified essentially as described in ref. 18. For the purification of M·HhaI-dod that purification was modified to use High-S fast protein liquid chromatography (FPLC) (Bio-Rad), with final purification obtained with a His-Trap (Pharmacia) affinity column (19).
Oligodeoxynucleotide Synthesis.
Oligodeoxynucleotides were synthesized on a Cyclone-Plus DNA synthesizer (Millipore). They were purified with Poly-Pak columns (Glen Research, Sterling, VA) as described (20).
Mobility Shift Assays.
Enzymes were incubated at 37°C for 2.5 hr with 2 μM 32P-end-labeled oligodeoxynucleotide duplex, 80 μM S-adenosyl-l-methionine (New England Biolabs), 50 mM Tris·HCl (pH 7.5), 10 mM EDTA, and 5 mM 2-mercaptoethanol. Reactions with M·HhaI or M·HhaI-dod alone contained no added NaCl, those with M·MspI contained 100 mM NaCl. NaCl (50 mM) was used when both were incubated together. The products were separated on native protein gels poured with an exponential gradient (6–20% arcylamide) and run at 25 mA for 3 hr (14). The gel was then exposed to x-ray film directly for autoradiography. 32P-end-labeled, TaqI-digested, ΦX174RF (BRL) provided molecular weight markers. M·MspI was purchased from New England Biolabs.
Western Blot Analysis.
After electrophoresis, gels were soaked in Western blot transfer buffer (39 mM glycine/48 mM Tris/20% methanol) for 20 min, then transferred overnight to polyvinylidene difluoride membrane (Millipore) at 50 mA, 4°C. To measure the extent of transfer the membrane was subjected to autoradiography. The membrane was blocked with 3% BSA in a buffer containing 10 mM Tris·HCl (pH 7.4) and 150 mM NaCl, exposed to anti-MRGShis6-antibody (Qiagen), and then to an anti-mouse IgG conjugated to alkaline phosphatase (Sigma) before staining with 5-bromo-4-chloro-3-indoyl phosphate/Nitro blue tetrazolium (BCIP/NBT) alkaline phosphate substrate (Sigma).
RESULTS
Size Limitations on Spacing Between Proteins.
To address multiple methyltransferases to a linear DNA molecule, one must first determine how far apart recognition sequences should be spaced for efficient linking. To approach this question, we measured the rate of the enzymatic reaction as a function of DNA length. The rate was observed to increase rapidly after a threshold length is reached for each of the two enzymes (Fig. 2A). Thus, both M·HhaI and M·MspI require ≈25–30 bp of duplex for significant activity with further stimulation of activity by additional DNA beyond this length. One can view this number as a function of the physical extent of protein–DNA contact along the DNA (i.e., its kinetic footprint; see ref. 20). Although its exact physical meaning is open to interpretation, one must consider the kinetic footprint as an operationally important parameter in this system for the design of macromolecular assemblies. Thus, for the fabrication of an addressable assembly, recognition sites for M·HhaI and M·MspI would have to be placed at least 25 bp (i.e., about 8.5 nm) apart.
Figure 2.

Spacing in linear molecules. (A) An homologous series of oligodeoxynucleotides each with a single centrally located HhaI recognition site (GCGC/GMGC) was labeled with M·HhaI (2 U) using 4 μM oligodeoxynucleotide at each length. M indicates the position of 5-methylcytosine. These results are compared with data from a second homologous series with the same DNA sequence except that the centrally located tetramer was replaced with the (CCGG/MCGG) sequence recognized by M·MspI. M·MspI (2 U) was used to label 4 μM oligodeoxynucleotide at each length. (B) Three oligodeoxynucleotides were tested for efficiency of M·HhaI assembly. Boxes indicate positions of recognition sites. (C) Nondenaturing gel (using a 6–20%, exponential acrylamide gradient) of the assemblies formed between M·HhaI and the oligodeoxynucleotides depicted in B. 0/30, the 30-mer in B with a single recognition site for M·HhaI (GFGC/GMGC); 6/31, the 31-mer in B with two recognition sites for M·HhaI spaced so that the distance between 5-fluorocytosine residues is 6 bp; 35/60, the 60-mer in B with two recognition sites for M·HhaI spaced so that the distance between 5-fluorocytosine residues is 35 bp. Several minor bands are present in these gels at low concentration. These bands appear to be due to the intrinsic property of oligodeoxynucleotides to form unusual DNA structures, since they are generally present in the absence of added enzyme (e.g., 30-mer lane on the far left). They are not due to impurities in the enzyme preparation because it contained only a single species with a mobility corresponding to the apparent molecular weight of 40,000 kD observed in SDS/gel electrophoresis.
Stable Complex Formation.
To confirm the kinetic results, we constructed DNA molecules (Fig. 2B) with single or double recognition sites for M·HhaI at intervals that were closer than the minimum spacing predicted by the kinetic analyses (6 bp), or farther apart than the minimum spacing (35 bp). In this case we used 5-fluorocytosine at the sites targeted for methylation to form stable covalent complexes that are retarded during gel electrophoresis. 32P-end-labeled DNA molecules were incubated with HhaI methyltransferase. Coupling was detected by retardation of the labeled DNA during electrophoresis under nondenaturing conditions. Complexes with one linked enzyme moiety migrated more slowly than the free oligodeoxynucleotide, but more rapidly than complexes with two linked enzyme moieties.
In the experiment in Fig. 2, the input enzyme was about 4 μM and the input oligodeoxynucleotide was about 4 μM. The identity of each species can be inferred from plots of calculated molecular weight against mobility for each assembly. Based on this analysis, these gels are easily capable of resolving assemblies containing 1, 2, and 3 copies of methyltransferase from one another. The yield of complex (based on the ratio of the scanned area of the relevant band to the total area of DNA-containing bands scanned in the lane) was about 46.5% of the input 32P-end-labeled DNA for those oligodeoxynucleotides in which one coupling site was available (Fig. 2C, 30-mer or 31-mer lanes). The yield of the complex in which one enzyme moiety is present on the 60-mer with two M·HhaI coupling sites was reduced to about 32% of input DNA because the enzyme is distributed between molecules that have only one coupling site occupied (Fig. 2C, 60-mer lane, middle band) and those molecules in which two sites are occupied (Fig. 2C, 60-mer lane, upper band). Complexes containing two copies of the enzyme were obtained in good yield under these conditions because they made up 5% of the input DNA. An idealized molecular model of the complex formed between two M·HhaI molecules and the 60-mer with recognition sequences spaced 35 bp apart is depicted in Fig. 3. For M·HhaI recognition sequences, the closest center-to-center juxtaposition of nonoverlapping GCGC recognition sites that can be constructed has a spacing of 6 bp. With the 30-mer containing recognition sites separated by 6 bp, the first coupling occurred with about the same efficiency as observed with the 35-bp spacing, but the second coupling occurred with almost negligible efficiency (<0.5% of input DNA).
Figure 3.

Molecular model of the 60-mer assembly. Molecular models were constructed in biograf 3.21 (Molecular Simulations, San Diego). The initial conformation of M·HhaI was that determined for the crystalline protein complexed with 5-fluorocytosine at its target site (21). The structure was minimized in molecular mechanics to 0.1 (kcal/mol)/Å, and rendered in midasplus (Computer Graphics Laboratory, University of California, San Francisco). The model assumes a linear DNA molecule with the pitch of 10.0 bp/turn (i.e., a helical twist of 36.0°/bp) derived from fiber diffraction. With this assumption the C termini of the two enzymes assume the 180° dihedral angle, measured down the helical axis, shown in the model. Unwinding of the helix within the DNA binding site has been observed in the M·HhaI DNA complex. Although the DNA is not bent by the enzymes this unwinding could result in a twist angle of 31.6°/bp for those base pairs in the binding site. This consideration, coupled with the possibility that the twist angles for the DNA outside the binding sites could be as low as 34.3°/bp based on solution measurements, suggests that the true dihedral angle could be as low as 80° or as high as the 180° shown in the model above.
DNA Recognition Specificity.
The selectivity of individual methyltransferases has been extensively studied (11). The experiment depicted in Fig. 4 illustrates this selectivity for M·HhaI and M·MspI. 32P-end-labeled retardation products were formed only between an enzyme and its homologous recognition site. Cross-reactivity was not observed. This was not only true of the native enzymes but it was also true of the chimeric protein containing the dodecapeptide at its C terminus. The gel also reflects differences in relative affinity displayed by M·MspI, M·HhaI, and M·HhaI-dod for their respective binding sites under these conditions. Scans of gels of this type were used to determine the ratios of bound-to-free enzyme for the various complexes to gauge the relative affinities. By this analysis, M·HhaI was found to have an apparent association constant for its recognition sequence that was about 3 times the affinity of the M·HhaI-dod fusion for the same sequence. Moreover the affinity of the M·HhaI for its recognition sequence (GFGC/GMGC) was about 2-fold greater than the affinity of M·MspI for its sequence (FCGG/MCGG).
Figure 4.

Addressing specificity of M·HhaI, M·MspI, and M·HhaI-dod fusion protein. Oligodeoxynucleotide sequences carrying the centrally located recognition sequence for M·MspI (Left) or M·HhaI (Right) were synthesized with 5-fluorocytosine substituted for cytosine at the target site. After electrophoresis through polyacrylamide gel, 32P-end-labeled DNAs in complexes were visualized by autoradiography. 32P-end-labeled retardation products are formed only between an enzyme and its homologous recognition site. Cross-reactivity was not observed. This was not only true of the native enzymes but it was also true of the chimeric protein containing the dodecapeptide extension at its C terminus.
5-Fluorocytosine Substitution.
While 5-fluorocytosine does not alter the recognition specificity of the methyltransferases that have been tested, it dramatically slows the rate of the reaction. The measured kcat value from enzyme saturation curves (data not shown) for methyltransfer to cytosine in the oligodeoxynucleotide 30-mer used in Fig. 2A was 82.2 hr−1, while that for 5-fluorocytosine in the same oligodeoxynucleotide background was 0.8 hr−1. This reduction in reaction rate (>100-fold) occurs under conditions where complex formation can be as high as 95% of input enzyme (13, 21). In the experiments reported here, where the complexes were incubated for 2.5 hr before separation, they are expected to be 100% covalent at C6 but only about 38% irreversible due to the slow rate of methyltransfer at C5 (see Fig. 1).
Addressing Heterologous Proteins.
Because the three enzymes used in this study retain sequence specificity, gel retardation can be used to identify complexes containing two heterologous proteins. M·HhaI-dod and M·HhaI retardation products are not well resolved on these gels. The 32P-end-labeled band corresponding to the control oligodeoxynucleotide containing M·HhaI-dod alone migrated with a mobility corresponding to that of a 370-bp marker DNA duplex.
The retardation product formed with M·MspI migrated with a mobility corresponding to a DNA of about 520 bp. The data in the autorad depicted in Fig. 5 also demonstrate the coupling of one copy of M·MspI and one copy of M·HhaI at a center-to-center spacing of 30 bp because the new retardation product at about 900 bp (rightmost lane) must contain two different protein moieties: M·MspI and M·HhaI-dod, bound at their respective sites. Cross-reactivity between sites is ruled out by the data in Fig. 4. Adventitious association of proteins with the complex through protein–protein interaction to produce complexes with the mobility of a single DNA linked to two coupled proteins is also ruled out by the data in Fig. 4 for like enzyme molecules, because DNAs with only one recognition site produce only one retardation product. Additional controls (data not shown) similar to those in Fig. 4 demonstrate that unlike pairs (e.g., M·HhaI and M·MspI) do not form adventitious complexes through protein–protein interaction.
Figure 5.

Heterologous assemblies containing a fusion protein. A 64-mer (Lower) carrying a recognition site for M·MspI and a recognition site for M·HhaI spaced at a distance of 30 bp was used. (Left) The 32P-end-labeled reaction products were separated by electrophoresis through polyacrylamide gel. (Right) The gel was transferred to polyvinylidene difluoride membrane, and the western blots were probed with anti-MRGShis6-antibodies to detect the M·HhaI-dod fusion protein.
It is of interest that the affinities of the several enzymes for their respective sites dictate the details of the results depicted in Fig. 5. M·HhaI-dod and M·MspI produce single retardation products at their respective mobilities with the yield of M·HhaI-dod and M·MspI complexes roughly corresponding to the differences in affinities for their coupling sites. The mixture of the two enzymes produces an oligodeoxynucleotide in which the enzyme with the higher affinity (M·MspI) occupies a higher proportion of its coupling site than the enzyme with lower affinity (M·HhaI-dod). The data also suggest competition between the two enzymes since the yield of doubly substituted complex is reduced relative to the yield of singly substituted complexes. This is particularly apparent in the rightmost lane in the gel where both M·HhaI and M·MspI are addressed to the 64-mer at 30-bp spacing. The higher affinity enzyme occupies nearly all of the available oligodeoxynucleotide; no complex is present at the mobility of the 64-mer singly substituted with M·MspI, suggesting that M·HhaI can displace it unless the assembly adopts a conformation that permits both enzymes to occupy their respective sites.
The apparent competition is consistent with the kinetic footprinting data since a 30-bp spacing approaches the minimal permissible distance for joint occupancy at adjacent sites. It is also consistent with the enhanced detection of the fusion peptide seen when M·MspI and M·HhaI-dod are adjacent with a 30-bp spacing on the 64-mer, but not when M·HhaI-dod is attached to the 64-mer in isolation. This enhancement of detection can be quantified from scans of the autoradiographic signal as a measure of the relative amount of DNA in the complex and scans of the Western blot signal as a measure of the relative amount of antibody bound to the complex. For the 64-mer occupied by M·HhaI-dod alone, the mean ratio of scanned area under the peak in the Western blot to the area under the peak in the autoradiograph was 1.69 based on the results of four independent experiments. For the 64-mer carrying a single copy of M·HhaI-dod and a single copy of M·MspI, the mean ratio was 0.1 in these same four experiments. Thus the capacity of the antibody to detect the dod peptide was enhanced about 16.9-fold in the complex containing both proteins.
DISCUSSION
Based on these results, the key finding in this report is that a system for molecular addressing in the construction of macromolecular assemblies can be based on the biospecificity of the DNA methyltransferases. We anticipate that these assemblies will be useful in the construction of regular protein arrays for structural analysis, in the construction of protein–DNA systems as models of chromatin and the synaptonemal complex, and in the construction of macromolecular devices.
Several principles governing this approach to macromolecular assembly are apparent from the results. First, the coupling reaction between methyltransferases and the 5-fluorocytosine provides a mechanism for stable attachment of methyltransferases and chimeric methyltransferase fusion proteins in a preselected arrangement along a DNA molecule. The formation of stable complexes between DNA methyltransferases and 5-fluorocytosine is well known, and the crystal structures of two of these complexes have now been determined (21, 22).
Several reports suggest that the generation of the 5-methyl form of the 5-fluorocytosine complex requires a slow methyltransfer step (12, 20, 23). Nevertheless, freshly prepared assemblies are stable enough for manipulation under nondenaturing conditions, and any functional group (e.g., the dodecamer used here or a contemplated functional fusion protein) attached via molecular cloning would, in general, require manipulation under such conditions. This, in turn, implies that other mechanism-based inhibitors [e.g., the 2-pyrimidinone studied by Hornby and coworkers (24)] may be used in place of 5-fluorocytosine in the construction of macromolecular assemblies. With 2-pyrimidinone, the formation of the covalent intermediate between the enzyme nucleophile and C6 of the pyrmidine ring is rapid but methyltransfer is not observed. In fact, this inhibitor has already been successfully used to couple an M·MspI-Gst fusion protein to an oligodeoxynucleotide (24, 25).
Quantum chemical calculations performed on models of the intermediates in the reaction were quite accurate in their general predictions. In spite of the simplifications in the theoretical model, the ab initio approach correctly predicts each of the salient features of the reaction: (i) the most likely points of nucleophilic attack are C4 and C6, (ii) the geometry of the saturated ring is non-planar, (iii) C5 is the most likely site of methyltransfer, and (iv) the presence of a significant thermodynamic barrier (0.2104 Hartree) to β-elimination with 5-fluorocytosine. The calculation also shows that the highest occupied molecular orbital (HOMO) energy of the model describing the activated intermediate formed after nucleophilic attack is decreased by 0.00635 Hartree due to the substitution of fluorine at C5. Thus, a kinetic barrier is predicted by the theory at the methyltransfer step as indicated in Fig. 1. This is an attractive explanation for the 100-fold reduction in reaction rate in light of the success of the calculation overall. The kinetic barrier predicted by Hartree–Fock is also attractive in light of the crystal structures reported for methyltransferases complexed with 5-fluorocytosine-containing DNAs, since they do not detect F5-related structural changes or hydrogen bonding to F5 of the targeted cytosine that could slow the reaction rate (21, 22).
A second principle that emerges from the molecular modeling of the assemblies is that placement of proteins and methyltransferase fusions is constrained by the properties of B-DNA. Thus, the spacing of the methyltransferase recognition sites along the helix results in axial rotation of the C terminus of the HhaI methyltransferase with the period of the helix. That is to say, the bulk of the M·HhaI protein is confined to one side of the helix (Fig. 3) with the side used for attachment specified by the placement of the fluorocytosine residues (21). Thus, assuming the 10 bp/turn pitch for DNA determined from fiber diffraction, placing 5-fluorocytosine residues at odd multiples of 5 bp will put the C termini on opposite sides of the helix, while placing the 5-fluorocytosines at multiples of 10 bp will put the C termini on the same side of the helix. The accuracy of this expectation must be considered in terms of the torsional flexibility of the helix, its intrinsic curvature, the variations in pitch that have been reported for DNA in solution, and for unwinding due to methyltransferase binding (see Fig. 3).
A third principle that emerges from the work is that the properties of a fusion peptide can be altered by the placement of a second protein at an adjacent site. While this is anticipated for the assembly of transcription factors at a single site, and it has been known for some time in the cooperative interactions between the lambda repressor and operator (26), detection of these effects has not always been straightforward. The enhancement of antibody detection of the dodecamer-fusion by the placement of a second methyltransferase at an adjacent site on the helix suggests that the structure of the dodecamer has been altered in the two-enzyme complex. It is not possible to construct a reasonable model of the complex because both the structure of M·HhaI-dod and the structure of M·MspI are unknown. However, modeling the fusion peptide at its full extension can be used to define a spherical envelope centered at the C terminus of the M·HhaI structure (not shown). This model suggests that the peptide is unlikely to contact M·MspI directly because it is arrayed on the face of the M·HhaI protein opposite to the M·MspI coupling site when one assumes a linear DNA molecule with a pitch of 10 bp/turn. Because the 30-bp spacing between adjacent DNA recognition sites is very close to the kinetic footprint (25–30 bp), the observed competition between sites is not unexpected. This, coupled with the enhanced detection of the M·HhaI-dod fusion protein, suggests that the 64-mer may adopt a curved conformation in the complex, or that methyltransferase-induced unwinding places the enzymes in a position to somehow interact in the doubly substituted complex. Thus the assembly system described here may shed new light on the structure of protein–DNA complexes in solution, and on the intrinsic pitch and curvature of DNA in these complexes.
Acknowledgments
We thank Rosalina Hernandez for her help with these experiments. This work was supported by a grant from the Office of Naval Research (N100014-94-1-1116).
ABBREVIATION
- dod
dodecapetide
References
- 1.Kallenbach N R, Ma R-I, Seeman N C. Nature (London) 1983;305:829–831. [Google Scholar]
- 2.Duckett D R, Murchie A I H, Diekmann S, von Kitzing E, Kemper B, Lilley D M J. Cell. 1988;55:79–89. doi: 10.1016/0092-8674(88)90011-6. [DOI] [PubMed] [Google Scholar]
- 3.Robinson B H, Seeman N C. Protein Eng. 1987;1:295–300. doi: 10.1093/protein/1.4.295. [DOI] [PubMed] [Google Scholar]
- 4.Seeman N C. Clin Chem. 1993;39:722–724. [Google Scholar]
- 5.Zhang Y, Seeman N C. J Am Chem Soc. 1994;116:1661–1669. [Google Scholar]
- 6.Zhang Y, Seeman N C. J Am Chem Soc. 1992;114:2656–2663. [Google Scholar]
- 7.Alivisatos P A, Johnsson K P, Peng X, Wilson T E, Loweth C J, Bruchez M P, Jr, Schultz P G. Nature (London) 1996;382:609–611. doi: 10.1038/382609a0. [DOI] [PubMed] [Google Scholar]
- 8.Pei D, Corey D R, Schultz P G. Proc Natl Acad Sci USA. 1990;87:9858–9862. doi: 10.1073/pnas.87.24.9858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moser H E, Dervan P B. Science. 1987;238:645–650. doi: 10.1126/science.3118463. [DOI] [PubMed] [Google Scholar]
- 10.Smith S S. In: Biological and Biomedical Science and Technology Division 1995 Programs. Eisenstadt E, editor. Arlington, VA: Office of Naval Research; 1995. U.S. Navy Publ. ONR 34196-3, pp. 161–162. [Google Scholar]
- 11.McClelland M, Nelson M. Gene. 1988;74:291–304. doi: 10.1016/0378-1119(88)90305-8. [DOI] [PubMed] [Google Scholar]
- 12.Osterman D G, DePillis G D, Wu J C, Matusda A, Santi D V. Biochemistry. 1988;27:5204–5210. doi: 10.1021/bi00414a039. [DOI] [PubMed] [Google Scholar]
- 13.Chen L, MacMillan A M, Chang W, Ezaz-Nikpay K, Lane W S, Verdine G L. Biochemistry. 1991;30:11018–11025. doi: 10.1021/bi00110a002. [DOI] [PubMed] [Google Scholar]
- 14.Smith S S, Kaplan B E, Sowers L C, Newman E M. Proc Natl Acad Sci USA. 1992;89:4744–4748. doi: 10.1073/pnas.89.10.4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friedman S, Ansari N. Nucleic Acids Res. 1992;20:3241–3248. doi: 10.1093/nar/20.12.3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Higuchi R. In: PCR Technology. Erlich H A, editor. New York: Stockton; 1989. pp. 61–70. [Google Scholar]
- 17.Pogge von Strandmann E, Zoidl C, Nakhei H, Holewa B, Pogge von Strandmann R, Lorenz P, Klein-Hitpass L, Ryffel G U. Protein Eng. 1995;8:733–735. doi: 10.1093/protein/8.7.733. [DOI] [PubMed] [Google Scholar]
- 18.Kumar S, Cheng X, Pflugrath J W, Roberts R J. Biochemistry. 1992;31:8648–8653. doi: 10.1021/bi00151a035. [DOI] [PubMed] [Google Scholar]
- 19.Hochuli E, Bannwarth W, Döbeli H, Gentz R, Stüber D. Bio/Technology. 1988;6:1321–1325. [Google Scholar]
- 20.Laayoun A, Smith S S. Nucleic Acids Res. 1995;23:1584–1589. doi: 10.1093/nar/23.9.1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klimasauskas S, Kumar S, Roberts R J, Cheng X. Cell. 1994;76:357–369. doi: 10.1016/0092-8674(94)90342-5. [DOI] [PubMed] [Google Scholar]
- 22.Reinisch K M, Chen L, Verdine G L, Lipscomb W N. Cell. 1995;82:143–153. doi: 10.1016/0092-8674(95)90060-8. [DOI] [PubMed] [Google Scholar]
- 23.Som S, Friedman S. J Biol Chem. 1994;269:25986–25991. [PubMed] [Google Scholar]
- 24.Ford K, Taylor C, Connolly B, Hornby D P. J Mol Biol. 1993;230:779–786. doi: 10.1006/jmbi.1993.1200. [DOI] [PubMed] [Google Scholar]
- 25.Taylor C, Ford K, Connolly B A, Hornby D P. Biochem J. 1993;291:493–504. doi: 10.1042/bj2910493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson A D, Meyer B J, Ptashne M. Proc Natl Acad Sci USA. 1979;76:5061–5065. doi: 10.1073/pnas.76.10.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]