

Abstract
The SIR2, SIR3, and SIR4 silent information regulator proteins are involved in the assembly of silent chromatin domains in the budding yeast Saccharomyces cerevisiae. Using a series of biochemical experiments, we have studied protein–protein interactions involving these proteins. We found that yeast extracts contained a SIR2/SIR4 complex that was associated with little or no SIR3. However, truncations of the N-terminal two-thirds of the SIR4 protein allowed it to efficiently associate with SIR3, suggesting that the N-terminal domain of SIR4 inhibited its interaction with SIR3. We propose that the SIR3 and SIR4 proteins interact only during the assembly of the SIR protein complex at the silencer and that an early step in assembly unmasks the SIR4 protein to allow its association with SIR3. To test whether the interactions observed in yeast extracts were direct, we tested these SIR-SIR interactions using bacterially expressed SIR proteins. We observed direct interactions between SIR4 and SIR2, SIR4 and SIR3, SIR2 and SIR3, SIR2 and SIR2, and SIR4 and SIR4, indicating that the associations observed in yeast extracts were direct.
Keywords: RAP1, macromolecular assembly, silencing, heterochromatin
Transcriptionally inactive heterochromatic domains of DNA are a feature of eukaryotic chromosomes conserved in organisms from human to yeast (reviewed in ref. 1). These specialized chromatin domains are involved both in the regulation of gene expression and in the maintenance of chromosome stability (2–4). In the budding yeast Saccharomyces cerevisiae, transcriptionally silent domains of chromatin are located at the silent mating type loci, HML and HMR, and at the telomeres. Silencing in yeast shares many features with heterochromatic inactivation of genes in larger eukaryotes. Like heterochromatin, silent domains in yeast are epigenetically inherited (5), replicate late in the S phase (6), and appear clustered in certain regions within the nucleus (7). Whether or not the yeast silent chromatin domains also share specific structural components with heterochromatin of multicellular eukaryotes remains unknown.
Silencing is directed by specific regulatory sites termed silencers (8, 9). The HMR silencers are tripartite elements that contain binding sites for the abundant nuclear factors RAP1 and ABF1, and the origin recognition complex (ORC) (10–13). At the telomeres, specific RAP1-binding sites are found within the telomeric [(C)1–3A]n tracts, and the identification of rap1 mutants defective in telomeric silencing indicates that RAP1 is involved in this process (14, 15). The SIR2, SIR3, and SIR4 silent information regulator proteins are required for silencing at both the silent mating type loci and telomeres (16, 17). Some of the SIR proteins interact with silencer binding proteins in two hybrid experiments. SIR3 and SIR4 proteins interact with RAP1 (18), and SIR4 interacts with SIR1 in this assay (19). SIR1, which is required for the establishment of silencing at the HM loci, but not at telomeres (5, 17), can interact with a subunit of ORC, ORC1, in two-hybrid experiments and under certain in vitro conditions (19).
Histones also have been shown to play an essential role in silencing; the N-termini of histones H3 and H4 are required for silencing and interact with the SIR3 and SIR4 proteins in vitro (20–22). Consistent with this observation, SIR3 and SIR4 copurify with nucleosomal histones (23), although it is possible that some of these interactions are mediated by DNA. Based on these observations, Grunstein and colleagues have proposed that after associating with silencer-bound proteins SIR3 and SIR4 interact with the N-termini of histones H3 and H4 and that these interactions allow for the spreading of the SIR proteins along chromatin (23). Whether a complex containing all three SIR proteins exists and is recruited to the silencer, or whether each protein is recruited independently and assembled in stepwise fashion is not known. Furthermore, two-hybrid studies, either as general screens or as direct assays for protein–protein interactions, have not uncovered an interaction between SIR2 and other silencing proteins. SIR2-like sequences have been identified in both eukaryotes, including Caenorhabditis elegans and human, and in Staphylococcus aureus (24). This evolutionary conservation suggests that interactions involving SIR2 are likely to play a central role in silencing.
We previously have shown that the SIR2 and SIR3 proteins are specifically retained by an affinity column composed of the C-terminal half of the SIR4 protein, indicating that these proteins can physically associate (25). Here we report on studies involving the interactions of the SIR2, SIR3, and SIR4 proteins in yeast extracts and also examine these interactions using SIR proteins purified from Escherichia coli. We found that in yeast extracts the SIR2 and SIR4 proteins were tightly associated, as determined by both coimmunoprecipitation and glutathione S-transferase (GST) affinity copurification under stringent conditions. The SIR2/SIR4 complex contained only trace amounts of SIR3, but truncations of the SIR4 protein that removed all but its C-terminal 244 amino acids allowed its efficient association with SIR3. These results, together with the interactions observed by others, suggest that the SIR3–SIR4 interaction may be a regulated step in the assembly of the SIR protein complex that takes place only during assembly of the complex at the silencer. Finally, using bacterially expressed and purified SIR proteins, we showed that the SIR2, SIR3, and SIR4 proteins could interact with each other directly, and that the SIR2 and SIR4 proteins could form dimers in vitro.
MATERIALS AND METHODS
Expression Plasmids, Strains, and Antibodies.
GST fusion proteins were expressed in yeast using a plasmid (pRD56, a generous gift from R. Deshaies, California Institute of Technology) that contains the EcoRI-BamHI fragment of the GAL1/10 promoter followed by the GST coding region in the polylinker of pRS316 (26). An EcoRI site was inserted after the ATG start codon of SIR4 as previously described (pDM118) (25). An EcoRI–XhoI fragment containing the entire SIR4 open reading frame and 200 bp downstream of the stop codon was ligated into the EcoRI–XhoI site of pRD56 to produce pDM118a (GST-SIR4). pDM118a was digested with BglII and ClaI (to remove sequences encoding SIR4 amino acids 731-1358), blunt ended, and ligated to produce pGN4 (GST-SIR4N2). pGC4–1 (GST-SIR4C1) was produced by digestion of pDM118a with EcoRI and BglII (to remove amino acids 1–730 of SIR4), followed by filling in with Klenow enzyme and ligating. pG4C-2 (GST-SIR4C2) was generated by ligation of a PvuII SIR4 fragment (encoding amino acids 1115–1358 of SIR4 and the SIR4 3′ untranslated region) into the SmaI site of pRD56. To modify the endogenous SIR4 gene, pDM196 was constructed as described below. pDM196 contains 600 bp of DNA from the upstream and promoter region of the SIR4 gene followed by 2.5 kb of DNA encoding the N-terminal half of the SIR4 protein; the N terminus of SIR4 in pDM196 is modified to insert six histidines followed by three hemagglutinin (HA) epitopes. The SIR4 upstream region was PCR-amplified using an oligonucleotide to insert six histidine codons after the SIR4 start codon followed by an EcoRI recognition site. The GAL1/10-GST region in plasmid pDM118a was then replaced with SIR4 promoter region-ATG-6His fragment by digesting pDM118a with EcoRI and XhoI and ligating this with the above PCR fragment, also digested with EcoRI and XhoI, to produce pDM151. An XbaI–BamHI fragment from pDM151 then was subcloned into the XbaI–BamHI site of pRS306ΔRI, pRS306 (26) in which the EcoRI site was destroyed, to produce pDM158. An EcoRI fragment containing the triple-HA epitope was PCR-amplified from YIplac111 (a plasmid that encodes three tandem HA epitopes in its polylinker) and inserted into the EcoRI site of pDM158. Correctly oriented inserts were identified by PCR, and a plasmid containing a single insert with the correct sequence was identified by DNA sequencing.
Plasmids for the expression of GST-SIR4 fusion proteins in E. coli were previously described (25). pDM111 (GST-SIR2) was produced by ligation of an EcoRI fragment containing the entire SIR2 coding region except for the ATG start codon into the EcoRI site of pGEX-1B. pDM113 was generated by subcloning of a 550-bp EcoRI–ClaI PCR fragment containing amino acids 2–183 of SIR3 into the EcoRI–ClaI site of pRD54a (identical to pRD56 except that the HA epitope replaces GST after the GAL1 promoter). pDM120a (GAL1-HA-SIR3) then was produced by ligation of the ClaI–HpaI fragment of SIR3 encoding the remainder of the SIR3 protein into pDM113. pDM135 (GST-SIR3) was produced by ligation of the SIR3 SmaI–BamHI fragment from pDM120a into the SmaI–BamHI site of the pGEX-1 expression vector (Pharmacia). pDM141 (GST-SIR3C) was constructed by ligation of a blunt-ended 1598-bp EcoRI fragment of SIR3, encoding amino acids 439–971 of SIR3, to the SmaI site of pGEX-2T. pDM168 (GST-RAP1C) was constructed by insertion of an EcoRI–XhoI PCR fragment encoding amino acids 563–827 of RAP1 into the EcoRI–XhoI site of pGEX-4T-1.
Strains JRY2334 (W303, 1a MATa ade2–1 can1–100 his3–11 leu2–3.112 trp1 ura3–1 GAL), JRY3411 (sir4Δ::HIS3 in JRY2334) and FM135 (MATa ura3–52 leu2–3.112 reg1–501 gal1 pep4–3 prb1–112; gift of R. Deshaies) (27) were used for the expression and purification of GST fusion proteins. Strain DM428 (6His-3HA-SIR4/URA3) was constructed by transforming BJ5459 (MATa ura3–52 trp1 lys2–801 leu2Δ1 his3Δ200 pep4::HIS3 prb1Δ1.6 can1 GAL) (28) with pDM196, digested with SphI, and screening the resulting URA+ transformants for correct integration using a PCR assay and for expression of HA-SIR4 by Western blotting. As determined by a quantitative mating assay (25), DM428 mated with wild-type efficiency, indicating that the tagged SIR4 protein was fully functional.
A trpE-SIR2 fusion protein was used to produce mouse monoclonal antibodies against the SIR2 protein (designated ADB6 and ADB9). Rabbit antibodies that recognize the SIR3 protein were produced against a β-galactosidase-SIR3 fusion protein. The SIR3 antiserum was affinity purified using GST-SIR3 crosslinked to Affigel 10 as described (29).
Expression and Purification of GST Fusion Proteins from Yeast.
Cultures of yeast cells expressing GST, GST-SIR4, GST-SIR4N2, GST-SIR4C1, or GST-SIR4C2 were grown in selective medium containing 2% raffinose and 1% galactose to an OD660 of 1–1.5. When induction was in strain FM135 (27), the carbon source was glucose. Cells were harvested, washed with ice-cold water, frozen in liquid nitrogen, and stored at −70°C. All of the remaining steps were performed at 4°C. Frozen cell pellets (≈15–20 g) were resuspended in 30 ml of lysis buffer A [50 mM Hepes, pH 7.6/10% glycerol (vol/vol)/10 mM EDTA/0.5 M NaCl/1% Triton X-100/5 mM DTT/2 μg/ml each leupeptin, bestatin, and pepstatin/5 mM benzamidine hydrochloride/1 mM phenylmethylsulfonyl fluoride]. Cells were disrupted by grinding with glass beads as described previously (25). After centrifugation at 15,000 rpm in a SS34 rotor (Sorvall) for 30 min, the supernatant was added to 5-ml packed volume of glutathione agarose beads (Sigma) that had been equilibrated with lysis buffer and incubated on a mixer at 4°C for 1 h. The beads were recovered by centrifugation at 500 × g for 2 min, resuspended in 5 ml of lysis buffer, transferred to a small column, and washed with 20 column volumes of wash buffer [50 mM Hepes, pH 7.6/10% glycerol/10 mM EDTA/0.5 M NaCl/5 mM Mg(OAc)2/1% Triton X-100/1 mM DTT/5 mM benzamidine hydrochloride, 1 mM phenylmethylsulfonyl fluoride/1 μg/ml each leupeptin, bestatin and pepstatin] and with 4 column volumes of the same buffer without Triton X-100 or the protease inhibitors. Bound protein then was eluted with 10 mM glutathione in 50 mM Hepes, pH 7.8/10% glycerol/200 mM NaCl/1 mM DTT. Peak protein fractions (0.7 ml each) were identified using the Bradford protein assay and pooled. Approximately equal amounts of each protein were loaded on SDS 8.5% polyacrylamide gels, and after electrophoresis and transfer to a poly(vinylidene difluoride) membrane (Millipore), probed sequentially or in parallel with appropriate antibodies. Proteins on Western blots were detected by using the Amersham Enhanced Chemiluminescence (ECL) system.
Immunoprecipitation of the SIR Proteins from Yeast Extracts.
Cultures (3 liters) of strain DM428 (6HIS-3HA-SIR4) were grown to an OD660 of 1–1.5 in rich medium (yeast extract/peptone/dextrose), harvested, washed with ice-cold water, frozen in liquid nitrogen, and stored at −70°C. Cell pellets (15–20 g) were resuspended in 40 ml of lysis buffer B (50 mM Hepes, pH 7.5/0.5 M NaCl/10% glycerol/0.5% Nonidet P-40/6 mM 2-mercaptoethanol/2.5 mM bezamidine·HCl/0.5 mM phenylmethylsulfonyl fluoride/2 μg/ml each leupeptin, bestatin, and pepstatin). After centrifugation at 12,000 rpm in an SS34 rotor for 20 min at 4°C, the supernatant either was used immediately for binding experiments or was snap-frozen and stored at −70°C. To obtain a fraction that is enriched for SIR4, 10 ml of the supernatant was added to 1 ml of Ni2+-NTA-agarose (Qiagen) and incubated by mixing at 4°C for 1 h. The agarose then was collected by centrifugation at 500 × g for 2 min and washed twice with 10 ml of lysis buffer containing 0.1% Nonidet P-40. Bound protein was eluted by incubating the agarose with 1.5 ml of 250 mM imidazole hydrochloride, pH 7.0/140 mM NaCl/10% glycerol, and protease inhibitors for 15 min at 4°C. The elution was repeated, and fractions were pooled. For immunoprecipitations, 200 μl of this supernatant was mixed with 2 μl of antibody (≈1 μg), Nonidet P-40 was added to give a final concentration of 0.1%, and reactions were incubated on ice for 1 h. After centrifugation for 10 min at 4°C, the supernatant was added to tubes containing 20 μl of a 50% slurry of protein A-Sepharose (Sigma) beads in PBS plus 0.1% Nonidet P-40. The reactions were incubated at 4°C on an end-over-end mixer for 1 h, then spun at 2000 rpm for 2 min in a Microfuge. The immunoprecipitates were washed twice with 1 ml of 50 mM Hepes, pH 7.6/150 mM NaCl/5 mM Mg(OAc)2, and protease inhibitor, resuspended in 15 μl of 1.5× sample buffer, and heated to 65°C for 10 min. Six microliters of each reaction was applied to a SDS 7–8.5% polyacrylamide gel and blotted to a membrane. Proteins on Western blots were detected by using the ECL system. The antibodies used for immunoprecipitation were a monoclonal anti-HA antibody (HA11, Babco, Richmond, CA), a monoclonal anti-SIR2 antibody, and an affinity-purified rabbit anti-SIR3 antibody. The 9E12 anti-Myc antibody (Babco), which does not recognize any of the proteins studied here, was used as a control for specificity. Similar results were obtained when immunoprecipitations were performed using a crude whole-cell extract, without the Ni2+-agarose step.
GST Spin Down Assays.
GST, GST-SIR2, GST-SIR3, GST-SIR3C, GST-SIR4C1, GST-SIR4C2, and GST-RAP1C fusion proteins were expressed in E. coli and purified as described previously (25). After purification from crude extract, proteins were either left bound to the glutathione-agarose or were rebound to it at a concentration of 0.1 mg/ml. SIR2, SIR3C, and SIR4C were prepared by the cleavage of GST-SIR2, GST-SIR3C, and GST-SIR4C1 with thrombin. Cleavage reactions contained 25 μg of GST-SIR2, GST-SIR3C, or GST-SIR4C1 and 0.4 μg of thrombin (Sigma) in 40 μl of 50 mM Tris·HCl, pH 7.5/250 mM NaCl/5 mM CaCl2 and were incubated at room temperature for 1 h. Thrombin was inactivated by addition of 4-(2-aminoethyl)benzenesulfonyl fluoride·HCl or phenylmethylsulfonyl fluoride to a final concentration of 1 mM. Uncleaved protein or free GST was removed by incubation of 3 μg of cleaved protein with 25 μl packed volume of glutathione agarose for 10 min at room temperature in 100 μl of binding buffer [20 mM Tris·HCl, pH 8.0/150 mM NaCl/1 mM DTT/0.1% Nonidet P-40/0.5 mg/ml insulin/0.1 mM 4-(2-aminoethyl)benzenesulfonyl fluoride], and this precleared protein was added to 10 μl of packed volume of glutathione-agarose beads loaded with GST or a GST fusion protein, which had been washed with 100 μl of binding buffer, and the reactions were incubated for 30 min at room temperature on an end-over-end mixer. The beads were recovered by centrifugation for 2 min at 2000 rpm in a Microfuge, washed twice with 200 μl of binding buffer, and resuspended in 20 μl of sample buffer (this is the bound fraction). After heating at 100°C for 3 min, 6 μl of each sample was loaded onto an SDS 8.5% polyacrylamide gel, and protein was visualized by staining with Coomassie brilliant blue. For Western analysis, 2 μl of each supernatant (unbound fraction) or 6 μl of 1:10 dilution of each bound fraction was loaded per lane.
RESULTS
Association of SIR2 and SIR3 with GST-SIR4 Fusion Proteins Purified from Yeast.
We used GST affinity chromatography to study SIR–SIR protein–protein interactions in yeast. The full-length SIR4 open reading frame, the N-terminal half, and the C-terminal half of SIR4 were fused to the GST gene and expressed in yeast under the control of the GAL1 promoter (Fig. 1A). Each fusion protein, along with a GST protein control, was purified from yeast extracts using glutathione-agarose, and in each case the associated proteins were examined. Extracts were prepared using a buffer that contained 0.5 M NaCl and 1% detergent. Both the high salt and detergent concentrations were required for optimal solubility of the SIR proteins, although a fraction of each protein was soluble when cells were lysed in buffers containing 0.5 M salt but no detergent or 0.15 M salt and 1% detergent (data not shown, see ref. 30). The high salt and detergent concentrations used in the purifications described here also reduce nonspecific interactions. Yeast extracts were incubated with glutathione-agarose beads, and after extensive washing with a buffer containing 0.5 M NaCl and 1% detergent, bound protein was eluted with free glutathione. Fig. 2A shows a Western blot of various eluates probed with an anti-GST antibody to visualize the GST, GST-SIR4, GST-SIR4N2 (N-terminal 730 amino acids of SIR4 fused to GST), and GST-SIR4C1 (C-terminal 628 amino acids of SIR4 fused to GST, lanes 1–4). Probing with an anti-SIR2 antibody showed that the SIR2 protein was associated with full-length SIR4 or its C-terminal half (Fig. 2B, lanes 2 and 4) but not with GST (lane 1) or the N-terminal half of SIR4 (lane 3). SIR3 also was found associated with full-length SIR4 and with its C-terminal half (Fig. 2C, lanes 2 and 4). However, whereas the SIR2 signal associated with GST-SIR4 and GST-SIR4C1 was greatly enriched when compared with the SIR2 signal in the input extract (data not shown, but see Fig. 3), the SIR3 signal associated with these same fusion proteins was not significantly enriched compared with the SIR3 signal in the input extract, suggesting that only small amounts of SIR3 were associated with these SIR4 fusions.
Figure 1.

Tagged proteins used in the experiments described here. (A) GST fusion proteins expressed under the control of the GAL1 promoter in yeast. (B) Modification of the endogenous SIR4 gene to produce 6His-3HA-SIR4 (DM428).
Figure 2.
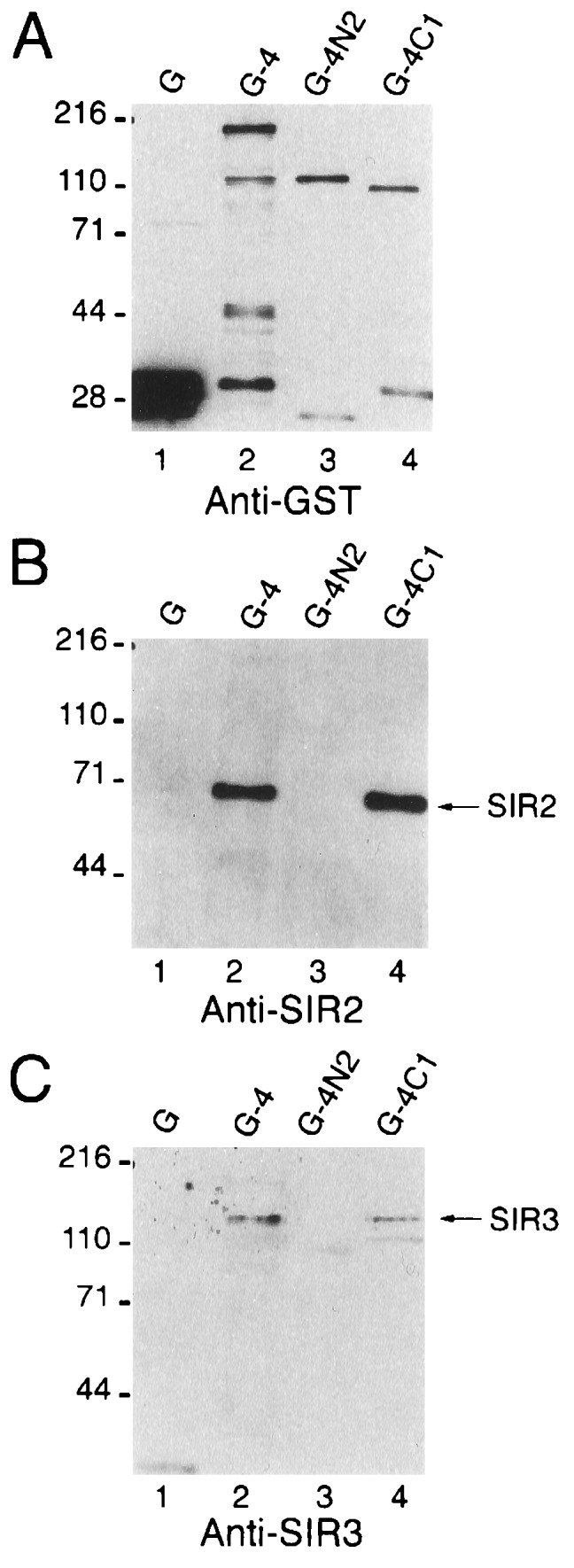
Western blots showing the association of SIR2 and SIR3 with GST-SIR4 fusion proteins in yeast. Proteins were expressed in JRY3411 (sir4Δ::HIS3), purified using glutathione-agarose, and analyzed by Western blotting using anti-GST (A), anti-SIR2 (B), and anti-SIR3 (C). G, GST; G-4, GST-SIR4; G-4N2, GST-SIR4N2, G-4C1, GST-SIR4C1. Positions of prestained molecular size markers (kDa) are indicated on the left of each panel.
Figure 3.

Preferential association of SIR3 with the C terminus of SIR4. G-4C2, GST-SIR4C2; G-4C1, GST-SIRC1. Proteins were expressed in strain FM135, and after purification with glutathione-agarose, detected by Western blotting. (A, B, and C) Western blots of the input and bound fractions probed with anti-GST, anti-SIR2, and anti-SIR3 antibodies, respectively. (D) Effect of overexpression of GST (pRD56), GST-SIR4C1 (pGC4–1), and GST-SIR4C2 (pGC4–2) on the mating ability of a wild-type SIR4 strain (FM135).
An N-Terminal Domain in SIR4 Inhibits Its Interaction with SIR3.
Overexpression of the extreme C terminus (amino acids 1115–1358) of the SIR4 protein in vivo has been shown to disrupt silencing (31, 32). This dominant negative activity is suppressed by overexpression of SIR3, suggesting that this domain of SIR4 interacts with SIR3 (31, 32). We fused the C-terminal 244 amino acids of SIR4 to GST (GST-SIR4C2, Fig. 1) and compared this protein to GST-SIR4C1 both for its dominant negative activity (in vivo as determined by a mating assay) and for its interaction with SIR2 and SIR3 in vitro. In agreement with previous results (31, 32), overexpression of GST-SIR4C2 from the GAL1 promoter drastically reduced the mating ability of a wild-type SIR4 strain, indicating that silencing had been disrupted, but overexpression of GST-SIR4C1 had no effect in this qualitative assay (Fig. 3D). GST-SIR4C1 and GST-SIR4C2 were purified using glutathione-agarose, and similar amounts of each protein were analyzed by Western blotting (Fig. 3A, lanes 1 and 2). In contrast to GST-SIR4C1, GST-SIR4C2 was efficiently associated with SIR3 (Fig. 3C, compare lanes 3 and 4); by comparing the intensity of the SIR3 signal in the input extract with the fraction bound to the various GST-SIR4 fusions, and with the unbound fraction, we estimate that in these extracts >50% of SIR3 was associated with GST-SIR4C2 and that ≈1% of SIR3 in the extract was associated with GST-SIR4 or GST-SIR4C1 (Figs. 2 and 3 and data not shown). Only very small quantities of SIR2 were associated with GST-SIR4C2, suggesting that the SIR2 binding site was absent or defective in this fusion protein (Fig. 3B, lane 3). These biochemical results are consistent with the dominant negative activity of each protein and suggest that severe N-terminal truncations of SIR4 are released from inhibitions that normally prevent their interaction with SIR3.
Coimmunoprecipitation of the SIR2 and SIR4 Proteins.
To provide an independent evaluation of the protein–protein associations that we observed by purifying overexpressed GST-SIR4 fusion proteins, we performed conventional coimmunoprecipitation experiments without overexpressing any of the SIR proteins. To facilitate these experiments, the endogenous SIR4 open reading frame was replaced by a modified gene encoding a polyhistidine tag followed by three HA epitopes at its N terminus. The resulting strain mated with wild-type efficiency in a quantitative mating assay, indicating that the tagged protein was fully functional in the cell. Extracts from the HA-SIR4 strain were immunoprecipitated with antibodies directed against the HA epitope, SIR2, or SIR3. Precipitation of HA-SIR4 with the anti-HA antibody (Fig. 4A, lanes 2 and 6) also efficiently precipitated SIR2 (Fig. 4B, lanes 2 and 6), but not SIR3 (Fig. 4C, lane 2). Consistent with these results, immunoprecipitation of these extracts with an anti-SIR2 antibody (Fig. 4B, lane 7) efficiently precipitated HA-SIR4 (Fig. 4A, lane 7), but not SIR3 (data not shown). Little SIR2 and HA-SIR4 was observed in the SIR3 immunoprecipitates (Fig. 4 A and B, lane 3); however, these proteins were detectable on long exposures of the blots to film (Fig. 4 A and B, lanes 4 and 9). Similar results were obtained when immunoprecipitations were carried out with partially solubilized SIR proteins from extracts prepared by lysis of cells at 150 mM instead of 500 mM NaCl (data not shown). The results of the immunoprecipitation experiments are consistent with those of the GST fusion protein purification experiments; both lines of evidence indicate that the primary SIR complex in yeast extracts is composed of SIR2 and SIR4.
Figure 4.

Coimmunoprecipitation of the SIR2 and SIR4 proteins from yeast extracts. Proteins were immunoprecipitated with the indicated antibody (IP) and detected by Western blotting using anti-HA (A), anti-SIR2 (B), or anti-SIR3 (C) antibodies. Input, 1–5% of the extract used for each immunoprecipitation reaction; ∗IgG, indicates the position of the immunoglobulin heavy chain. Lanes 4 and 9 show longer exposures to film to visualize protein that is less efficiently immunoprecipitated.
Direct Physical Association of the SIR2, SIR3, and SIR4 Proteins.
To determine whether the associations observed in the experiments described above are direct, we tested them using SIR proteins expressed in E. coli and purified. In these experiments one protein was fused to GST and was immobilized on glutathione-agarose beads. Protein–protein interactions then were tested by incubating the glutathione-agarose-GST fusion complex with an untagged protein, also produced in E. coli, followed by an analysis of the proteins that remain bound to the glutathione-agarose after extensive washing. Protein was detected either by staining directly with Coomassie brilliant blue or, after transfer to a membrane, by Western blotting. Western blotting provides a sensitive assay, whereas staining with Coomassie brilliant blue allows the simple detection of strong and efficient interactions. As shown in Fig. 5A, the C-terminal half of SIR4, SIR4C1 (lane 2), bound to both GST-SIR2 (lane 5) and GST-SIR3 (lane 11), but not to GST alone (lane 8). In addition, this experiment showed that the SIR2 protein (lane 1) binds to GST-SIR2 and GST-SIR3 (lanes 4 and 10), but not to GST (lane 7). The smaller bands in the GST-SIR3 lane (Fig. 5, lane 9) were degradation products of SIR3 (data not shown). Using this assay, we also found that the SIR4 protein interacted with itself through its C terminus (data not shown).
Figure 5.

Direct physical association of bacterially produced SIR2, SIR3, and SIR4 proteins. (A) Coomassie brilliant blue-stained SDS/polyacrylamide gel showing the binding of SIR2 and SIR4C1 (lanes 1 and 2) to GST-SIR2 (G-2, lanes 3–5) and GST-SIR3 (G-3, lanes 9 and 10) proteins. (B) Specific association of SIR2 with GST-SIR4C1 (G-4C1), but not with GST-SIR4C2 (G-4C2), detected by Western blotting with anti-SIR2. (C) Specific association of the C-terminal half of SIR3 (SIR3C) with a GST-RAP1 (G-RAP1C) fusion protein detected by Western blotting with anti-SIR3.
The RAP1 protein previously has been shown to interact with the SIR3 and SIR4 proteins in a two-hybrid assay, and a direct interaction between RAP1 and an in vitro-translated SIR3 protein also has been observed (18). By Coomassie brilliant blue staining, we were unable to detect an interaction between a C-terminal fragment of RAP1 and either GST-SIR3C (C-terminal 432 amino acids of SIR3 fused to GST) or GST-SIR4C1. However, when monitored using the Western assay, the C-terminal half of SIR3 (SIR3C) could be shown to bind preferentially to GST-RAP1C (fusion of the C-terminal 264 amino acids of RAP1 to GST) when compared with GST alone (Fig. 5C, compare lanes 2 and 4). Using Western blotting as a detection assay, we also could demonstrate that SIR2 interacted specifically with GST-SIR4C1, but not with GST-SIR4C2 (containing only the C terminal 244 amino acids of SIR4) by comparing the binding of the SIR2 protein to either empty glutathione-agarose beads or to beads loaded with GST or GST-SIR4 fusion proteins (Fig. 5B, compare bound and unbound fractions in lanes 1–4). This result agrees with the GST purification experiments (Figs. 2 and 3) and with previous affinity chromatography results (25) and indicates that the SIR2 interaction domain is absent in the GST-SIR4C2 protein. Therefore, in these experiments, we observed a strong and specific association between SIR2 and the C-terminal half of SIR4 (amino acids 743-1358), SIR3 and the C-terminal half of SIR4 (amino acids 743-1358), SIR2 and SIR3, SIR2 and SIR2, and SIR4 (amino acids 743-1358) and SIR4 (amino acids 743-1358). In addition, as previously reported (18), we could detect a specific, but weaker, interaction between the C-terminal half of SIR3 (amino acids 439–971) and the C terminus of RAP1 (amino acids 563–827).
DISCUSSION
The experiments reported here demonstrate that the silencing proteins SIR2, SIR3, and SIR4 are physically associated in the cell and directly interact with each other. Both GST affinity purification and coimmunoprecipitation experiments reveal a tight association between the SIR2 and SIR4 proteins in yeast extracts. The SIR3 protein, on the other hand, is found efficiently associated with SIR4 only if the N-terminal two-thirds of SIR4 is absent, suggesting that the SIR3-SIR4 interaction normally is inhibited by N-terminal sequences in SIR4. It seems likely that the relief of this inhibition contributes to a regulatory step in the assembly of silent chromatin. Using bacterially expressed and purified proteins, we also show that the SIR2, SIR3, and SIR4 proteins interact directly and that SIR2 and SIR4 can form at least dimers in vitro. These results agree with and extend previous two-hybrid studies showing that the SIR3 and SIR4 proteins interact with each other and that each protein can interact with itself (18, 33).
Overexpression of the C terminus of the SIR4 protein in yeast has been shown to interfere with silencing. This dominant negative activity is suppressed by the simultaneous overexpression of SIR3 and has been proposed to result from the titration of the SIR3 protein by overexpressed SIR4 (31, 32). The results reported here provide strong support for this hypothesis. In our experiments, the GST-SIR4 truncations with the strongest dominant negative phenotype also associate with SIR3 the most efficiently. These results suggest that the disruption of silencing observed by overexpression of SIR4 or its truncations results from premature association of SIR4 with SIR3 into a complex that cannot be recruited to the silencer.
Silencing could initiate either by the recruitment of a preassembled SIR protein complex to the silencer or by the stepwise assembly of such a complex at the silencer. In the latter model (Fig. 6), interactions with the DNA-bound proteins promote assembly of the SIR protein complex through allosteric conformational changes in one or more of these proteins. Two lines of evidence in the present study point to such a stepwise assembly mechanism. First, in our purifications we detect a tightly associated SIR2/SIR4 complex that contains very little SIR3, and second, truncations of SIR4 that retain only its extreme C terminus allow it to associate efficiently with SIR3. This latter result suggests that the N-terminal domain of SIR4, either directly or indirectly, inhibits its association with SIR3. Therefore, the SIR3–SIR4 interaction may be a regulated step in the assembly of the SIR protein complex that occurs only at the silencer. For example, the SIR2/SIR4 complex may be recruited to the silencer through interactions with DNA-binding proteins. These initial interactions might then promote the assembly of the full complex by inducing a conformational change in SIR4 that allows its interaction with SIR3. This model (Fig. 6) also provides an explanation for the inability of otherwise functional SIR2, SIR3, and SIR4 to establish silencing from isolated LexA or GAL4 binding sites when tethered to DNA by the DNA-binding domains of LexA or GAL4 (34, 35). According to the model, the necessary conformational change would not occur unless the SIR proteins are bound to DNA by their normal partner proteins. We further propose that the stepwise assembly of SIR3 and the SIR2/SIR4 complex on DNA could be important for the accurate initiation of silencing.
Figure 6.

A model for the stepwise assembly of the SIR protein complex at a silencer. The SIR2/SIR4 complex and SIR3 are independently recruited to the silencer (black and white shaded bar) through interactions with silencer-bound proteins. ORC, origin recognition complex. Interactions at the silencer unmask a SIR3 interaction domain in SIR4 and promote the assembly of the SIR2/SIR3/SIR4 complex, which may now be able to interact with histones and spread along chromatin. Each SIR2, SIR3, and SIR4 is depicted as a dimer.
Acknowledgments
We thank Elizabeth Blackburn, Burk Braun, Rebecca Smith, and José Pérez-Martin for comments on the manuscript. This work was supported by National Institutes of Health grants GM53452–01 (A.D.J.), GM31105 (J.R.), and T32 CA09270 (Department of Biochemistry and Biophysics, University of California, San Francisco).
ABBREVIATIONS
- SIR
silent information regulator
- GST
glutathione S-transferase
- HA
hemagglutinin
References
- 1.Laurenson P, Rine J. Microbiol Rev. 1992;56:543–560. doi: 10.1128/mr.56.4.543-560.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allshire R C, Nimmo E R, Ekwall K, Javerzat J P, Cranston G. Genes Dev. 1995;9:218–233. doi: 10.1101/gad.9.2.218. [DOI] [PubMed] [Google Scholar]
- 3.Dernburg A F, Sedat J W, Hawley R S. Cell. 1996;86:135–146. doi: 10.1016/s0092-8674(00)80084-7. [DOI] [PubMed] [Google Scholar]
- 4.Kellum R, Alberts B M. J Cell Sci. 1995;108:1419–1431. doi: 10.1242/jcs.108.4.1419. [DOI] [PubMed] [Google Scholar]
- 5.Pillus L, Rine J. Cell. 1989;59:637–647. doi: 10.1016/0092-8674(89)90009-3. [DOI] [PubMed] [Google Scholar]
- 6.Reynolds A E, McCarroll R M, Newlon C S, Fangman W L. Mol Cell Biol. 1989;9:4488–4494. doi: 10.1128/mcb.9.10.4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palladino F, Laroche T, Gilson E, Axelrod A, Pillus L, Gasser S M. Cell. 1993;75:543–555. doi: 10.1016/0092-8674(93)90388-7. [DOI] [PubMed] [Google Scholar]
- 8.Brand A H, Breeden L, Abraham J, Sternglanz R, Nasmyth K. Cell. 1985;41:41–48. doi: 10.1016/0092-8674(85)90059-5. [DOI] [PubMed] [Google Scholar]
- 9.McNally F J, Rine J. Mol Cell Biol. 1991;11:5648–5659. doi: 10.1128/mcb.11.11.5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shore D, Nasmyth K. Cell. 1987;51:721–732. doi: 10.1016/0092-8674(87)90095-x. [DOI] [PubMed] [Google Scholar]
- 11.Halfter H, Kavety B, Vandekerckhove J, Kiefer F, Gallwitz D. EMBO J. 1989;8:4265–4272. doi: 10.1002/j.1460-2075.1989.tb08612.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rhode P R, Sweder K S, Oegema K F, Campbell J L. Genes Dev. 1989;3:1926–1939. doi: 10.1101/gad.3.12a.1926. [DOI] [PubMed] [Google Scholar]
- 13.Bell S P, Stillman B. Nature (London) 1992;357:128–134. doi: 10.1038/357128a0. [DOI] [PubMed] [Google Scholar]
- 14.Buchman A R, Kimmerly W J, Rine J, Kornberg R D. Mol Cell Biol. 1988;8:210–225. doi: 10.1128/mcb.8.1.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kyrion G, Liu K, Liu C, Lustig A J. Genes Dev. 1993;7:1146–1159. doi: 10.1101/gad.7.7a.1146. [DOI] [PubMed] [Google Scholar]
- 16.Rine J, Herskowitz I. Genetics. 1987;116:9–22. doi: 10.1093/genetics/116.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aparicio O M, Billington B L, Gottschling D E. Cell. 1991;66:1279–1287. doi: 10.1016/0092-8674(91)90049-5. [DOI] [PubMed] [Google Scholar]
- 18.Moretti P, Freeman K, Coodly L, Shore D. Genes Dev. 1994;8:2257–2269. doi: 10.1101/gad.8.19.2257. [DOI] [PubMed] [Google Scholar]
- 19.Triolo T, Sternglanz R. Nature (London) 1996;381:251–253. doi: 10.1038/381251a0. [DOI] [PubMed] [Google Scholar]
- 20.Thompson J S, Ling X, Grunstein M. Nature (London) 1994;369:245–247. doi: 10.1038/369245a0. [DOI] [PubMed] [Google Scholar]
- 21.Kayne P S, Kim U J, Han M, Mullen J R, Yoshizaki F, Grunstein M. Cell. 1988;55:27–39. doi: 10.1016/0092-8674(88)90006-2. [DOI] [PubMed] [Google Scholar]
- 22.Hecht A, Laroche T, Strahl-Bolsinger S, Gasser S M, Grunstein M. Cell. 1995;80:583–592. doi: 10.1016/0092-8674(95)90512-x. [DOI] [PubMed] [Google Scholar]
- 23.Hecht A, Stahl-Bolsinger S, Grunstein M. Nature (London) 1996;383:92–96. doi: 10.1038/383092a0. [DOI] [PubMed] [Google Scholar]
- 24.Brachmann C B, Sherman J M, Devine S E, Cameron E E, Pillus L, Boeke J D. Genes Dev. 1995;9:2888–2902. doi: 10.1101/gad.9.23.2888. [DOI] [PubMed] [Google Scholar]
- 25.Moazed D, Johnson A D. Cell. 1996;86:667–677. doi: 10.1016/s0092-8674(00)80139-7. [DOI] [PubMed] [Google Scholar]
- 26.Sikorski R S, Hieter P. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hovland P, Flick J, Johnston M, Sclafani R A, Sikorski R S, Hieter P. Gene. 1989;83:57–64. doi: 10.1016/0378-1119(89)90403-4. [DOI] [PubMed] [Google Scholar]
- 28.Jones E W. Methods Enzymol. 1991;194:428–453. doi: 10.1016/0076-6879(91)94034-a. [DOI] [PubMed] [Google Scholar]
- 29.Kellogg D R, Alberts B M. Mol Biol Cell. 1992;3:1–11. doi: 10.1091/mbc.3.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cockell M, Palladino F, Laroche T, Kyrion G, Liu C, Lustig A J, Gasser S M. J Cell Biol. 1995;129:909–924. doi: 10.1083/jcb.129.4.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ivy J M, Klar A J, Hicks J B. Mol Cell Biol. 1986;6:688–702. doi: 10.1128/mcb.6.2.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marshall M, Mahoney D, Rose A, Hicks J B, Broach J R. Mol Cell Biol. 1987;7:4441–4452. doi: 10.1128/mcb.7.12.4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chien C T, Bartel P L, Sternglanz R, Fields S. Proc Natl Acad Sci USA. 1991;88:9578–9582. doi: 10.1073/pnas.88.21.9578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chien C T, Buck S, Sternglanz R, Shore D. Cell. 1993;75:531–541. doi: 10.1016/0092-8674(93)90387-6. [DOI] [PubMed] [Google Scholar]
- 35.Lustig A J, Liu C, Zhang C, Hanish J P. Mol Cell Biol. 1996;16:2483–2495. doi: 10.1128/mcb.16.5.2483. [DOI] [PMC free article] [PubMed] [Google Scholar]