

Abstract
The tobacco budworm, Heliothis virescens (Noctuidae), a destructive insect pest, is remarkably resistant to l-canavanine, l-2-amino-4-(guanidinooxy)butyric acid, an arginine antimetabolite that is a potent insecticide for nonadapted species. H. virescens employs a constitutive enzyme of the larval gut, known trivially as canavanine hydrolase (CH), to catalyze an irreversible hydrolysis of l-canavanine to l-homoserine and hydroxyguanidine. As such, it represents a new type of hydrolase, one acting on oxygen–nitrogen bonds (EC 3.13.1.1). This enzyme has been isolated from the excised gut of H. virescens and purified to homogeneity; it exhibits an apparent Km value for l-canavanine of 1.1 mM and a turnover number of 21.1 μmol·min−1·μmol−1. This enzyme has a mass of 285 kDa and is composed of two subunits with a mass of 50 kDa or 47.5 kDa. CH has a high degree of specificity for l-canavanine as it cannot function effectively with either l-2-amino-5-(guanidinooxy)pentanoate or l-2-amino-3-(guanidinooxy)propionate, the higher or lower homolog of l-canavanine, respectively. l-Canavanine derivatives such as methyl-l-canavanine, or l-canaline and O-ureido-l-homoserine, are not metabolized significantly by CH.
Keywords: allelochemical detoxification
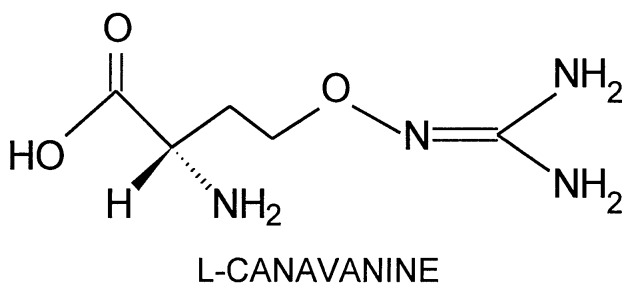
l-Canavanine, the l-2-amino-4-(guanidinooxy)butyric acid structural analog of l-arginine synthesized by leguminous plants, is normally a potent insecticide (1, 2); it provides a formidable chemical barrier against predation by nonadapted species (3).
In contrast, insects such as the bruchid beetle, Caryedes brasiliensis and the weevil, Sternechus tuberculatus, which feed exclusively on canavanine-containing seeds, are adapted to this potentially poisonous natural product (4, 5). Indeed, C. brasiliensis employs l-canavanine to provide the nitrogen for the biosynthesis of many essential amino acids (5, 6).
The tobacco budworm, Heliothis virescens (Noctuidae), exhibits a remarkable degree of tolerance to dietary canavanine as the larvae can be reared on a diet containing 300 mM canavanine, or nearly 40% nonprotein amino acid by dry weight, with only a marginal effect on larval growth and without eliciting discernible developmental aberrations (7). In contrast, tobacco hornworm larvae, Manduca sexta, maintained on a 2.5 mM canavanine-containing diet develop massive developmental aberrations (1, 2). The appreciable natural resistance of H. virescens to this protective allelochemical instigated earlier studies to elucidate the biochemical basis for canavanine tolerance by H. virescens (7–9).
These studies established that H. virescens actively metabolizes canavanine under the control of a constitutive enzyme that has been isolated from the larval gut (8). Administration of l-[guanidinooxy-14C]canavanine to fifth instar H. virescens larvae disclosed that [14C]guanidine was the principal in vivo larval radiolabeled canavanine catabolite (9). Subsequent in vivo studies employing l-[1,2,3,4-14C]canavanine identified l-[1,2,3,4-14C]homoserine as the preponderant radiolabeled catabolite (9). These studies of radiolabeled metabolite disposition implicated a larval reductase in the detoxification of canavanine via homoserine and guanidine.
More recent experiments revealed a larval gut reductase able to catalyze an NADH-dependent reduction of hydroxyguanidine to guanidine (10). This finding suggested that canavanine was catabolized initially to homoserine and hydroxyguanidine by a hydrolase able to cleave the O–N bond of the guanidinooxy moiety of the substrate. While this metabolic ability has been observed in a soil-borne Pseudomonas (11), the responsible enzyme was not isolated nor has this metabolic capacity been described from an eukaryotic organism.
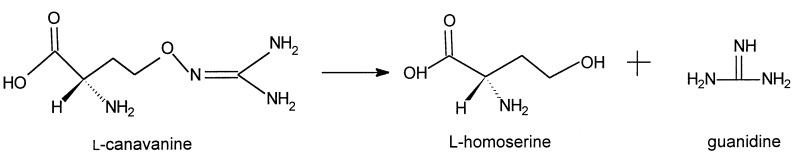
This communication details purification and characterization of this canavanine-degrading enzyme, which we have named canavanine hydrolase (CH). This protein mediates an irreversible hydrolysis of l-canavanine to l-homoserine and hydroxyguanidine.
In this reaction, CH functions as a hydrolase that cleaves an oxygen–nitrogen bond; this enzyme is the only protein known to demonstrate this ability. (12). As such it represents a new type of hydrolase, one that acts on oxygen–nitrogen bonds (EC 3.13.1.1).
MATERIALS AND METHODS
Substrates.
l-Canavanine free base was obtained by recrystallization of material isolated from a methanolic extract of Jack bean seed, Canavalia ensiformis (13). l-Canaline was synthesized from canavanine by arginase-mediated hydrolysis and subsequent isolation of the dipicric acid salt of canaline; canaline free base was prepared via canaline sulfate (13). O-Ureido-l-homoserine was made from canaline by carbamylation with KCNO (14). l-Homocanavanine was prepared from l-2-amino-5-hydroxypentanoic acid, the higher homolog of l-homoserine, as described (15). The lower homolog of l-canavanine was synthesized from l-cycloserine by ring opening followed by zinc-mediated guanidination with cyanamide (16). The methyl, ethyl, and butyl ester of canavanine were produced from the parent compound (15). Barium [14C]cyanamide (2.15 GBq/mmol) was synthesized by Amersham. All remaining reagents were secured from Sigma/Aldrich.
Insects.
H. virescens terminal instar larvae were reared in a continuous colony maintained at the University of Kentucky as described (17). All insects were anesthetized by chilling on ice before gut collection.
Canavanine Hydrolase Assay.
Because of their equivalent basicity, hydroxyguanidine cannot be separated effectively from canavanine by ionic-exchange chromatography. Moreover, hydroxyguanidine reacts with pentacyanoammonioferrate (PCAF), the reagent that is also used for colorimetric analysis of canavanine, to generate a stable chromogen. The PCAF colorimetric assay is the only suitable reaction for colorimetric analysis of hydroxyguanidine, but the canavanine–PCAF chromogen, produced from unreacted substrate, masks the hydroxyguanidine–PCAF chromogen. To overcome this difficulty, unreacted canavanine was removed by treatment with excess Jack bean leaf arginase.
CH (7.5 milliunits) was reacted with 25 mM l-canavanine (pH 7.4) and 80 mM sodium tricine buffer (pH 7.4) containing 30% (vol/vol) glycerol in a final volume of 0.5 ml for 120 min. The reaction was terminated by placing the reaction mixture in a boiling water bath for 3 min. The turbid reaction mixture was treated with 100 μl purified Jack bean leaf arginase (see below) for at least 7 h, but often overnight at 37°C. A unit of CH is that amount of enzyme that catalyzed the formation of 1 μmol hydroxyguanidine/min−1 under the described assay conditions.
The hydroxyguanidine-PCAF chromogen was produced by adding 1.0 ml of 200 mM potassium phosphate buffer (pH 7.0) to the arginase-treated CH reaction mixture followed by 0.2 ml of 1.0% (wt/vol) potassium persulfate and 0.2 ml of 1.0% (wt/vol) PCAF as described (18). The turbid colorimetric assay tube was clarified by centrifugation with an International Equipment (Needham, MA) table-top centrifuge for 1 min. At least 3 min but not more than 5 min after the addition of PCAF, the hydroxyguanidine–PCAF chromogen was read at 475 nm.
Purified Arginase.
Jack bean leaf arginase, purified through the ammonium sulfate precipitation step and dialysis, was prepared as described and stored at −80°C (13, 19). The thawed protein preparation, obtained from 120 g of 12-day old jack bean leaves, was placed on a 90 × 300 mm column of DEAE-cellulose equilibrated with 25 mM sodium tricine (pH 7.6) containing 0.1% (vol/vol) 2-mercaptoethanol and 0.5 mM dithiothreitol (standard buffer). The column was washed with 200 ml of 50 mM standard buffer. Arginase was obtained by gradient elution employing 300 ml of 50 mM standard buffer and an equal volume of standard buffer with 250 mM NaCl. The effluent was collected as 7 ml fractions in test tubes containing 7 μmol MnCl2; the flow rate was 105 ml/h.
The column fractions were assayed for arginase activity by treating 50 μl of sample with 25 mM l-arginine (pH 9.7), 1 mM MnCl2, and 50 mM sodium glycylglycine buffer (pH 9.7) in a final volume of 0.5 ml for 10 min at 37°C. The assay was terminated by transferring 100 μl of the reaction mixture to a tube containing 2.9 ml of a solution of 1.25% (wt/vol) ninhydrin in glacial acetic/6 M phosphoric acid (4:1, vol/vol) after the method of Chinard (20). After standing in boiling water for 10 min, the tubes were inspected visually for the orange-red chromogen produced by ornithine. The 45 most active fractions were pooled, concentrated to 20 ml by ultrafiltration with an Amicon P-10 membrane, and stored as 1.0 ml samples at −80°C.
Every arginase preparation was evaluated for canavanine-degrading ability by reacting 100 μl of purified arginase with 25 mM l-canavanine, under the conditions of the CH assay, at 37°C for 7 h. In all instances, the above procedure provided arginase of sufficient activity to degrade all canavanine as determined by the PCAF colorimetric assay.
Protein Assay.
Protein was determined in crude preparations by the Bio-Rad protein assay mixture. The absorbance of homogeneous canavanine hydrolase was determined as described (21). A 0.1% solution (wt/vol) had an A280 of 1.35.
Amino Acid Evaluations.
Canaline was monitored by a colorimetric procedure after its conversion to O-ureidohomoserine by reaction with 20 mM KCNO in 100 mM sodium acetate buffer (pH 4.5) (14). O-Ureidohomoserine was determined directly by the same colorimetric procedure (14). Homoserine was quantified by automated amino acid analysis employing a Dionex model D-300 automated amino acid analyzer with lithium citrate gradient elution and ninhydrin detection at 570 nm. Homoserine eluted after 17 min, ammonia eluted at 73 min, and canavanine after 105 min.
Kinetic Analysis.
The apparent Km value, determined by regression analysis, was taken from Hanes–Woolf modification of Lineweaver–Burk plots. Lines having an r value less than 0.999 were discarded. The turnover number was calculated from Vmax data obtained on these plots and were based upon a molecular mass of 285 kDa.
Synthesis of Radiolabeled Hydroxyguanidine.
Hydrogen cyanamide (0.45 mmol) was added to 0.55 mmol hydroxylamine hydrochloride in 1.6 ml of deionized water and the pH taken to 9.3 with NaOH. A total of 100 MBq barium [14C]cyanamide (2.15 GBq/mmol) was treated with a 2-fold excess of sulfuric acid in 0.4 ml of deionized water; barium sulfate was removed by centrifugation. The radiolabeled cyanamide was added to the reaction mixture and the pH re-adjusted to 8.9–9.1 with dilute NaOH. [14C]Hydroxyguanidine formation was allowed to continue for 48 h at 3°C. The radioactive reaction mixture was taken to pH 3.5 with 2 M HCl and placed on a 17 × 60 mm column of Dowex 50 (NH4+). After washing the column exhaustively with deionized water, radiolabeled hydroxyguanidine was obtained with 300 mM ammonia. The first 25 ml of effluent, containing radiolabeled hydroxyguanidine, was discarded to remove any unreacted cyanamide. The subsequent 10 ml of effluent (fraction 2) was dried to a residue by rotary evaporation in vacuo at a temperature not more than 23°C. Before drying, a sample of fraction 2 was used to determine the hydroxyguanidine content by colorimetric analysis. Sufficient carrier hydroxyguanidine, taken to pH 7.4, and deionized water were added to the residue to prepare 2 ml of 175 mM [14C]hydroxyguanidine that was stored at −20°C overnight before use. The specific activity of the [14C]hydroxyguanidine solution was 13.2 KBq/μmol−1.
Polyacrylamide Gel Electrophoresis.
SDS/PAGE was conducted under denaturing conditions and 15% polyacrylamide gel. Nondenaturing gels consisted of a 5–20% gradient of polyacrylamide. The running buffer was 100 mM Tris/glycine (pH 8.3). Proteins were stained with 0.1% (wt/vol) Coomassie blue R-250. Before electrophoresis, CH was treated with SDS in the presence of 2-mercaptoethanol for 3 min at 100°C. Bio-Rad high and low SDS/PAGE molecular weight standards and Sigma electrophoresis kit standards were used to determine CH mass.
Sequence Data.
Protein alkylation, enzymatic cleavage, peptide purification, and peptide sequence analysis were performed at the Macromolecular Structure Analysis Facility of the University of Kentucky. CH (≈0.1 mg) was desalted by adsorbing the protein onto a polyvinylidene fluoride membrane held in a ProSorb Apparatus (Perkin–Elmer). The polyvinylidene fluoride-bound protein was reduced by placing the membrane in 90 μl of 100 mM Tris·HCl buffer (pH 8.5) with 10 μl acetonitrile containing 7 mM dithiothreitol at 37°C for 1 h, and then alkylated with 20 mM iodoacetamide in the dark at 23°C for 15 min. After alkylation, the membrane-bound protein was washed with 1 ml of deionized water, 1 ml of 100 mM Tris·HCl (pH 8.5), and then treated with 10 μl acetonitrile plus 10 μl 10% hydrogenated Triton X-100 with 100 ng endoproteinase Lys-C (Boehringer Mannheim) in 100 mM Tris·HCl (pH 8.5) in a final volume of 100 μl for 18 h at 37°C.
The supernatant solution was saved and the membrane washed with 100 μl 40% (vol/vol) aqueous acetonitrile and then with the same solvent containing 0.05% (vol/vol) trifluoroacetic acid (TFA). The peptides of the supernatant solution and pooled washes were isolated by reverse-phase HPLC using a Hewlett–Packard model 1050 equipped with a Vydac (Hesperia, CA) 2.1 × 250 mm C18 TP silica column. Peptides were obtained by linear gradient elution with 0.06% (vol/vol) aqueous TFA and 70% (vol/vol) aqueous acetonitrile containing 0.054% (vol/vol) TFA over 40 min. The peaks were detected at 214 nm and the peptide fractions collected by hand.
All peptide sequence analysis was performed on an Applied Biosystems model 477A peptide sequencer using Polybrene-coated glass fiber discs and standard chromatography for phenylthiohydantoin-amino acid analysis.
Evaluation of the Reversibility of the Reaction.
Canavanine hydrolase (160 milliunits) was mechanically agitated with 25 mM hydroxyguanidine (13.2 KBq/μmol−1) and 25 mM l-homoserine in standard buffer containing 0.15 ml of Jack bean leaf arginase supplemented with 2 mg jack bean seed urease (Sigma; type III, 264 units/mg−1) in a final volume of 0.7 ml at 37°C overnight. The reaction was initiated by injecting 0.3 ml of CH into the reaction mixture and terminated by a similar administration of 1.0 ml of 2 M HCl. The acidified reaction mixture was shaken mechanically overnight to collect the evolved radioactive carbon dioxide. All assays were conducted in triplicate. This radiometric assay would convert any radiolabeled canavanine, formed in the reverse direction, to radiolabeled urea and then 14CO2. The latter is trapped in hydroxide of hyamine and quantified by liquid scintillation spectroscopy; this radiometric assay has been described (22).
Reaction Product Identification.
Hydroxyguanidine was established as a reaction product after isolating it by ionic-exchange chromatography using Dowex 50 (NH4+) after all detectable canavanine had been removed by enzymatic degradation. The isolated hydroxyguanidine gave an absorption spectrum, over the range of 400–600 nm, that was identical to authentic material and different from that of canavanine. In addition, the isolated hydroxyguanidine was stoichiometrically hydrogenated to guanidine using a palladium black catalyst in the presence of H2. Guanidine was isolated by ionic-exchange chromatography with Dowex 50 (NH4+). After washing the resin with 3 M ammonia, a solution able to elute all hydroxyguanidine and substances as basic as arginine, guanidine was obtained by developing the column with 5 M ammonia. The eluted guanidine was identified by colorimetric analysis using the diacetyl reaction and guanidine·HCl as a standard (23).
Homoserine was established as a reaction product by automated amino acid analysis of the reaction mixture. In addition, the reaction mixture was processed to convert homoserine to its corresponding lactone. To verify the identity of homoserine by way of its lactone, the deproteinized reaction mixture, after treatment with a vast excess of Jack bean arginase, was placed on a 10 × 25 mm column of Dowex 50 (NH4+). Homoserine was eluted with deionized water while any remaining canavanine was bound to the resin. The column effluent was refluxed with 19% (wt/vol) anhydrous HCl for 90 min. After removing the solvent by rotary evaporation in vacuo, the residue was allowed to dry in vacuo at 55°C for an additional 30 min after solvent removal. Deionized water was added to the residue and the drying process repeated twice. The residue was then dissolved in 3 M HCl and refluxed for 90 min at 115°C. Afterward, the HCl was removed by exhaustive rotary evaporation in vacuo and the residue dissolved in deionized water, taken to pH 3.5 with 1 M NH3, and placed on a 10 × 25 mm column of Dowex 50 (NH4+). The column was washed with 0.7 liter of deionized water and developed with 0.5 liter of 200 mM NH3; the effluent was concentrated by rotary evaporation in vacuo.
The above procedure converts homoserine stoichiometrically to homoserine lactone; unlike homoserine, the latter compound is basic. Free homoserine and any neutral or acidic components of the reaction mixture cannot bind to this resin in the ammonia form. Homoserine lactone is converted to homoserine in situ when the column is developed with 200 mM ammonia. Thus, this procedure provides a means of demonstrating unequivocally the production of homoserine by CH through its lactone. Ammonia was identified as a reaction product by automated amino acid analysis as described above.
Purification of Canavanine Hydrolase.
Preparation of the homogenate. H. virescens fifth instar larvae (n = 125) fresh weight, 300–360 mg/larva, were submerged under crushed ice for at least 15 min. The chilled larvae were opened with a small scissor, the midgut removed from the body cavity, and the gut contents flushed out by forcing 50 mM sodium tricine buffer (pH 7.4) containing 30% (vol/vol) glycerol (standard buffer) through the gut with a fine-needle syringe. The clean midguts were homogenized with 4 ml of the above buffer; the homogenizer was rinsed with 1–2 ml of the standard buffer. After clarifying the turbid solution by centrifugation at 12,000 × g for 15 min, the pellet was reextracted with 2–3 ml of standard buffer and processed as above. The combined supernatant solutions were poured over cheesecloth to remove fat and other floating debris.
Ammonium sulfate fractionation.
The crude homogenate was taken to 52% (vol/vol) saturation with liquid saturated ammonium sulfate (pH 7.2) and allowed to sit at 6°C for 60 min before centrifugation at 12,000 × g for 15 min. The pellet was discarded and the supernatant solution taken to 65% (vol/vol) saturation and allowed to sit on ice for 90 min before centrifugation.
CH was stored routinely at this stage at −80°C as a pellet under frozen liquid ammonium sulfate without loss in catalytic activity. Each pellet contained 3.8 mg protein obtained from 25 larval guts.
Acetone fractionation.
Two thawed CH pellets (50 larval guts) were centrifuged at 11,500 × g for 6 min before dissolving the pellets in 5.0 ml of 50 mM tricine (pH 7.4) containing 30% (vol/vol) glycerol. The protein was stirred mechanically in an ice bath and treated by the drop-wise addition of 4.0 ml (44.4% saturation) of freshly distilled acetone kept at −80°C immediately before use. After standing for exactly 7.0 min at −20°C, the turbid solution was clarified by centrifugation at 14,500 × g for 4 min at 1°C. The supernatant solution was treated with 2.2 ml of acetone (55.4% saturation) and processed as described above. The pellet was dissolved in 1.2 ml of 50 mM tricine (pH 7.4) containing 30% (vol/vol) glycerol, comminuted with a glass homogenizer, and centrifuged at 14,500 × g for 12 min. The supernatant solution was purified further by G-200 Sephadex chromatography.
G-200 Sephadex chromatography.
Canavanine hydrolase (1.0 ml), purified through acetone fractionation, was applied to a 1.4 × 83 cm column of G-200 Sephadex equilibrated with 50 mM tricine (pH 7.4) containing 30% (vol/vol) glycerol. Fractions (1.5 ml) were collected every 15 min (Fig. 1). The two most active fractions, ≈0.3 mg, were pooled and evaluated by HPLC (Table 1).
Figure 1.

Elution profile from G-200 Sephadex chromatography. The effluent protein was measured at 280 nm. CH eluted as the initial peak.
Table 1.
Purification of CH from H. virescens
| Fraction | Total activity, milliunits | Protein, mg | Specific activity, milliunits/mg | Recovery, % |
|---|---|---|---|---|
| Larval gut extract | 690 | 187 | 3.7 | |
| Ammonium sulfate precipitation (52–65%) | 505 | 19 | 26.6 | 73 |
| Acetone fractionation (44.5–55.4%) | 385 | 4.8 | 80.2 | 56 |
| G-200 Sephadex chromatography | 115 | 0.8 | 143.7 | 17 |
Experimental details are provided in the text.
HPLC.
Trace impurities of CH, obtained by G-200 chromatography, were removed from CH by injecting 250 μl of the pooled sample into a C18-WP column (Bakerbound, Beckman); the protein was eluted with a linear gradient of 0.1% (vol/vol) aqueous TFA and 80% (vol/vol) aqueous acetonitrile containing 0.02% (vol/vol) TFA over 40 min. The effluent was monitored at 280 nm with an Scientific Systems (State College, PA) model 500 variable wavelength spectrophotometer. The digitalized data were stored and processed by a Dionex advanced computer interface. Homogeneous CH was collected by hand employing analog output detection obtained with a Spectra-Physics SP-4270 automated area integrator.
Criteria of purity.
The purity of the H. virescens CH was established by PAGE at acid, neutral, and basic pH. CH purity was confirmed by HPLC analysis which revealed a single peak using a variety of elution profiles.
RESULTS
Electrophoretic Analysis.
PAGE procedures revealed that under reduced, denaturating conditions, CH ran as two bands possessing molecular masses of 50 or 47.5 kDa. Under nondenaturing conditions, CH exhibited a mass of 285 kDa. These data suggest that CH is a hexameric protein composed of two different subunits.
CH Stability.
CH was stable for as long as 10 days at 3°C in the presence of 50 mM tricine buffer (pH 7.4) supplemented with 30% (vol/vol) glycerol, but underwent a rapid loss in activity at 3°C when standing in buffer alone (t½ ≈ 8 h). Dialysis of CH, against glycerol-supplemented buffer, caused a complete loss in catalytic activity (t½ ≈ 5 h). This loss also occurred when CH was processed by ultrafiltration with an Amicon P-10 membrane. The marked lability of CH undoubtedly resulted from a dissociation of the native enzyme which can be prevented with glycerol.
Microsome Purification.
To determine if CH was a soluble protein rather than one associated with the microsomal fraction, a freshly prepared gut extract was centrifuged at 12,000 × g for 15 min, and the supernatant solution was recentrifuged at 105,000 × g for 60 min. Assay of the resulting supernatant solution and microsomal pellet revealed that all the detectable enzymic activity resided solely in the supernatant solution.
Characterization of the CH-Mediated Reaction.
The ability of CH to cleave hydrolytically the O–N bond of the guanidinooxy moiety of l-canavanine to generate l-homoserine and hydroxyguanidine with the expected stoichiometric yields was established by direct isolation, identification, and then quantitation of the reaction products by the procedures described in full above. These procedures demonstrated that reaction of 10.0 μmol of l-canavanine with CH (200 milliunits), overnight under standard assay conditions, yielded 9.62 ± 0.4 μmol l-homoserine and 9.71 ± 0.3 μmol hydroxyguanidine. These values are within 3–4% of the expected stoichiometric yields.
Kinetic Analysis.
Kinetic analysis of CH revealed an apparent Km value of 1.1 mM for l-canavanine (Fig. 2). The highest specific activity CH gave a turnover number of 21.1 ± 0.2 μmol·min−1·μmol−1.
Figure 2.

Apparent Km determination for CH. The Km value for CH was determined under standard assay conditions with 0.267 mg CH purified through the G-200 Sephadex step.
Substrate Specificity.
We determined whether CH could attack the O–N bond of the aminooxy moiety of canaline, presumably to yield homoserine and hydroxylamine. While CH degraded canaline [l-2-amino-4-(aminooxy)butyric acid], as determined by colorimetric analysis of the reaction mixture, canaline loss was 4.5 × 10−3 less than that observed for comparably processed canavanine. We also assessed the ability of CH to attack O-ureido-l-homoserine [l-2-amino-4-(ureidooxy)butyric acid; UHS] to yield homoserine and hydroxyurea. UHS was such a poor substrate that compound loss was barely detectable; UHS decomposition was at least 10−3 less than that of canavanine. This finding undoubtedly reflects steric hindrance of the bulky ureidooxy moiety of UHS in failing to gain access to the active site of the enzyme.
Recent development of chemical methods for the synthesis of the lower and higher homolog of l-canavanine, 2-amino-3-(guanidinooxy)propionate, and 2-amino-5-(guanidinooxy)hexanoate (homocanavanine), respectively, permitted an assessment of the effect of aliphatic chain length on CH activity. Neither canavanine derivative supported detectable hydroxyguanidine formation. In a similar vein, neither methyl, ethyl, or butyl ester of l-canavanine yielded hydroxyguanidine on treatment with CH. Our structure–activity studies established the rigid requirement for canavanine’s unique structural features for a compound to serve as an active substrate for CH.
Reversibility of the Reaction.
The reversibility of the CH-mediated cleavage of l-canavanine to l-homoserine and hydroxyguanidine was evaluated with a highly sensitive radiometric assay described above. This radiometric assay revealed that, even under conditions in which arginase pulled the reaction in the reverse direction, only 1.2% of the [14C]hydroxyguanidine was converted to l-[guanidinooxy-14C]canavanine overnight at 37°C. Thus, CH mediated essentially an irreversible reaction.
Prior analysis of hydroxyguanidine metabolism in H. virescens demonstrated that this nitrogen-rich compound is reduced to guanidine which is the end product of canavanine catabolism (10). Guanidine evidently has no discernible consequence on larval growth and development.
Primary Structural Analysis.
To further characterize this larval enzyme, portions of its primary structure were determined. This analysis revealed structural information for three peptidal fragments: peptide 1, KFVPISCPMPTNRQP; peptide 2, KSLVVNINENSVK; and peptide 3, KMPWFTNXXTYFGXT. Comparison of these sequences with primary structural information stored in the nonredundant protein database at GenBank, the Saccharomyces genome database, and the Haemophilus influenzae genome database failed to disclose significant structural homology with a known macromolecule.
DISCUSSION
H. virescens larvae are able to tolerate extraordinarily high levels of dietary canavanine even though this nonprotein amino acid is highly insecticidal to nonadapted animals. This ability results primarily from a gut enzyme, canavanine hydrolase, that mediates an irreversible hydrolysis of l-canavanine to l-homoserine and hydroxyguanidine. This detoxification mechanism stands in contrast to canavanine catabolism by larvae of the bruchid beetle, C. brasiliensis, who develop within and are nurtured by the canavanine-laden seeds of the legume, Dioclea megacarpa (4, 5). This neotropical insect hydrolytically cleaves l-canavanine to l-canaline and urea (5). At the same time, these larvae have extraordinarily high levels of urease (24) which enables the developing larvae to generate ammoniacal nitrogen for the synthesis of a number of amino acids (6).
The gut of H. virescens also contains an active l-arginine kinase (adenosine 5′-triphosphate: l-arginine phosphotransferase, EC 3.5.3.1) which catalyzes an ATP-dependent phosphorylation of l-canavanine to yield the novel phosphagen, l-canavanine phosphate (25). These two enzymes account for virtually all of the canavanine catabolism fostered by an extract of the larval gut.
H. virescens larvae are generalist feeders—i.e., they consume a diversified array of plants. Presently, this insect feeds predominately on canavanine-free plants (26, 27), but many leguminous plants, particularly members of the Fabaceae—the group of legumes most noted for their ability to store canavanine (28, 29)—are consumed by the larvae. It is not know how this insect evolved this detoxification enzyme; perhaps, this evolved when the larvae relied more heavily on canavanine-containing plants as a food resource.
Given that the gut was cleansed thoroughly before extraction, CH is probably an enzyme of the gut wall rather than a protein secreted into the gut cavity. The stabilizing effect of glycerol on this otherwise highly labile protein undoubtedly results from its ability to mimic conditions existing within the gut wall matrix that stabilize and support CH.
Finally, prior studies of canavanine disposition in the hemolymph of H. virescens larvae established that an injected dose of 5 g l-canavanine/kg−1 fresh body weight is cleared with a t½ of 135 min by active metabolic processes (7). This route of administration bypassed the gut initially and such canavanine clearance from the hemolymph might be achieved by a CH of the fat body.
The fat body, in simple terms, is the metabolic equivalent of the vertebrate liver. It is the site of synthesis of complex carbohydrates, lipids, nonessential amino acids, and a host of structural and enzymic proteins (30). Analysis of the CH activity of the fat body revealed that this organ does not have any significant CH activity. While movement of CH from the lumen of the digestive tract into the hemolymph has not been established, enzymes can move unchanged across the gut wall into the hemolymph (30). Such transport may account for the ability of H. virescens to efficiently clear the hemolymph of canavanine.

Acknowledgments
We gratefully acknowledge National Science Foundation Grant IBN-9302875 and support provided by Consejo de Desarrollo Cientifico y Humanistico and Fundacion Polar, Maracay, Venezuela. This is paper 96–08-035 of the Kentucky Agricultural Experimental Station, Lexington.
ABBREVIATION
- CH
l-canavanine hydrolase
- PCAF
pentacyanoammonioferrate
- TFA
trifluoroacetic acid
References
- 1.Dahlman D L, Rosenthal G A. Comp Biochem Physiol A. 1975;51:33–36. doi: 10.1016/0300-9629(75)90409-0. [DOI] [PubMed] [Google Scholar]
- 2.Rosenthal G A. Q Rev Biol. 1977;52:155–178. doi: 10.1086/409853. [DOI] [PubMed] [Google Scholar]
- 3.Rosenthal G A. In: Nonprotein Amino Acids as Protective Allelochemicals. Rosenthal G A, Berenbaum M, editors. San Diego: Academic; 1991. pp. 1–11. [Google Scholar]
- 4.Bleiler J, Rosenthal G A, Janzen D H. Ecology. 1988;69:427–433. [Google Scholar]
- 5.Rosenthal G A. Sci Am. 1983;249:164–171. [Google Scholar]
- 6.Rosenthal G A, Hughes C G, Janzen D H. Science. 1982;217:353–355. doi: 10.1126/science.217.4557.353. [DOI] [PubMed] [Google Scholar]
- 7.Berge M A, Rosenthal G A, Dahlman D L. Pestic Biochem Physiol. 1986;25:319–326. [Google Scholar]
- 8.Berge M, Rosenthal G A. J Food Agric Chem. 1990;38:2061–2065. [Google Scholar]
- 9.Berge M A, Rosenthal G A. Chem Res Toxicol. 1991;4:237–240. doi: 10.1021/tx00020a018. [DOI] [PubMed] [Google Scholar]
- 10.Rosenthal G A. Bioorg Chem. 1992;20:55–61. [Google Scholar]
- 11.Kalyankar G D, Ikawa M, Snell E E. J Biol Chem. 1958;233:1175–1178. [PubMed] [Google Scholar]
- 12.Webb E C. Enzyme Nomenclature. San Diego: Academic; 1992. [Google Scholar]
- 13.Bass M, Harper L, Rosenthal G A, NaPhuket S, Crooks P. Biochem Syst Ecol. 1995;23:717–721. [Google Scholar]
- 14.Rosenthal G A. Anal Biochem. 1973;56:435–439. doi: 10.1016/0003-2697(73)90209-1. [DOI] [PubMed] [Google Scholar]
- 15.Rosenthal G A, Dahlman D L, Crooks P A, NaPhuket S, Trifonov L S. J Food Agric Chem. 1995;43:2728–2734. [Google Scholar]
- 16.Ozinskas A J, Rosenthal G A. Bioorg Chem. 1986;14:157–162. [Google Scholar]
- 17.Berger R S. USDA Res Serv. 1963;33:84. [Google Scholar]
- 18.Rosenthal G A. Anal Biochem. 1977;77:147–151. doi: 10.1016/0003-2697(77)90299-8. [DOI] [PubMed] [Google Scholar]
- 19.Kavanaugh D, Berge M, Rosenthal G A. Plant Physiol. 1990;94:67–70. doi: 10.1104/pp.94.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chinard F P. J Biol Chem. 1952;199:91–94. [PubMed] [Google Scholar]
- 21.Rosenthal G A, Dahlman D L. J Biol Chem. 1991;266:15684–15687. [PubMed] [Google Scholar]
- 22.Rosenthal G A, Thomas D. Anal Biochem. 1985;147:428–431. doi: 10.1016/0003-2697(85)90292-1. [DOI] [PubMed] [Google Scholar]
- 23.Bonas J E, Cohen B D, Natelson S. Microchem J. 1978;7:63–77. [Google Scholar]
- 24.Rosenthal G A. In: Bruchids and Legumes: Economics, Ecology and Coevolution. Fujii K, Gatehouse A M R, Johnson C D, Mitchell R, Yoshida T, editors. Amsterdam: Kluwer; 1990. pp. 161–170. [Google Scholar]
- 25.Gindling H L, Rosenthal G A, Dahlman D L. Insect Biochem Mol Biol. 1995;25:933–938. [Google Scholar]
- 26.Bell E A, Lackey J A, Polhill R M. Biochem Syst Ecol. 1978;6:201–212. [Google Scholar]
- 27.Natelson S. J Agric Food Chem. 1985;33:413–419. [Google Scholar]
- 28.Pearson O. The Insect Pests of Cotton in Tropical Africa. London: Commonwealth Inst. Entomol.; 1958. [Google Scholar]
- 29.Zulucki M P, Daglish G, Firempong S, Twine P. Aust J Zool. 1986;34:779–814. [Google Scholar]
- 30.Friedman S. In: Fundamentals of Insect Physiology. Blum M S, editor. New York: Wiley; 1985. pp. 467–506. [Google Scholar]