

Abstract
Significant cleavage by hammerhead ribozymes requires activation by divalent metal ions. Several models have been proposed to account for the influence of metal ions on hammerhead activity. A number of recent papers have presented data that have been interpreted as supporting a one-metal-hydroxide-ion mechanism. In addition, a solvent deuterium isotope effect has been taken as evidence against a proton transfer in the rate-limiting step of the cleavage reaction. We propose that these data are more easily explained by a two-metal-ion mechanism that does not involve a metal hydroxide, but does involve a proton transfer in the rate-limiting step.
Hammerhead ribozymes are one of several types of RNA enzymes (1). The term hammerhead refers to a small domain, found in some viral satellite RNAs, that is required for RNA self-cleavage (2). This small domain is necessary for cleavage activity on RNA sequences that form complementary base pairs with the hammerhead (3). Crystal structures have been obtained for hammerhead ribozymes complexed with different noncleavable substrate analogues (4, 5). Because of its small size and simple structure, the hammerhead has been extensively studied as a prototypic RNA enzyme. Fig. 1 depicts a hammerhead ribozyme that is base paired to a 17-nucleotide substrate RNA. This hammerhead, called HH16 (6), has 8 bp in both recognition helices that flank the cleavage site on the substrate. Cleavage occurs on the 3′ side of a cytosine residue in the substrate strand.
Figure 1.

The hammerhead ribozyme HH16–substrate complex. Putative base pairing in the hammerhead ribozyme HH16–substrate complex. The hammerhead hairpin (Stem II) is shown with two G·A base pairs, as observed in the hammerhead crystal structures (4, 5).
Several models have been proposed for the cleavage of RNA phosphodiester bonds by hammerhead ribozymes; most models include a solvated metal hydroxide ion at the catalytic center (4, 7, 8). In these metal-hydroxide-ion models (Fig. 2A) a divalent metal hydroxide binds to the pro-R oxygen on the phosphate at the site of cleavage and activates the ribozyme primarily by removing a proton from the 2′-OH of the attacking nucleotide residue. The activated 2′-O− species then acts as the nucleophile by attacking the phosphodiester bond.
Figure 2.

Two possible models for the hammerhead ribozyme chemical mechanism. (A) A one-metal-hydroxide-ion model. In this model, the metal hydroxide acts as a Brønsted base by accepting a proton from the 2′-OH. The activated 2′-O− acts as a nucleophile by attacking the phosphate at the site of cleavage. (B) A two-metal-ion model. In this model, the metal ion in site A acts as a Lewis acid by accepting electrons from the 2′-oxygen, which polarizes and weakens the OH bond and makes the 2′-OH proton dissociate more readily. The metal ion in site B also acts as a Lewis acid by accepting electrons from the 5′-oxygen, which polarizes and weakens the O-P bond and makes this oxygen a better leaving group. In both models the reaction is shown as concerted, with the rate-limiting step being the removal of the 2′-OH proton.
A two-metal-ion model has also been proposed for the hammerhead ribozyme (9). This model is based on mechanisms that have been put forward to explain the activities of alkaline phosphatase and the 3′-5′ exonuclease of DNA polmerase I (10, 11). Similar two-metal-ion models have been proposed for a wide variety of protein and RNA enzymes that mediate phosphoryl transfer reactions (9). In one such model for the hammerhead ribozyme (Fig. 2B), both divalent metal ions bind to the pro-R oxygen on the phosphate at the site of cleavage. The first metal ion (in site A) interacts directly with the 2′-oxygen of the attacking ribonucleotide residue, while the second metal ion (in site B) interacts with the 5′-oxygen leaving group. In a two-metal-ion mechanism, the metal ion in site A is thought to facilitate the formation of the 2′-oxyanion that acts as the nucleophile, and the metal ion in site B is considered to stabilize the oxyanion leaving group (11). Both metal ions may also stabilize the specific geometry of the transition state.
Although both types of models have been suggested for the hammerhead ribozyme, several papers in the literature have presented data that have been taken as evidence in support of the single-metal-hydroxide-ion model (8). These include an inverse correlation between the pKa values of different metal ions and their relative cleavage activities (7), as well as the effects of phosphorothioate replacements at the 5′-internucleotide bridging oxygen (12). In addition, an observed solvent isotope effect has been interpreted as evidence that a proton transfer step does not occur in the transition state for the hammerhead ribozyme (13).
Recently we have obtained data on the relationship between metal ion concentration and hammerhead activity that is consistent with the presence of two metal ion sites on the hammerhead–substrate complex that are critical for cleavage activity (unpublished data). Our results support and extend a separate study suggesting that two divalent metal ions are especially important in the hammerhead ribozyme cleavage reaction (14). These results led us to reexamine some of the conclusions drawn from the literature that have been taken as evidence in support of a single-metal-hydroxide-ion model, and against a two-metal-ion model. In contrast to most earlier interpretations, this reexamination has suggested to us that the literature provides support for a two-metal-ion mechanism for the hammerhead ribozyme, and that these data are not easily explained by a single-metal-hydroxide-ion model. Furthermore, we have concluded that the observed solvent isotope effect is consistent with a proton transfer step occurring in the transition state in a two-metal-ion model, in contrast to the interpretation supported by Sawata et al. (13). In this paper we present our rationale for these conclusions.
RESULTS AND INTERPRETATIONS
The Correlation Between Metal Ion pKa and Cleavage Activity.
The hammerhead cleavage reaction can be facilitated by a variety of divalent metal ions, including Mg2+, Co2+, Mn2+, and Ca2+ (7, 15). For some of these ions an inverse correlation between metal ion pKa and the ability to promote RNA cleavage has been noted—i.e., the lower the pKa, the higher the cleavage rate at a given metal ion concentration. This pKa dependence has been taken as evidence that a metal hydroxide [Me2+(OH)−] participates in the cleavage reaction by abstracting a proton from the 2′-OH of the attacking nucleotide residue [(7), Fig. 2A]. This role is analogous to that proposed for the metal ion in the Pb2+-catalyzed cleavage of phosphodiester bonds in tRNA (16), and is consistent with the role of histidine groups that act as Lewis bases in other ribonucleases, including RNase A and RNase T1. According to this model, abstraction of the 2′-OH proton forms an activated 2′-O−, which then acts as a nucleophile by attacking the phosphodiester linkage.
Careful consideration is required to understand how metal ion pKa values might influence catalysis. The pKa is, of course, a measure of the equilibrium constant for the release of a hydrogen ion from water. For example, the pKa of a water molecule in bulk solution is ≈16. Thus, for the reaction H2O ⇔ OH− + H+, the pKa is defined as pKa = −log {([H+] [OH−])/[H2O]}. For metal ions, the pKa is the negative logarithm of the equilibrium constant for the loss of a proton from a water molecule in the first hydration sphere surrounding the metal ion in solution. For Mg2+, which has six water molecules in the first hydration sphere, the pKa is ≈11.4 for the loss of the first proton from an inner-sphere water, where Mg2+ (H2O) ⇔ Mg2+ (OH−) + H+, and pKa = −log{[Mg2+ OH−] [H+]/[Mg2+ (H2O)]}.
A water molecule directly coordinated with a Mg2+ ion in solution is more likely to be missing a proton than is a water molecule in bulk solution. The lower the pKa of a particular divalent ion, the greater the fraction of this ion that will be in the form of metal hydroxide at a given pH. At pH values well below the pKa of a given metal ion, the concentration of metal hydroxide in solution is approximated by:
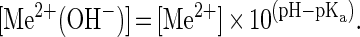 |
For the hammerhead, if a metal hydroxide is the active species and the concentration of metal hydroxide in the active site is proportional to the concentration of metal hydroxide in solution, then metal ions with lower pKa values will be in the form of the [Me2+ (OH−)] species at higher concentrations in the active site. If the ability of a metal ion to stimulate catalysis is simply proportional to the concentration of [Me2+ (OH−)] at the active site, then metal ions with lower pKa values should have higher cleavage activities due to the increased presence of the [Me2+ (OH−)] species. This argument, coupled with the observed relationship between metal ion pKa values and metal ion cleavage activities, has been taken as evidence that a solvated metal hydroxide participates in catalysis (7).
However, the ability of a [Me2+ (OH−)] to activate cleavage should not be the same for metal ions with different pKa values according to the metal-hydroxide-ion model. In removing a proton from the 2′-OH of the nucleotide sugar, the metal ion is acting as a Brønsted base. Of course, the conjugate base of a strong acid is a weak base. Thus metal ions with lower pKa values (stronger acids) may be present at higher concentrations in the active site in the form of a solvated metal hydroxide (at a given pH), but such ions should be correspondingly weaker bases and therefore be less able to remove the 2′-OH proton, since the 2′-OH group has a relatively high pKa. [We estimate the pKa of the 2′-OH at greater than 13 based on the literature values of ≈12.3 for the analogous proton in the free nucleotides and sugars (17). The cost of placing an additional negative charge in such close proximity to the already negatively charged phosphate diester moiety should drive the pKa of this proton considerably higher. Indeed we have been unable to find a value for this pKa in the literature, presumably because it is too high to be readily measured. For the purposes of this discussion it need only be assumed that the pKa of the 2′-OH is above the pKa of the metal ions from which the pKa activity correlation was determined.]
We consider two possible mechanisms, both of which are examples of specific base catalysis involving a metal hydroxide. One role of the metal ion could be to increase the equilibrium concentration of the 2′-O− species, under conditions where the nucleophilic attack of the 2′-O− on the phosphorus center occurs within the rate determining step. In this case an increase in the equilibrium concentration of the 2′-O− would increase the rate of cleavage according to the proposed rate law: rate = k [2′-O−]. A metal ion with a lower pKa could indeed be present in the active site as a metal ion hydroxide at a higher concentration (7), but it should be a weaker base and therefore less effective in driving the equilibrium to favor the 2′-O− species. Indeed there should be virtually no correlation between metal ion pKa and cleavage activity because these two effects (greater basicity and reduced occupancy of the metal hydroxide species) must cancel.† We suggest that the experimentally observed inverse correlation between metal ion pKa and cleavage rate should not be obtained if this is the operative mechanism.
A similar argument applies when deprotonation of the 2′-OH occurs as part of the rate determining step. In this mechanism the metal ion affects the kinetics of the reaction by lowering the activation energy barrier to deprotonation, since the rate at which the proton is removed from the 2′-OH directly contributes to the overall rate of the reaction. Of course, the pKa of a solvated metal ion is a thermodynamic parameter and does not necessarily correlate directly with the kinetic profile of the ribozyme cleavage reaction. In general the rate of deprotonation of an acid by base will increase with base strength when the pKa of the acid is significantly higher than the pKa of the abstracting base (18). This should apply for the hammerhead, since the pKa of the 2′-OH is significantly higher than the pKa values of the different divalent metal ions examined (7). Again, low pKa metal ions should be present at higher concentrations as metal ion hydroxides (7), but they will remove the 2′-OH proton at reduced rates. This mechanism also should not produce the observed correlation between metal ion pKa and cleavage activity. We conclude that a simple model involving a metal hydroxide acting as a base to abstract the 2′-OH proton is not supported by the metal ion pKa dependence that is observed.
In contrast, the observed pKa dependence is easily explained by a direct coordination between a divalent metal ion and the 2′-oxygen of the attacking nucleotide residue, as shown for site A in the two-metal-ion model in Fig. 2B. In this situation, a divalent metal ion, not a metal hydroxide, associates with the 2′-oxygen. This inner sphere association by the metal ion to the 2′-oxygen withdraws electrons from the oxygen, which polarizes and weakens the 2′-OH bond. This increases the acidity of the 2′-OH proton in the same way that a proton on a metal-coordinated inner sphere water molecule becomes more acidic. Because this activity for a metal ion should be directly related to the pKa of the metal ion, metal ions with lower pKa values will polarize the OH bond to a greater extent and thus activate the ribozyme to a greater extent, provided that the removal of this proton is rate limiting (see below). Thus a model in which a metal ion is coordinated directly to the 2′ oxygen is consistent with the observed correlation between the pKa values of different metal ions and their ability to activate the ribozyme.
The pH-Rate Profile.
Hammerhead cleavage activity increases linearly with pH (i.e., with log [OH−]) from pH 6.5 to 9.0 (7). Such a pH-rate profile is consistent with any model in which an increase in the OH− concentration stimulates catalysis, including both models presented in Fig. 2. In Fig. 2A, a higher OH− concentration increases the fraction of metal ion that is present in the form of the metal hydroxide. The increased metal hydroxide concentration in the catalytic center facilitates catalysis by deprotonating the nucleophilic 2′-OH, leading to an increased concentration of the attacking 2′-O− species. In Fig. 2B a hydroxide directly deprotonates the 2′-OH. In this case a higher hydroxide concentration generates more of the attacking 2′-O− species directly. The observed pH-rate profile is therefore consistent with both models presented in Fig. 2 and cannot be used to distinguish between them.
The Deuterium Isotope Effect.
A solvent deuterium isotope effect has been observed for the hammerhead ribozyme, in that the cleavage reaction is 4-fold faster in the presence of H2O than in the presence of D2O under the same conditions (13). Isotope effects of this kind and magnitude can be due to a proton transfer step occurring in the transition state for catalysis, because chemical bonds to deuterons are broken more slowly than chemical bonds to protons. However, this isotope effect for the hammerhead has been taken as evidence against a proton transfer step occurring in the transition state. The observed solvent isotope effect was instead interpreted as being consistent with a change in the equilibrium concentration of metal hydroxide in the catalytic center (13). The supposed lack of a proton transfer step in the transition state was further interpreted as indicating that the transition state occurs late in the reaction pathway and probably involves a metal ion that stabilizes the leaving 5′-oxygen, as depicted for the metal ion located in site B in Fig. 2B.
However, in the “standard” two-metal-ion model (Fig. 2B) it is a divalent metal ion, rather than a metal hydroxide in site A, that facilitates the removal of the proton from the 2′-OH. In this version of the two-metal-ion model the observed isotope effect could not be due to a change in the metal hydroxide concentration, because metal hydroxide does not participate in catalysis and should be present at very low concentrations at neutral pH. There can be no compensating change in metal hydroxide concentration that could account for the isotope effect that is observed. Thus the observed isotope effect is consistent with the occurrence of a proton transfer step in the transition state, provided that a metal ion participates as shown in site A of Fig. 2B.
If a metal ion does participate in site A (Fig. 2B), and if a proton transfer step does occur in the transition state, then an argument for a late transition state based on the observed solvent isotope effect is invalid. The observed isotope effect is simply explained by a model in which there is an early transition state involving a proton transfer. Thus the isotope effect does not directly argue in favor of a metal ion in site B, as has been suggested (13). However, other arguments based on ab initio calculations have been presented that support the presence of a metal ion in site B (ref. 19; see below).
Phosphorothioate Replacement in the Catalytic Center.
Several phosphate oxygens in the ribozyme-substrate complex have been replaced by sulfur (12, 20–22). These phosphorothioate replacement studies are used to identify oxygen atoms that interact directly with divalent metal ions (23). The logic behind these experiments is that oxygen atoms coordinate “soft” metal ions (such as Mn2+) and “hard” metal ions (such as Mg2+) with similar affinities, while sulfur atoms bind soft metal ions more tightly than hard ones. When oxygen is replaced by sulfur at a particular position, a lower catalytic activity in the presence of Mg2+ relative to Mn2+ has been interpreted to mean that this position is coordinated with a divalent metal ion that is important for hammerhead activity. Conversely if no relative difference in activity is observed, then this position is considered unlikely to be coordinated to a divalent ion that is important for catalysis. Thio-replacement experiments suggest that the natural oxy-form of the ribozyme has a metal ion in the identified position, provided that the thio- and oxy-forms of the ribozyme are mechanistically similar (23).
For example, when the pro-S terminal oxygen on the phosphate at the site of cleavage (Fig. 2A) is replaced by sulfur, the cleavage rate is similar regardless of whether Mn2+ or Mg2+ are present (22). This result suggests that a sulfur in this position (and, by analogy, the oxygen it replaces) is not directly coordinated with a divalent ion. In contrast, when the pro-R oxygen is replaced by sulfur, Mn2+ mediates cleavage much more effectively than does Mg2+. This result suggests that the pro-R sulfur (and, by analogy, the oxygen it replaces) is directly coordinated with a divalent metal ion. Taken together these results suggest that the pro-R, but not the pro-S, phosphate oxygen is directly coordinated with a divalent metal ion that is required for cleavage. Both models presented in Fig. 2 include the coordination of one or more metal ions to the pro-R and not to the pro-S phosphate oxygen. The results of thiophosphate replacement experiments at this position are therefore consistent with both models.
The internucleotide-bridging 5′-oxygen (Fig. 2A) has also been replaced by sulfur. Cleavage of the 5′-sulfur substrate is greatly stimulated even in the absence of the ribozyme and divalent metal ions (12). For the sulfur-containing substrate, the cleavage rate is increased by the addition of Mn2+ and Mg2+, but no significant difference in cleavage activity is observed for these two metal ions. These data were taken as evidence that a metal ion is not coordinated at the 5′-sulfur and, by analogy, also not coordinated at the naturally-occurring 5′-oxygen (12). The absence of a metal ion at this position was taken as direct evidence against a two-metal-ion model, since the two-metal-ion model includes a metal ion coordinated to the 5′-oxygen.
In contrast, ab initio calculations predict that the bond breaking portion of the cleavage reaction should provide the largest contribution to the activation energy barrier in hammerhead catalysis (19). Stabilization of the leaving group (the 5′-oxygen) with a metal cation would therefore be expected to lower the activation barrier and increase the cleavage rate.
We suggest that actually the 5′-sulfur replacement studies strongly support a model in which a metal ion coordinated to the 5′-oxygen plays a critical role in catalysis. Unlike thiophosphate replacements made in other positions, the 5′-sulfur replacement substrate is activated ≈106-fold for cleavage (relative to the 5′-oxygen substrate) even in the absence of divalent ions (12). This substrate is prone to cleavage even in the absence of ribozyme (12). Because sulfur is a much better leaving group than is oxygen, this result suggests that cleavage of the 5′-oxygen∼phosphorus bond is normally part of the rate-limiting step. Only if this step is rate limiting should providing a better leaving group greatly increase the cleavage rate.
The observation that the 5′-sulfur replacement is greatly activated for cleavage also suggests that a metal ion coordinated to the 5′-oxygen should activate the ribozyme, since a metal ion will act to stabilize the developing negative charge on the leaving 5′-oxygen in the transition state, thus making it a better leaving group. This affect might even explain the observed metal ion pKa dependence (see above), since metal ions with lower pKa values will weaken the 5′-oxygen∼phosphorus bond, and thereby activate the ribozyme, to a greater extent.
An earlier step in the reaction pathway is likely to be rate limiting for the sulfur-containing substrate. Otherwise some thio-effect at the 5′-position should still be observed. Moreover no solvent deuterium isotope effect is seen for the 5′-sulfur-containing substrate (12). This lack of a solvent isotope effect can be explained by a model in which the cleavage reaction does not require a deprotonation of the 2′-OH in the transition state. It may be that, because the 5′-sulfur is such a good leaving group, the 2′-OH species can act directly as the nucleophile, in which case a proton transfer at this position will not be required for cleavage, and no solvent isotope effect will be observed. This would place the transition state very early in the reaction pathway for the 5′-sulfur-containing substrate, possibly corresponding to a rate-limiting conformational change within the ribozyme-substrate complex. Such an early transition state must still be facilitated by divalent metal ions, since the 5′-sulfur-containing substrate can still be activated for cleavage (12), but the metal ion must act at a position other than the 5′-sulfur. In the two metal model, this would presumably be at site A. In this case, however, the primary role of the metal ion could not be to deprotonate the 2′-OH or else an isotope affect would be seen. We suggest that a metal ion in this position may play an important structural role in orienting the 2′-oxygen so that it can act as the nucleophile in catalysis.
X-Ray Structures.
An x-ray crystal structure of a hammerhead-substrate complex with an apparent bound divalent metal ion near the cleavage site has been advanced as evidence in support of the single-metal-hydroxide-ion model (8). However, crystal structures necessarily represent energy minima and generally do not provide direct detailed structural information about transition states unless the structural data represent a designed transition state analog. Significant molecular reorganization is required to reach the proposed transition state structure from the observed hammerhead crystal structures, and possible pathways for this reorganization have been proposed (4, 5). If the ribozyme complex depicted in the available x-ray structures must proceed through one or more intermediates before the cleavage event, it is difficult to interpret the absence of one or more metal ions bound near the catalytic center. The essential metal ion binding site (or sites) may exist transiently in a shallow and relatively high energy well located further along the reaction pathway from the observed crystal structure and closer to the transition state. Similarly, it cannot be assured at this time that any particular divalent metal ion observed in the ribozyme crystal structure actually plays a direct role in catalysis. The currently available x-ray structures do not provide definitive evidence for the number or function of metal ions in hammerhead catalysis.
CONCLUSIONS
We conclude that the correlation between cleavage activity and metal ion pKa, the solvent deuterium isotope effect, and the results of 5′-sulfur replacement experiments, can all be explained by a mechanism that falls within the framework of a general two-metal-ion model proposed for a number of phosphoryl transfer reactions (9–11). The data available in the literature are consistent with a two-metal-ion model in which metal ions in sites A and B both play a role in the transition state. According to our simple model, a Mg2+ ion in site B is directly coordinated to the 5′ oxygen of the leaving group and stabilizes the negative charge buildup on the 5′-oxygen in the transition state, lengthening and weakening the bond between this oxygen and the phosphorus center. A Mg2+ ion in site A is directly coordinated to the 2′-oxygen and increases the acidity of the 2′-OH proton. The 2′-O− acts as a nucleophile by displacing the leaving group from the electrophyllic phosphorus center. One or both metal ions may also play additional roles in organizing the transition state, and at least one metal ion interacts with the pro-R oxygen on the phosphate at the site of cleavage. In summary, and in contrast to several earlier reports, we believe that a two-metal-ion model most easily explains the available data on the hammerhead ribozyme and provides important mechanistic insight into the cleavage reaction. Experimental results from our laboratory further characterizing two divalent ions critical for catalysis will be presented elsewhere (unpublished data).
Acknowledgments
We thank Mark Young and other colleagues for helpful discussions. This work was supported in part by Public Health Service Research Grants GM-15792 and GM-29158 (to P.H.v.H.) and by a postdoctoral fellowship (to B.W.P.) from the Jane Coffin Childs Memorial Fund for Medical Research. P.H.v.H. is an American Cancer Society Research Professor of Chemistry.
Footnotes
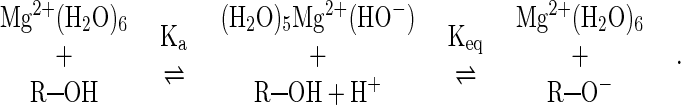

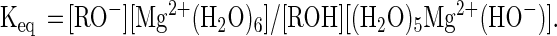

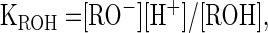
References
- 1.Christoffersen R E, Marr J J. J Med Chem. 1995;38:2023–2037. doi: 10.1021/jm00012a001. [DOI] [PubMed] [Google Scholar]
- 2.Keese P, Symons R H. In: Viroids and Viroid-like Pathogens. Semancik J S, editor. Boca Raton, FL: CRC; 1987. pp. 1–47. [Google Scholar]
- 3.Forster A C, Symons R H. Cell. 1987;50:9–16. doi: 10.1016/0092-8674(87)90657-x. [DOI] [PubMed] [Google Scholar]
- 4.Scott W G, Finch J T, Klug A. Cell. 1995;81:991–1002. doi: 10.1016/s0092-8674(05)80004-2. [DOI] [PubMed] [Google Scholar]
- 5.Pley H W, Flaherty K M, McKay D B. Nature (London) 1994;372:68–74. doi: 10.1038/372068a0. [DOI] [PubMed] [Google Scholar]
- 6.Hertel K J, Herschlag D, Uhlenbeck O C. Biochemistry. 1994;33:3374–3385. doi: 10.1021/bi00177a031. [DOI] [PubMed] [Google Scholar]
- 7.Dahm S C, Derrick W B, Uhlenbeck O C. Biochemistry. 1993;32:13040–13045. doi: 10.1021/bi00211a013. [DOI] [PubMed] [Google Scholar]
- 8.Scott W G, Klug A. Trends Biochem Sci. 1996;21:220–224. [PubMed] [Google Scholar]
- 9.Steitz T A, Steitz J A. Proc Natl Acad Sci USA. 1993;90:6498–6502. doi: 10.1073/pnas.90.14.6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Freemont P S, Friedman J M, Beese L S, Sanderson M R, Steitz T A. Proc Natl Acad Sci USA. 1988;85:8924–8928. doi: 10.1073/pnas.85.23.8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beese L S, Steitz T A. EMBO J. 1991;10:25–33. doi: 10.1002/j.1460-2075.1991.tb07917.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuimelis R G, McLaughlin L W. Biochemistry. 1996;35:5308–5317. doi: 10.1021/bi952994p. [DOI] [PubMed] [Google Scholar]
- 13.Sawata S, Komiyama M, Taira K. J Am Chem Soc. 1995;117:2357–2358. [Google Scholar]
- 14.Hendry P, McCall M J. Nucleic Acids Res. 1995;23:3928–3936. doi: 10.1093/nar/23.19.3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dahm S C, Uhlenbeck O C. Biochemistry. 1991;30:9464–9469. doi: 10.1021/bi00103a011. [DOI] [PubMed] [Google Scholar]
- 16.Brown R S, Dewan J C, Klug A. Biochemistry. 1985;24:4785–4801. doi: 10.1021/bi00339a012. [DOI] [PubMed] [Google Scholar]
- 17.Dean J A. Lange’s Handbook of Chemistry. New York: McGraw–Hill; 1992. pp. 8.19–8.71. [Google Scholar]
- 18.Eigen M. Angew Chem Int Ed. 1964;3:1–72. [Google Scholar]
- 19.Uebayasi M, Uchimaru T, Koguma T, Sawata S, Shimayama T, Taira K. J Org Chem. 1994;59:7414–7420. [Google Scholar]
- 20.Koizumi M, Ohtsuka E. Biochemistry. 1991;30:5145–5150. doi: 10.1021/bi00235a005. [DOI] [PubMed] [Google Scholar]
- 21.Ruffner D E, Uhlenbeck O C. Nucleic Acids Res. 1990;18:6025–6029. doi: 10.1093/nar/18.20.6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Slim G, Gait M J. Nucleic Acids Res. 1991;19:1183–1188. doi: 10.1093/nar/19.6.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herschlag D, Piccirilli J A, Cech T R. Biochemistry. 1991;30:4844–4854. doi: 10.1021/bi00234a003. [DOI] [PubMed] [Google Scholar]