

Abstract
Protein phosphatase 1 (PP-1) is known to be a critical component of eukaryotic cell cycle progression. In vitro, our previous studies showed that cdc2 kinase phosphorylates Thr-320 (T320) in PP-1, and that this leads to inhibition of enzyme activity. To examine directly the phosphorylation of PP-1 in intact mammalian cells, an antibody has been prepared that specifically recognizes PP-1Cα phosphorylated at T320. Cell synchronization studies revealed in a variety of cell types that T320 of PP-1 was phosphorylated to high levels only during early to mid-mitosis. The phosphorylation of T320 of PP-1 was reduced by the cyclin-dependent protein kinase inhibitor, olomoucine, and increased by the PP-1/PP-2A inhibitor, calyculin A. Immunofluorescence microscopy using phospho-T320 antibody indicated that in NIH 3T3 cells the phosphorylation of PP-1 began to increase from basal levels in prophase and to peak at metaphase. Immunostaining indicated that phospho-PP-1 was localized exclusively to nonchromosomal regions. Furthermore, in cell fractionation studies of mitotic cells, phospho-PP-1 was detectable only in the soluble fraction. These observations suggest that phosphorylation by cdc2 kinase in early to mid-mitosis and inhibition of PP-1 activity is likely to contribute to the increased state of phosphorylation of proteins that is critical to the initiation of normal cell division.
Protein phosphorylation is widely recognized as the major mechanism that controls cell cycle progression. A family of cyclin-dependent protein kinases (CDKs) have been identified and found to be enzymes critical for the initiation and completion of DNA replication and cell division from yeast to mammals (1–5). The activity of CDKs is modulated through phosphorylation of their catalytic subunits and by association with activating or inhibiting proteins. For example, mitotic transition is mediated by the cdc2 kinase/cyclin B complex, and activated cdc2 kinase has been found to drive dramatic structural reorganizations in the nuclear envelope, spindle apparatus, and chromosomal DNA by phosphorylation of a variety of substrates including histone, Eg5, lamin, vimentin, and plectin (6–12).
Evidence suggests that serine/threonine protein phosphatases function as crucial regulators of cell proliferation (4, 13–15). In particular, protein phosphatase 1 (PP-1), which is highly conserved in all eukaryotes, has been found to play a pivotal role in the cell cycle. Genetic studies have indicated that mutation of the enzyme in yeast, Aspergillus, and in Drosophila leads to a variety of defects in mitosis (16–18). Microinjection of anti-PP-1 antibody into B cells before cell division arrests cells at metaphase, whereas injection of PP-1 into anaphase cells accelerates cytokinesis (19). Expression of inhibitor-2, a specific PP-1 inhibitor, changes during the cell cycle, peaking during S phase and mitosis (20). Thus, the tightly balanced activity of CDKs and PP-1 appears to be required for normal cell division. However, the substrates that are targets for PP-1 action remain to be identified and characterized. Potential substrates include the tumor suppressor, p110 retinoblastoma (RB) protein, and cdc25 phosphatase. The catalytic subunit of PP-1 physically associates with RB during mitosis and G1 and is believed to act as a positive regulator of its function, in direct opposition to the cdk/cyclin action (21). PP-1 is believed to dephosphorylate and inactivate cdc25 phosphatase, which activates cdc2 kinase by dephosphorylating Tyr-15 at the onset of mitosis (15, 22).
Recently, our studies of mammalian PP-1 have shown in vitro that the catalytic subunit of PP-1 is phosphorylated at Thr-320 (T320) by cdc2 kinase and that this results in its inhibition (23). Similar results have been observed for an isoform of PP-1 from Schizosaccharomyces pombe (24). Thus, it seemed of considerable interest to determine at exactly what stage of the cell cycle phosphorylation of PP-1 occurs in growing and dividing cells. Our previous studies revealed the usefulness of phosphorylation state-specific antibodies for the analysis of in situ phosphorylation of various substrates (25–28). In the present study, we have developed such an antibody that specifically recognizes PP-1 phosphorylated at T320. Indirect immunofluorescence and cell fractionation studies using the phospho-T320 antibody have shown that PP-1 is phosphorylated in intact cells predominantly during early and mid-mitosis by mitotic CDKs. Phosphorylation of T320 in PP-1 is observed in many cell types arrested at mitosis, indicating that this phosphorylation is a general regulatory mechanism in mammalian cells. These results, together with our previous studies, suggest that phosphorylation and the associated inhibition of PP-1 activity are likely to contribute to the increased phosphorylation of substrates for cdc2 kinase/cyclin B that are necessary for entry into mitosis. Moreover, the subsequent dephosphorylation and activation of PP-1 are likely to contribute to completion and exit from mitosis.
MATERIALS AND METHODS
Antibodies.
Rabbit polyclonal PP-1Cα antibody was prepared as described (29). PP-1Cα phosphorylation-state-specific rabbit antisera, G-97 and G-98, were raised against the chemically phosphorylated synthetic peptide, Gly-Arg-Pro-Ile-(phospho-Thr)-Pro-Pro-Asn (residues 316–323 of PP-1Cα). Serum antibodies were prepared by injecting New Zealand White rabbits with phosphopeptide coupled to thyroglobulin. The IgG fraction was affinity-purified on a Sepharose-4B column (Pharmacia) coupled to antigen peptide. Characterization of antibody on immunoblots was carried out using nonphosphorylated or phosphorylated PP-1C. PP-1Cα and PP-1Cγ1 were expressed in Sf9 cells using baculovirus and purified (unpublished data). PP-1Cα and PP-1Cγ1 were incubated with purified cdc2/cyclin complex for up to 90 min in 50 mM Tris·HCl (pH 7.5), 10 mM MgCl2, 100 mM NaCl, 0.1 mM EDTA, 0.1 mM ATP (or [32γ-P]ATP). Proteins were separated by SDS/PAGE [10% acrylamide (wt/vol)] and transferred electrophoretically to immobilon-P (Millipore). For analysis of PP-1 phosphorylation in cells, cells were suspended in 50 mM Tris·HCl (pH 7.0) containing 0.1 M β-glycerolphosphate, 15 mM sodium pyrophosphate, 150 mM NaCl, 10 mM sodium fluoride, 4 mM benzamidine, 1 mM EDTA, 0.5 mM EGTA, 1% SDS, and protease inhibitors. After gentle sonication, proteins [bicinchoninic acid (BCA) assay; Pierce] were resolved by SDS/PAGE (10% acrylamide) and transferred to immobilon-P. Membranes were probed with phospho-PP-1C antibody (0.3 μg/ml), and either 125I-labeled protein A (New England Nuclear) or ECL (Amersham), and autoradiography.
Transfection and Retroviral Infection.
PP-1 was expressed with an N-terminal tag (ID4) that encoded part of rhodopsin (30). Briefly, the EcoRI–XhoI fragment of rabbit PP-1Cα cDNA was subcloned into a shuttle vector plasmid pBluescript SK (Stratagene). The T320A mutant cDNA was prepared by replacing the codon for T320 with that of alanine, using standard PCR methods. PCR products were excised and subcloned into the ID4 plasmid. The resulting tagged wild-type and mutant PP-1Cα cDNA were excised from the plasmid and ligated into the XhoI and BamHI sites of the retroviral expression vector, pLXSN, for the transfection experiments. DNA sequencing analysis indicated that the correct mutation was obtained. The retroviral vectors expressing wild-type PP-1Cα and the T320A mutant were transiently transfected into a “packaging” cell line, BOSC-23, as described (31). After 2 days transfection, recombinant virus was collected and used to infect the T cell hybridoma, KMls-8.3.5. Transfectants were selected as single colonies in 96-well plates in the presence of 500 μg/ml G418. Each of the 15 different colonies that survived the G418 treatment were isolated, and the expression of recombinant PP-1Cα was analyzed by immunoblotting using PP-1Cα polyclonal antibody. The transfectants showing highest expression of PP-1Cα were used for further analysis.
Cell Synchronization and Fractionation.
KMls cells were grown in suspension culture at 1–4 × 105 cells/ml in RPMI 1640 medium supplemented with 10% fetal bovine serum. Synchronization at the G1/S boundary was achieved with a sequential thymidine and aphidicolin block as described (32). Cells (2 × 105) were blocked with 2 mM thymidine for 12 h, released from the block by three washes in fresh media devoid of serum, and resuspended in complete media containing 240 μM thymidine. After 9 h, aphidicolin was added to a final concentration of 5 μg/ml for an additional 12 h. The G1/S cells were released from the block by three washes with fresh media. Synchronization of cells at the G2/M boundary was carried out by incubating 2 × 105 cells with 400 ng/ml nocodazole for 16 h. The cell synchronization stage was determined using a FACScan flow cytometer (Becton Dickinson). For subcellular fractionation, 2 × 108 KMls cells synchronized at the G2/M phase were washed with PBS, pelleted at 2000 × g, and resuspended in 10 ml of hypotonic homogenization buffer (20 mM Hepes, pH 7.5/10 mM KOAc/1 mM MgSO4/0.5 mM EDTA/1 mM DTT/10 mM NaF/0.6 mM phenylmethylsulfonyl fluoride/0.1 μM microcystin/5 μg/ml leupeptin/5 μg/ml pepstatin A/10 μg/ml chymostatin). Cells were allowed to swell for 10 min on ice and broken with a Dounce homogenizer (10 strokes). The homogenate was centrifuged at 15,000 × g for 15 min to separate the soluble fraction from the particulate fraction. The particulate fraction was resuspended in homogenization buffer containing 1% SDS plus 150 mM NaCl and completely dissolved by brief sonication on ice.
Indirect Immunofluorescence.
NIH 3T3 cells were grown overnight on 30 mm Nunclon tissue culture dishes, fixed for 15 min in 4% paraformaldehyde-PBS, permeabilized by incubation for 5 min at −20°C in methanol/acetone (50:50; vol/vol) (or alternatively, permeabilized for 10 min in PBS containing 0.1% Triton X-100), and air dried. Cells were washed and blocked by PBS containing 1% BSA and 5% fetal bovine serum. Cells were incubated with primary antibody for 2 h at room temperature, washed with PBS, and incubated for 45 min with Texas red-conjugated goat anti-rabbit immunoglobulin (Rockland, Gilbertsville, PA). After washing three times with PBS, cells were stained for DNA with 4′,6′-diamidino-2-phenylindole (DAPI) for 10 min and the coverslips were mounted with Fluoromount (Fisher).
RESULTS
Characterization of Phospho-T320-Specific Antibodies.
To assess the specificity of the phosphorylation-state-specific antisera, PP-1Cα and PP-1Cγ isoforms were phosphorylated in vitro by purified cdc2/cyclin B. Both isoforms share the consensus sequence for cdc2 phosphorylation (TPPR) but differ slightly (α, GRPITPPRN; γ, TRPVTPPRG). Serum G-97 (Fig. 1A) and G-98 (data not shown) both equally detected phosphorylated PP-1. PP-1Cα was also incubated with cdc2 kinase for various times. Phosphorylation was monitored by [32P]phosphate incorporation and G-98 immunoreactivity was compared (Fig. 1B). Immunoreactivity increased with time of incubation reaching a maximum at ≈60 min, a result that paralleled that measured by [32P]phosphate incorporation. Quantitative analysis indicated that the antibodies could detect nanogram quantities of phosphorylated PP-1C with several hundred-fold specificity over nonphosphorylated PP-1C (data not shown).
Figure 1.

Phosphorylation of PP-1C in vitro and detection by phosphorylation-state-specific antibodies. (A) Sf9 PP-1Cα (Left) and PP-1Cγ (Right) were phosphorylated by purified cdc2/cyclin B (New England Biolabs) and nonradioactive ATP for 90 min. The samples were resolved by SDS/PAGE (10% acrylamide) and immunoblotted with G-97 antibody (anti-T320 phosphorylation-state-specific PP-1Cα antibody). (B) Sf9 PP-1Cα was incubated with cdc2/cyclin B for various times in the presence of [γ-32P]ATP (Left) or nonradioactive ATP (Right). Samples were resolved by SDS/PAGE. [32P]-Labeled PP-1Cα was detected by autoradiography. Samples incubated with nonradioactive ATP were transferred to Immobilon-P, followed by immunoblotting with G-98 antibody and autoradiography. The region of the gel or immunoblot containing PP-1C is shown. No immunoreactivity was observed in any other part of the immunoblot.
PP-1C Is Phosphorylated During the G2/M Phase of the Mammalian Cell Cycle.
We analyzed phosphorylation of PP-1C using the phospho-T320 antibodies in nonsynchronized cells and in cells synchronized at various stages of the cell cycle. KMls cells were synchronized at the G1/S phase using thymidine–aphidicolin block. Cells were then released through S phase for up to 4 h. G2/M phase cells were synchronized using nocodazole and then released through the G1 phase for 4 h. Immunoblotting using an antibody which recognizes PP-1Cα irrespective of its phosphorylation state indicated that the total amount of PP-1Cα did not change during the cell cycle (Fig. 2B). The highest level of phosphorylation of T320 of PP-1C, as measured by the G-97 antibody, was detected in G2/M phase cells arrested with nocodazole (Fig. 2A). When cells were released from nocodazole block, the levels of phosphorylated PP-1 gradually decreased as the number of mitotic cells diminished. Similar results were obtained using the G-98 antibody. However, in addition to PP-1C, several cross-reacting bands were detected in the G2/M cell lysates (data not shown). The additional phosphoproteins detected by G-98 appear to be unidentified mitotic phosphoproteins that presumably have phosphorylation site sequences similar to that of PP-1C. Phosphorylation of PP-1C at the G2/M phase was found to be common to other cells in addition to KMls cells (NIH 3T3, CHO, COS-7, and PC-12). These results indicated that phosphorylation of PP-1 at T320 by cdc2 during the mitotic phase of the mammalian cell cycle is a ubiquitous regulatory mechanism. Quantitation of the results obtained in the KMls cells, using standardization with known amounts of phosphorylated and nonphosphorylated PP-1, indicated that phosphorylation was increased up to ≈25-fold and that the maximal stoichiometry of phosphorylation of phospho-T320 at the G2/M phase of the cell cycle was ≈0.1 mol/mol. However, because nocodazole treatment did not arrest all cells and cells were not arrested at exactly the same point, we consider this to be an underestimate of the maximal level of phosphorylation. Thus the degree of synchrony of the various cell preparations influenced the fold-increase in phosphorylation observed.
Figure 2.

Phosphorylation of PP-1C during the cell cycle. T cell hybridoma KMls-8.3.5 were synchronized at the G1/S or G2/M boundaries, and arrested cells were released by incubating with fresh medium for the indicated time periods. Equal amounts of protein (20 μg) were resolved by SDS/PAGE (10% acrylamide) and transferred to Immobilon-P. (A) Lysates from asynchronous (lane C), G1/S, G2/M phase cells, or cells released from drug treatment were analyzed by immunoblotting using G-97 antibody (PP-1C, Mr ≈ 37). (B) Total PP-1Cα was measured in the same samples using an anti-PP-1Cα antibody. The stages of the cell cycle were confirmed by fluorescence-activated cell sorter analysis (data not shown). PP-1Cα is present in ≈5-fold higher amounts than PP-1Cα in KMls cells (data not shown). The signal obtained with the G-97 antibody in KMls cells therefore represents predominantly PP-1Cα.
Phosphorylation of PP-1Cα in Cells Stably Expressing Wild-Type PP-1Cα or PP-1Cα T320A.
To distinguish recombinant proteins from endogenous PP-1Cα, an ID4-tagged PP-1Cα expression vector was used. The epitope tag, which was fused to the N terminus of PP-1Cα, did not affect the activity of the enzyme or phosphorylation of the C terminus by cdc2 kinase when expressed and purified in Escherichia coli (data not shown). We obtained several clones, most of which expressed the recombinant wild-type and mutant proteins at low levels (≈1–5%) compared with that of endogenous PP-1Cα. In a few clones, which were used for subsequent analysis, recombinant wild-type and mutant proteins were expressed at the same level which was ≈10% of that found for endogenous PP-1Cα (Fig. 3B). Little phosphorylation of transfected wild-type PP-1Cα or PP-1Cα T320A was detected in unsynchronized cells (Fig. 3A Left) (longer exposure of the blot showed that transfected wild-type PP-1Cα was phosphorylated to a low extent). In cells synchronized at the G2/M phase of the cell cycle, a significant increase in phosphorylation of T320 of recombinant wild-type PP-1Cα was detected (Fig. 3A Right). However, no phosphorylation of the PP-1Cα T320A mutant could be detected with the G-97 antibody. The pattern of phosphorylation of endogenous PP-1Cα was similar to that obtained in the studies shown above. However, phosphorylation of the endogenous protein was consistently slightly lower in cells expressing PP-1CαT320A (Fig. 3A Right, and data not shown), suggesting that the nonphosphorylatable T320A mutant may dephosphorylate endogenous PP-1Cα due to its higher activity during the mitotic phase. Notably, the low expression of the T320A mutant precluded an analysis of the functional consequences of the mutation, and the growth characteristics of cells stably expressing the T320A mutant were apparently normal.
Figure 3.

T320 phosphorylation in cells stably transfected with wild-type PP-1Cα or a PP-1CαT320A mutant. T cell hybridoma KMls were infected with retrovirus stocks expressing either ID4-tagged wild-type PP-1Cα (T320) or ID4-tagged PP-1Cα in which T320 was mutated to alanine (T320A). Single clones that expressed similar levels of ID4-tagged PP-1Cα or PP-1Cα T320A were analyzed in this experiment. (Left) Control asynchronous cells. (Right) Untransfected (−), wild-type PP-1Cα-transfected (T320), and PP-1Cα T320A-transfected (T320A) cells were synchronized at the G2/M phase using 400 ng/ml nocodazole treatment for 16 h. Whole cell lysates were prepared as described and aliquots (20 μg protein) were resolved on SDS/PAGE, followed by immunoblotting with either G-97 antibody (A) or anti-PP-1Cα antibody (B). The ID4-tagged PP1C (T-PP-1Cα) migrated with a slightly higher molecular weight than endogenous PP1Cα.
PP-1 Phosphorylation Is Blocked by a CDK Inhibitor and Stimulated by the PP-1/2A Inhibitor, Calyculin A.
The phosphorylation state of PP-1 seemed likely to reflect the relative activities of cdc2 kinase and protein phosphatase(s) at all stages of the cell cycle. To investigate this further, NIH 3T3 cells were synchronized at the G2/M phase of the cell cycle and incubated with either olomoucine, a specific inhibitor of CDK activity (33), okadaic acid or calyculin A, specific inhibitors of PP-1 and PP-2A, or a combination of olomoucine and calyculin A (Fig. 4). Treatment with olomoucine significantly reduced the level of phospho-T320 of PP-1C. Okadaic acid at a concentration of 100 nM had no significant effect on phosphorylation of T320. Higher concentrations of okadaic acid (up to 1 μM) also had little effect (data not shown). In contrast, treatment with 100 nM calyculin A stimulated phosphorylation of T320 ≈3-fold. When both calyculin A and olomoucine were added simultaneously, the result obtained was similar to that observed with calyculin A alone. Based on the lack of effect of okadaic acid and the sensitivity to low concentrations of calyculin A, these results raise the possibility that PP-1 is responsible for its own dephosphorylation following the decrease in cdc2 kinase activity at anaphase (34).
Figure 4.

Effect of phosphatase and cdk kinase inhibitors on the T320 phosphorylation of PP-1C. Asynchronous cells (lane 1) or NIH 3T3 cells synchronized at the G2/M phase using 400 ng/ml nocodazole treatment for 16 h (lanes 2–6) were incubated for an additional 1 h in the absence of inhibitors (lanes 1 and 2), in the presence of 300 μM olomoucine (lane 3), 100 nM okadaic acid (lane 4), 100 nM calyculin A (lane 5), or 300 μM olomoucine plus 100 nM calyculin A (lane 6). Whole cell lysates were prepared as described, and aliquots (15 μg protein) were resolved on SDS/PAGE and immunoblotted with either G-97 antibody (A) or anti-PP-1Cα antibody (B). Quantitation of three individual experiments indicated the following averages: control, 2 (arbitrary) units; nocodazole, 45 units; olomoucine, 4 units; okadaic acid, 46 units; calyculin A, 141 units; calyculin A plus olomoucine, 140 units.
Subcellular Localization of T320 Phosphorylation of PP-1C in Mitotic Cells.
The high level of specificity of the G-97 antibody allowed a detailed analysis of the subcellular localization of phosphorylated PP-1C in cells using indirect immunofluorescence microscopy (Fig. 5). This type of analysis had the further advantage that the distribution of phosphorylated PP-1C could be studied in populations of growing cells that had not been exposed to any drug treatment. Immunofluorescence analysis indicated that the 2 cells (of 13 cells shown in Fig. 5A) that were mitotic as revealed by DAPI staining exhibited a high level of staining using the G-97 antibody. Comparison of these two labeled cells indicated that a higher level of staining was present in the cell in metaphase compared with the late anaphase cell. Further examples of individual cells at various stages of mitosis are shown in Fig. 5 B–F. G-97 immunofluorescence was first detected at prophase (Fig. 5B), peaked at metaphase (Fig. 5D), and faded through late anaphase (Fig. 5F). At prophase, phospho-PP-1 was detected in the nucleus most likely between condensing chromosomes. In later mitotic stages, including prometaphase, metaphase, and anaphase, immunofluorescence was present mainly in nonchromosomal regions. To further analyze the subcellular localization of phosphorylated PP-1, KMls cells were arrested at the G2/M phase of the cell cycle, and soluble and particulate fractions were prepared. Immunoblotting analysis indicated that both α and γ isoforms of nonphosphorylated PP-1 were present in both soluble and particulate fractions (Fig. 6). In contrast, phosphorylated PP-1 was present only in the soluble fraction.
Figure 5.
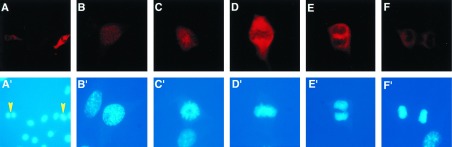
T320 phosphorylation of PP-1Cα analyzed in NIH 3T3 cells by indirect immunofluorescence. NIH 3T3 cells were fixed, permeabilized, and stained with G-97 antibody followed by anti-rabbit IgG conjugated with Texas red as secondary antibody. (A) Low magnification view showing several cells at various stages of the cell cycle. (B–F) Higher magnification showing individual cells in prophase (B), prometaphase (C), metaphase (D), anaphase (E), and late anaphase (F). (A′–F′) DAPI-stained DNA of cells shown in A–F. The yellow arrowheads in A′ indicate 2 of 13 cells that are mitotic. The differential staining of mitotic cells is not due to cell thickness because identical results were obtained using confocal microscopy (data not shown). The results shown are representative of many individual experiments in which the same results were obtained.
Figure 6.

Subcellular localization of PP-1Cα. KMls cells were synchronized at the G2/M phase, pelleted, and fractionated; W, S, and P indicate whole cell lysates, soluble, and particulate fractions, respectively. Aliquots (20 μg protein) were subjected to SDS/PAGE and immunoblotting with either G-97 antibody (Left) or anti-PP-1Cα antibody (Right). Identical results were obtained for the subcellular distribution of PP-1Cγ.
DISCUSSION
Using immunoblotting and immunofluorescence methods, together with a phosphorylation-state-specific antibody that recognizes only the phosphorylated form of T320, we have observed that PP-1 is maximally phosphorylated during early and mid-mitosis. Because any attempt to block dephosphorylation in cell extracts by phosphatase inhibitors would lead to inhibition of PP-1 itself, it is not feasible to directly study the consequences of the phosphorylation of PP-1 in intact cells. However, the immunofluorescence analysis used here allows a quantitative assessment of the cellular pool of PP-1 that would be subject to regulation by phosphorylation. Our results indicated that ≈10% of PP-1C in KMls cells synchronized with nocodazole was phosphorylated at the G2/M phase. However, we believe that this is an underestimate of the true stoichiometry because the cells were not completely synchronized. Moreover, the immunofluorescence studies indicated how precisely the peak of PP-1C phosphorylation occurred during mitosis. Finally, the cell fractionation studies indicated that phosphorylated PP-1C was present only in the soluble fraction (which represents ≈40% of the total). Our previous in vitro studies (23) as well as those of Yanagida and colleagues (24) indicated that phosphorylation of PP-1 at T320 by cdc2 kinase resulted in inhibition of enzyme activity. Together then, the results suggest that the activity of a significant fraction of the soluble pool of cellular PP-1 is decreased during mitosis.
Several genetic studies have indicated that PP-1 activity is critical for exit from mitosis. In Aspergillus, the bimG11 gene is necessary to complete the separation of daughter nuclei at anaphase (17). In Drosophila, mutation of one of four PP-1 isoforms, PP-1 87B, delays progress through mitosis, and the mutation is characterized by defective spindle elongation, defective chromosome segregation, and excessive chromosome condensation (18). In S. pombe, mutation of one of two PP-1 isoforms, the dis2 gene, results in a failure to exit from mitosis (16). Similar results were found when the one PP-1 gene, GLC7, was mutated in Saccharomyces cerevisiae (35). Several biochemical studies have also examined the role of PP-1 in cell cycle progression. In Xenopus egg extracts, PP-1 activity was found to be high during interphase, to decrease at the onset of mitosis, and to increase slightly during mid-mitosis (36). In this study, injection of inhibitor-2, a specific inhibitor of PP-1, resulted in early entry into mitosis, while injection of PP-1 lengthened interphase. In a study using REF-52 fibroblasts, injection of inhibitory PP-1 antibodies into late G2 cells blocked the cells at metaphase, while injection of PP-1 into anaphase cells accelerated cytokinesis and completion of daughter cell separation (19).
Based on the present study, together with the genetic and biochemical studies, we propose a potential model for the role of phosphorylation of PP-1 by cdc2 kinase. PP-1 phosphorylation begins to increase from a very low basal level in prophase, to peak in metaphase, to decrease in anaphase, and to return to basal levels by telophase. Thus, phosphorylation of PP-1 parallels the activity of cdc2 kinase through M phase (4). Phosphorylation of PP-1 at T320 by cdc2 kinase inhibits enzyme activity in vitro (23, 24). We speculate that increased phosphorylation and inactivation of PP-1 during early to mid-mitosis, together with activation of CDK, is likely to contribute to the maintenance of high levels of phosphorylation of mitotic phosphoproteins that are necessary for the transition through early to mid-mitosis. The decrease in phosphorylation of PP-1 and its subsequent activation during anaphase would then be required for the dephosphorylation of certain mitotic phosphoproteins that are required for exit from mitosis.
An important feature of PP-1 isoforms is that most but not all contain the C-terminal site of phosphorylation for cdc2 kinase (37). All mammalian PP-1 isoforms, two of the four Drosophila genes (96A and 9C), and one of the S. pombe genes (dis2) contain the site, but sds21, the other PP-1 gene in S. pombe, and GLC7, the one PP-1 gene in Saccharomyces cerevisiae, do not. In S. pombe dis2 is phosphorylated in vitro by cdc2 kinase and inhibited, and is phosphorylated in intact cells at the C-terminal site, T316 (homologous to T320 of mammalian PP-1Cα) (24). In wild-type cells, dis2 is much more abundant than sds21. Overexpression of sds21 resulted in inhibition of cell growth, and a mutation that introduces the cdc2 phosphorylation site into the C terminus of sds21 resulted in normal cell growth (24). In contrast, overexpression of wild-type dis2 did not disrupt cell growth. However, when T316 of dis2 was replaced by alanine, colony formation was severely inhibited (24). Thus, PP-1 phosphorylation by cdc2 kinase is important for regulation of isoforms of PP-1 that are expressed at high levels. In mammalian cells, PP-1 is an abundant multifunctional protein phosphatase whose activity is required for the regulation of many cell processes during interphase as well as in late mitosis. Therefore, in mammalian cells, phosphorylation/dephosphorylation of PP-1 appears to provide a mechanism that balances the requirement for active PP-1 during late mitosis and interphase with a requirement for a pool of inactive PP-1 that is necessary during early to mid-mitosis.
The expression of cdc2 kinase is fairly constant, and the protein is present in both the cytosol and nucleus during the cell cycle, whereas cyclin B translocates from the cytosol to the nucleus at the onset of mitosis (38, 39). PP-1 is present in both the cytosol and nucleus in interphase cells (16, 40). It has also been suggested that PP-1 may be recruited from the cytosol to the nucleus at the G2 phase (19). Our immunofluorescence results indicate that phospho-PP-1 is present within the nucleus in early mitotic cells. Thus, at least initially, PP-1 is phosphorylated within the nucleus, suggesting that inhibition of PP-1 activity might be relevant to nuclear substrates for cdc2 kinase. A variety of proteins are phosphorylated by cdc2 kinase as a requirement for entry into mitosis. These include components of the kinase/phosphatase cascade that regulates cdc2 kinase, nuclear envelope proteins such as lamins, and microtubule-associated proteins. Other proteins such as histones and transcription factors are also phosphorylated (6–8, 10–12, 15, 22). In turn, dephosphorylation of critical phosphoproteins is required for mitosis to end. PP-1 substrates at the end of mitosis could be identical to those whose phosphorylation was required for mitosis to begin. However, there may be independent groups of PP-1 substrates that are involved only in entry or in exit from mitosis. Our present results indicate that while phospho-PP-1 is first observed within the nucleus, it exists as a soluble protein after breakdown of the nuclear envelope, being localized to nonchromosomal regions. Notably, the transcription factor Oct-1 is highly phosphorylated during mitosis, resulting in translocation from the chromosome to the cytosol (41). In addition, many other mitotic phosphoproteins exhibit a localization pattern similar to that observed for the G-97 PP-1 antibody (42, 43).
PP-1 is highly regulated by the interaction of the catalytic subunit with a variety of targeting proteins that serve to localize the enzyme to discreet subcellular compartments as well as to influence substrate specificity (13, 14, 44, 45). These include proteins that target PP-1 to glycogen, myofibrils, and the nucleus. Interestingly, in S. pombe, a protein termed sds22 that is required in late mitosis has been shown to bind both the dis2 and sds21 PP-1 isoforms, to activate PP-1, and to alter its substrate specificity (46, 47). The RB protein (p110RB), a tumor suppressor, is known to be hyperphosphorylated by cdc2, reaching a peak at mid-mitosis, and to be physically associated with PP-1 from mid-mitosis to early G1 (21). Moreover, RB-specific mitotic phosphatase activity, which is believed to be primarily PP-1, was found to be higher at anaphase than at metaphase (48). It is therefore possible that in addition to inhibiting PP-1 activity, phosphorylation may modulate the interaction between PP-1 and its associated proteins, resulting in alteration of the substrate specificity of PP-1.
In conclusion, the present results indicate that PP-1 phosphorylation at T320 by cdc2 kinase occurs during early to mid-mitosis and that the associated inhibition of PP-1 activity is likely to be an important factor in the balance between protein phosphorylation and dephosphorylation that accompanies, respectively, entry into and exit from mitosis. This regulatory mechanism is only one of many that are critical to maintaining the correct progression through mitosis as well as other phases of the cell cycle. PP-1 activity is also likely to be regulated during the cell cycle by other mechanisms. For example, inhibitor-2, sds22 or its homologs, or any one of many PP-1 targeting proteins likely to exist could be involved in this process. Future studies will be needed to identify substrates for PP-1 that are important for the different phases of mitosis, as well as the role that PP-1 phosphorylation might play in regulating its intracellular localization and interactions with other proteins.
Acknowledgments
We thank Dr. Edgar da Cruz e Silva for the PP-1 antibodies, Dr. Hsien-bin Huang for PP-1, Dr. Chae Gyu Park for help with preparation of the ID4 plasmid, Gloria Bertuzzi for technical assistance, and Dr. James Bibb for assistance in figure preparation. Y.C. is an Assistant Investigator of the Howard Hughes Medical Institute. This research was supported by National Institutes of Health Grant MH 40899 (to A.C.N. and P.G.).
ABBREVIATIONS
- PP-1
protein phosphatase 1
- CDK
cyclin-dependent protein kinase
- RB
retinoblastoma
- DAPI
4′,6′-diamidino-2-phenylindole
Note Added in Proof
Similar results have been recently obtained in studies of phosphorylation of PP-1 in S. pombe (49).
References
- 1.Reed S I. Annu Rev Cell Biol. 1992;8:529–561. doi: 10.1146/annurev.cb.08.110192.002525. [DOI] [PubMed] [Google Scholar]
- 2.Coleman T R, Dunphy W G. Curr Opin Cell Biol. 1994;6:877–882. doi: 10.1016/0955-0674(94)90060-4. [DOI] [PubMed] [Google Scholar]
- 3.King R W, Jackson P K, Kirschner M W. Cell. 1994;79:563–571. doi: 10.1016/0092-8674(94)90542-8. [DOI] [PubMed] [Google Scholar]
- 4.Hunter T, Pines J. Cell. 1994;79:573–582. doi: 10.1016/0092-8674(94)90543-6. [DOI] [PubMed] [Google Scholar]
- 5.Elledge S, Harper J W. Curr Opin Cell Biol. 1994;6:847–852. doi: 10.1016/0955-0674(94)90055-8. [DOI] [PubMed] [Google Scholar]
- 6.Langan T A, Gautier J, Lohka M, Hollingsworth R, Moreno S, Nurse P, Maller J, Sclafani R A. Mol Cell Biol. 1989;9:3860–3868. doi: 10.1128/mcb.9.9.3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heald R, McKeon F. Cell. 1990;61:579–589. doi: 10.1016/0092-8674(90)90470-y. [DOI] [PubMed] [Google Scholar]
- 8.Chou Y-H, Bischoff J R, Beach D, Goldman R D. Cell. 1990;62:1063–1071. doi: 10.1016/0092-8674(90)90384-q. [DOI] [PubMed] [Google Scholar]
- 9.Reeves R. Curr Opin Cell Biol. 1992;4:413–423. doi: 10.1016/0955-0674(92)90006-x. [DOI] [PubMed] [Google Scholar]
- 10.Nigg E A. Curr Opin Cell Biol. 1993;5:1187–1193. doi: 10.1016/0955-0674(93)90101-u. [DOI] [PubMed] [Google Scholar]
- 11.Blangy A, Lane H A, d’Hérin P, Harper M, Kress M, Nigg E A. Cell. 1995;83:1159–1169. doi: 10.1016/0092-8674(95)90142-6. [DOI] [PubMed] [Google Scholar]
- 12.Malecz N, Foisner R, Stadler C, Wiche G. J Biol Chem. 1996;271:8203–8208. doi: 10.1074/jbc.271.14.8203. [DOI] [PubMed] [Google Scholar]
- 13.Shenolikar S, Nairn A C. Adv Second Messenger Phosphoprotein Res. 1991;23:1–121. [PubMed] [Google Scholar]
- 14.Shenolikar S. Annu Rev Cell Biol. 1994;10:55–86. doi: 10.1146/annurev.cb.10.110194.000415. [DOI] [PubMed] [Google Scholar]
- 15.Hunter T. Cell. 1995;80:225–236. doi: 10.1016/0092-8674(95)90405-0. [DOI] [PubMed] [Google Scholar]
- 16.Ohkura H, Kinoshita N, Miyatani S, Toda T, Yanagida M. Cell. 1989;57:997–1007. doi: 10.1016/0092-8674(89)90338-3. [DOI] [PubMed] [Google Scholar]
- 17.Doonan J, Morris N R. Cell. 1989;57:987–996. doi: 10.1016/0092-8674(89)90337-1. [DOI] [PubMed] [Google Scholar]
- 18.Axton J M, Dombrádi V, Cohen P T W, Glover D M. Cell. 1990;63:33–46. doi: 10.1016/0092-8674(90)90286-n. [DOI] [PubMed] [Google Scholar]
- 19.Fernandez A, Brautigan D L, Lamb N J C. J Cell Biol. 1992;116:1421–1430. doi: 10.1083/jcb.116.6.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brautigan D L, Sunwoo J, Labbe J-C, Fernandez A, Lamb N J C. Nature (London) 1990;344:74–78. doi: 10.1038/344074a0. [DOI] [PubMed] [Google Scholar]
- 21.Durfee T, Becherer K, Chen P-L, Yeh S-H, Yang Y, Kilburn A E, Lee W-H, Elledge S J. Genes Dev. 1993;7:555–569. doi: 10.1101/gad.7.4.555. [DOI] [PubMed] [Google Scholar]
- 22.Izumi T, Walker D H, Maller J L. Mol Biol Cell. 1992;3:927–939. doi: 10.1091/mbc.3.8.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dohadwala M, da Cruz e Silva E F, Hall F L, Williams R T, Carbonaro-Hall D A, Nairn A C, Greengard P, Berndt N. Proc Natl Acad Sci USA. 1994;91:6408–6412. doi: 10.1073/pnas.91.14.6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yamano H, Ishii K, Yanagida M. EMBO J. 1994;13:5310–5318. doi: 10.1002/j.1460-2075.1994.tb06865.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Czernik A J, Girault J-A, Nairn A C, Chen J, Snyder G, Kebabian J, Greengard P. Methods Enzymol. 1991;201:264–283. doi: 10.1016/0076-6879(91)01025-w. [DOI] [PubMed] [Google Scholar]
- 26.Snyder G L, Girault J-A, Chen J Y C, Czernik A J, Kebabian J W, Nathanson J A, Greengard P. J Neurosci. 1992;12:3071–3083. doi: 10.1523/JNEUROSCI.12-08-03071.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fisone G, Cheng S X-J, Nairn A C, Czernik A J, Hemmings H C, Jr, Hoog J-O, Bertorello A M, Kaiser R, Bergman T, Jornvall H, Aperia A, Greengard P. J Biol Chem. 1994;269:9368–9373. [PubMed] [Google Scholar]
- 28.Jovanovic J, Benfenati F, Siow Y L, Sihra T S, Sanghera J S, Pelech S L, Greengard P, Czernik A J. Proc Natl Acad Sci USA. 1996;93:3679–3683. doi: 10.1073/pnas.93.8.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.da Cruz e Silva E F, Fox C A, Quimet C C, Gustafson E, Watson S J, Greengard P. J Neurosci. 1995;15:3375–3389. doi: 10.1523/JNEUROSCI.15-05-03375.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.MacKenzie D, Arendt A, Hargrave P, McDowell J H, Molday R S. Biochemistry. 1984;23:6544–6549. doi: 10.1021/bi00321a041. [DOI] [PubMed] [Google Scholar]
- 31.Pear W S, Nolan G P, Scott M L, Baltimore D. Proc Natl Acad Sci USA. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heintz N, Sive H L, Roeder R G. Mol Cell Biol. 1983;3:539–550. doi: 10.1128/mcb.3.4.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park D S, Farinelli S E, Greene L A. J Biol Chem. 1996;271:8161–8169. doi: 10.1074/jbc.271.14.8161. [DOI] [PubMed] [Google Scholar]
- 34.Murray A W, Solomon M J, Kirschner M W. Nature (London) 1989;339:280–286. doi: 10.1038/339280a0. [DOI] [PubMed] [Google Scholar]
- 35.Hisamoto N, Sugimoto K, Matsumoto K. Mol Cell Biol. 1994;14:3158–3165. doi: 10.1128/mcb.14.5.3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walker D H, DePaoli-Roach A A, Maller J L. Mol Biol Cell. 1992;3:687–698. doi: 10.1091/mbc.3.6.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barton G J, Cohen P T W, Barford D. Eur J Biochem. 1994;220:225–237. doi: 10.1111/j.1432-1033.1994.tb18618.x. [DOI] [PubMed] [Google Scholar]
- 38.Gallant P, Nigg E A. J Cell Biol. 1992;117:213–224. doi: 10.1083/jcb.117.1.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pines J, Hunter T. EMBO J. 1994;13:3772–3781. doi: 10.1002/j.1460-2075.1994.tb06688.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bullens M, Eynde A V, Stalmans W, Bollen M. J Biol Chem. 1992;267:16538–16544. [PubMed] [Google Scholar]
- 41.Segil N, Roberts S B, Heintz N. Science. 1991;254:1814–1816. doi: 10.1126/science.1684878. [DOI] [PubMed] [Google Scholar]
- 42.Butschak G, Harborth J, Osborn M, Karsten U. Acta Histochem. 1995;97:19–31. doi: 10.1016/S0065-1281(11)80203-5. [DOI] [PubMed] [Google Scholar]
- 43.Masumoto-Taniura N, Pirollet F, Monroe R, Gerace L, Westendorf J M. Mol Biol Cell. 1996;7:1455–1469. doi: 10.1091/mbc.7.9.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hubbard M J, Cohen P. Trends Biochem Sci. 1993;18:172–177. doi: 10.1016/0968-0004(93)90109-z. [DOI] [PubMed] [Google Scholar]
- 45.Faux M C, Scott J D. Trends Biochem Sci. 1996;21:279–323. [PubMed] [Google Scholar]
- 46.Ohkura H, Yanagida M. Cell. 1991;64:149–157. doi: 10.1016/0092-8674(91)90216-l. [DOI] [PubMed] [Google Scholar]
- 47.Stone E M, Yamano H, Kinoshita N, Yanagida M. Curr Biol. 1993;3:13–26. doi: 10.1016/0960-9822(93)90140-j. [DOI] [PubMed] [Google Scholar]
- 48.Ludlow J W, Glendening C L, Livingston D M, DeCaprio J A. Mol Cell Biol. 1993;13:367–372. doi: 10.1128/mcb.13.1.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ishii K, Kumada K, Toda T, Yanagida M. EMBO J. 1996;15:6629–6640. [PMC free article] [PubMed] [Google Scholar]