

Abstract
The 2.1-Å resolution crystal structure of wild-type green fluorescent protein and comparison of it with the recently determined structure of the Ser-65 → Thr (S65T) mutant explains the dual wavelength absorption and photoisomerization properties of the wild-type protein. The two absorption maxima are caused by a change in the ionization state of the chromophore. The equilibrium between these states appears to be governed by a hydrogen bond network that permits proton transfer between the chromophore and neighboring side chains. The predominant neutral form of the fluorophore maximally absorbs at 395 nm. It is maintained by the carboxylate of Glu-222 through electrostatic repulsion and hydrogen bonding via a bound water molecule and Ser-205. The ionized form of the fluorophore, absorbing at 475 nm, is present in a minor fraction of the native protein. Glu-222 donates its charge to the fluorophore by proton abstraction through a hydrogen bond network, involving Ser-205 and bound water. Further stabilization of the ionized state of the fluorophore occurs through a rearrangement of the side chains of Thr-203 and His-148. UV irradiation shifts the ratio of the two absorption maxima by pumping a proton relay from the neutral chromophore’s excited state to Glu-222. Loss of the Ser-205–Glu-222 hydrogen bond and isomerization of neutral Glu-222 explains the slow return to the equilibrium dark-adapted state of the chromophore. In the S65T structure, steric hindrance by the extra methyl group stabilizes a hydrogen bonding network, which prevents ionization of Glu-222. Therefore the fluorophore is permanently ionized, causing only a 489-nm excitation peak. This new understanding of proton redistribution in green fluorescent protein should enable engineering of environmentally sensitive fluorescent indicators and UV-triggered fluorescent markers of protein diffusion and trafficking in living cells.
The green fluorescent protein (GFP) from the jellyfish Aequorea victoria is the first known protein in which visible fluorescence is genetically encodable. The fluorophore is derived from natural residues present within the primary structure of GFP, so no exogenous cofactor or substrate is needed for fluorescence (1, 2). The tremendous potential of GFP as a reporter of gene expression, cell lineage, and protein trafficking and interactions has been extensively reviewed (3–5).
Wild-type (WT) GFP is a 238-aa protein (2). In vitro GFP is a particularly stable protease-resistant protein (6) and is only denatured under extreme conditions (7). The GFP chromophore, p-hydroxybenzylideneimidazolinone (8, 9), is formed by internal cyclization of a Ser-Tyr-Gly tripeptide and 1,2-dehydrogenation of the Tyr. This posttranslational modification is oxygen-dependent, requiring ≈2–4 h for the WT protein (10, 11). A mechanism for the fluorophore formation has been proposed (3) but needs to be confirmed by further studies.
GFP absorbs blue light at 395 nm, with a smaller peak at 475 nm, and emits green light at 508 nm with a quantum yield of 0.72–0.85 (12, 13). The ratio between the two absorption peaks is sensitive to factors such as pH, temperature, and ionic strength (7, 14), suggesting the presence of two different forms of the chromophore. It has been proposed that the larger 395-nm peak is due to a neutral chromophore and that the minor peak at 475 nm is caused by an ionized chromophore (10). Upon illumination with UV light, WT GFP undergoes photoisomerization, which increases the excitation absorbance at 475 nm at the expense of that at 395 nm (3). The diversion of excitation amplitude into two peaks whose ratio is dependent on past illumination history complicates the application of WT GFP for routine fluorescence observations, though it also raises the possibility of tagging GFP by localized UV irradiation (3, 15). Mutation of Ser-65 → Thr (S65T) completely suppresses the 395-nm peak and the photoisomerizability while amplifying the 475-nm excitation peak ≈6-fold and shifting it to 489 nm (11). Therefore, this substitution is highly advantageous and has been incorporated in many of the variants of GFP (16–18).
Recently the structures of S65T as a monomer and the WT as a dimer were separately determined (19, 20). No detailed comparison of the two structures has yet appeared. We have crystallized WT GFP as a monomer and solved its structure by x-ray crystallography using the S65T model. Comparison of the two monomeric structures suggests an explanation for why WT has such unusual dual wavelength absorption and photoisomerization properties and why the replacement of a proton in WT by a methyl group in S65T causes such profound spectroscopic alterations.
MATERIALS AND METHODS
The recombinant GFP was obtained from Escherichia coli strain BL21 (DE3) containing the WT GFP expression vector TU 58 as described (21). After the initial sonication and clarification steps (21), GFP was purified as previously described (9). Crystallization was carried out by the sitting-drop vapor diffusion technique at room temperature (22) in multiwell crystal growth chambers (C. Supper, Natick, MA). The WT crystals were grown mixing 5 μl of the protein solution (15 mg/ml GFP in 10 mM Tris, pH 8.0/0.02% NaN3) with 2 μl of the well solution containing 0.05 M KH2PO4 and 20% (wt/vol) polyethylene glycol (PEG) 8000 (pH 3.8). The crystals were fully grown within 4 days, having a long rod-like shape (0.3 × 0.3 × 1 mm3). The crystals diffracted x-rays to 2.1-Å resolution. They belong to space group P212121, with unit cell parameters of a = 51.9 Å, b = 63.2 Å, and c = 68.8 Å. The packing parameter (Vm; ref. 23) is 2.09 Å3/Da, assuming one molecule in the asymmetric unit and a solvent content of 41%. Table 1 shows the statistics on data collection and evaluation. Diffraction data were collected on a MAR image-plate scanner (MAR-Research, Hamburg, Germany), processed with mosflm (24), and scaled and reduced with the ccp4 crystallographic package (25).
Table 1.
Data collection and evaluation statistics for WT GFP
| Resolution Å | 2.13 |
| Total observations | 60,182 |
| Unique observations | 13,001 |
| Completeness,* % | 91.3/54.5 |
| I/σ(I)* | 7.1/3.9 |
| Rmerge† | 6.1/18.6 |
*Resolution range, 20–2.13 Å/2.20–2.13 Å.
Rmerge = ∑hkl ∑i (|Ii − I|)/∑hkl ∑iI.
The structure of WT GFP was determined by molecular replacement on the basis of the S65T model [Ormö et al. (19)]. The crystallographic refinement was carried out with the tnt package (26), and model building was performed using the program o (27). The final model of the WT GFP has an R value of 0.192 (free R value of 0.254) with rms deviations from target values of 0.011 Å for bond distances and 1.5° for bond angles (see Table 2 for refinement statistics). As shown by procheck (28), 92% of the residues in the model have main chain torsion angles in the most favored Ramachandran region. There are no residues in disallowed regions, and 16 residues are in generously allowed regions. Good electron density was visible for the entire molecule, except for the two N-terminal and the seven C-terminal residues. The side chains of Asp-117, Thr-118, Glu-172, Asp-190, and Thr-203 have been modeled in two different conformations as indicated by the electron density and refined with proper occupancies. Of these, only Thr-203 contacts the chromophore, so the relative occupancies of the two conformations of the Thr-203 side chain were investigated by an objective procedure. The side chain was deleted, and 10 cycles of refinement were performed to remove model bias. The omit map was then examined with the program chckdens (V. Lamzin, personal communication) to calculate the electron density for the side chain atoms in all possible orientations as defined by the χ1 torsion angle. From the density at the two maxima (for χ1 = −30° and χ1 = 80°), it was estimated that the relative occupancy of the two conformations is ≈6:1. Thus two conformations were modeled, with occupancies of 0.85 and 0.15, respectively. After 10 cycles of refinement, the B valuesrefined to 20.5 Å2 and 19.4 Å2 for the OG1 and CG2 atoms of the major conformation and 19.1 Å2 and 21.2 Å2 for the OG1 and CG2 atoms of the minor conformation, respectively. The refinement of occupancies set to different values (0.75:0.25, 0.5:0.5) resulted in larger differences in B factors between the two conformations.
Table 2.
Refinement statistics for WT GFP
| Protein atoms, no. | 1832 |
| Solvent atoms, no. | 74 |
| Resolution range, Å | 20–2.13 |
| R factor (R free) | 0.192 (0.254) |
| Mean B-value, Å2 | |
| All atoms | 33.3 |
| Protein atoms | 33.1 |
| Chromophore atoms | 19.5 |
| Solvent atoms | 38.8 |
| Deviations from ideality | |
| Bond lengths, Å | 0.011 |
| Bond angles,° | 1.534 |
| Restrained B values, Å2 | 2.32 |
The rms deviations between the WT and S65T GFP are 0.7 Å for 1755 atoms (excluding the chromophore atoms) and 0.23 Å after elimination of the 408 most deviating atoms. For 225 Cα atoms (excluding the chromophore atoms), the rms deviation is 0.28 Å, and it is 0.22 Å for Cα atoms after elimination of 23 atoms deviating by >0.4 Å. Atomic superpositions were performed with the program suppos from the biomol suite (W.G.J. Hol and B. Dijkstra, personal communication).
RESULTS AND DISCUSSION
The overall structure of monomeric WT GFP is essentially identical to that of monomeric S65T (19) and the recently reported dimeric WT (20). It consists of a unique and compact β-barrel fold (Fig. 1). An 11-stranded β-barrel forms a nearly perfect cylinder 42 Å long and 24 Å in diameter. A single helix with partial 310 character runs through the center of the cylinder. The chromophore intervenes in the middle of this central helix, where it is highly protected from bulk solvent by the surrounding β-strands. The positioning of the chromophore and its inaccessibility probably account for the small Stokes shift, high quantum yield of fluorescence, and inability of O2 to quench the excited state. Interestingly, qualitative calculation of the electrostatic potential on the WT GFP surface [as calculated by grasp (ref. 32; data not shown)] indicates a region on the surface of the β-barrel of a distinct negative potential that might be involved in an interaction with the aequorin molecule.
Figure 1.

Ribbon diagram of the WT GFP structure. The α-helices are shown in red, the β-strands are shown in green, and the chromophore is shown as a ball-and-stick model. The figure was produced by molscript (29) and raster3d (30, 31).
Differences Between WT and S65T in the Chromophore Environment.
Except for a shift of the three N-terminal residues and minor changes in the loop regions, the main differences between the WT and S65T structure are found in the chromophore environment (Fig. 2 a and b). Interestingly, all these differences are due to rearrangements caused by the presence or absence of a single methyl group in residue 65.
Figure 2.


Schematic diagram of the interactions between the chromophore and the surrounding residues and water molecules in (a) WT GFP and (b) S65T mutant. Hydrogen bonds are shown as dashed lines and have the indicated lengths in Å. The cut-off for the hydrogen bond distance is set to 3.2 Å. (c) Stereoview of the final 2Fo-Fc electron density map of the WT GFP contoured at 1.0 σ, showing chromophore and the surrounding residues. (d) Stereoview of the Thr-203 side chain with the omit electron density (2Fo-Fc) of the WT GFP contoured at 1.0 σ. The side chain was modeled as A (major) and B (minor) conformations with relative occupancies 0.85:0.15.
In the WT structure, the hydroxyl group of Ser-65 occupies a different position with respect to the equivalent oxygen in the S65T mutant. In WT, the oxygen is still hydrogen-bonded to Glu-222, but no longer donates a hydrogen bond to the main chain carbonyl oxygen of Val-61, as correctly predicted by Ormö et al. (19). Instead, this oxygen in WT forms a second hydrogen bond to a water molecule (W12). This arrangement allows Ser-65 to be either a hydrogen bond donor or acceptor with Glu-222, which can therefore be either charged or neutral in contrast to S65T, whereas Glu-222 has to be protonated to form a hydrogen bond to Thr-65. Consequently, the Glu-222 side chain has a somewhat different conformation in the WT structure, now displaying a hydrogen bond to Ser-205. Through this hydrogen bond there is a direct hydrogen bonding network from Glu-222 through Ser-205 and water W22 (Sol316 in S65T) to the hydroxyl group of Tyr-66 of the chromophore, which is not seen in the S65T structure (Fig. 2b). In addition to its short hydrogen bonds to Tyr-66 and Ser-205, water W22 has long hydrogen bonds to the carbonyl oxygens of Asn-146 and Thr-203, which are probably not present simultaneously. In the S65T structure, the Thr-203 carbonyl oxygen does not form a hydrogen bond to the water molecule (Fig. 2b).
Two more major changes are seen in the WT structure compared with S65T. The largest is the rearrangement of Thr-203, which has two conformers in WT, the predominant of which differs from the single conformation seen in S65T. Not only does its main chain move closer to the chromophore, allowing a hydrogen bond to water W22, but its side chain rotates to a position where it no longer hydrogen bonds to the Tyr-66 oxygen atom. Therefore, it cannot stabilize a negative charge on the chromophore. A similar effect is seen for the His-148 side chain which also moves away from the Tyr-66 hydroxyl in WT. The His-148 side chain, however, has poorly defined density in WT, indicating that it is flexible and may be protonated due to the low pH of the crystal.
Small shifts in the hydrogen bond distances are seen at the opposite side of the chromophore. Arg-96 moves away from the chromophore and toward Gln-183 in the WT structure (Fig. 2 a and b), again suggesting that the predominant form of the chromophore in WT is neutral.
The 395-nm absorbance peak of native GFP typically has about 3 times the amplitude of the 475-nm peak, but because the latter has about twice the extinction coefficient, the molar ratio of neutral to anionic chromophores is ≈6:1 (33). The WT structure should predominantly show the major fraction that is responsible for the 395-nm absorption. Evidence for a minor fraction seen in the Thr-203 side chain will be discussed below. Predictions that the 395-nm peak would be due to a neutral chromophore (10) fit well with the structure that we see in WT (Fig. 2c), with a negative Glu-222 stabilizing this state. In the S65T mutant the only absorption peak is at 489 nm, thought to be due to an ionized chromophore. This correlates well with the structure of this mutant, where the Glu-222 is forced into a protonated neutral state and a negative charge on the chromophore is stabilized by hydrogen bonds from Thr-203, His-148, and water molecule Sol316 (water W22). Excitation of phenolic chromophores normally entails migration of electron density from the phenol or phenolate toward the rest of the chromophore, which explains why protonation of the phenolate raises the energy required for excitation and why that proton becomes prone to leave in the excited state (34). The cause for the small shift in the absorption peak between the ionized states of WT (475 nm) and the S65T mutant (489 nm) is discussed below.
Proposed Explanation of the Dual Wavelength Absorption in WT GFP.
Based on the comparison of the WT and S65T structures, we propose an explanation for the unusual dual wavelength absorption and photoisomerization of the WT GFP at the molecular level (Fig. 3). In this model, we propose that the two structures, the predominant and the S65T-like conformations of WT, represent the states corresponding to the two absorption maxima, and we describe the rearrangements that are necessary to convert the predominant WT conformation to an S65T-like conformation. The equilibrium between these states appears to be governed by a hydrogen-bonding network that permits proton transfer between the chromophore and neighboring side chains.
Figure 3.
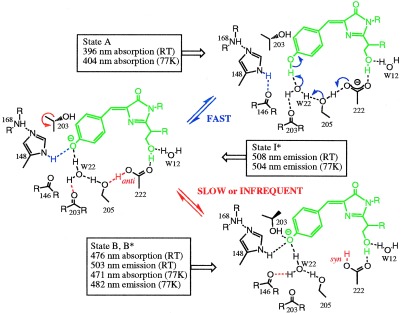
Proposed mechanism for the photoisomerization of WT GFP based on the structural data and the spectroscopic work (33). State A is the predominant form seen in the WT structure, state B is the form seen in the S65T structure, and state I is the intermediate not seen in either of the two structures. Changes from A to I are indicated in blue, and differences between I and B are in red. The side chains of Arg-96 and Gln-94, which make hydrogen bonds with the carbonyl of the imidazolinone ring, do not show major conformational changes and are not drawn in this figure.
The neutral form of the chromophore absorbs at 395 nm (Fig. 3, state A), as seen in the predominant WT structure. The neutral state is maintained by the carboxylate of Glu-222 through both electrostatic repulsion and hydrogen bonding via a bound water molecule (W22) and the side chain oxygen of Ser-205. In this state, water molecule W22 is in hydrogen bonding contact with the hydroxyl group of Tyr-66, main chain oxygen of Thr-203, and side chain oxygen of Ser-205. The side chain of His-148 is in hydrogen bond distance to the main chain nitrogen of Arg-168. The carboxylate of Glu-222 is additionally stabilized by the hydrogen bonding with the Ser-65, which makes a hydrogen bond to the water molecule W12.
The ionized form of the chromophore, absorbing at 475 nm, is detected in a minor fraction of the native protein. The conversion from the neutral (state A) to the ionized (state B) form involves an additional state I, which is a transient intermediate not seen in the crystal structures. In this state (Fig. 3, state I), Glu-222 donates its charge to the fluorophore by proton extraction through a hydrogen bond network involving Ser-205 and the bound water W22. The stabilization of the ionized state is achieved through the water molecule W22 and the His-148 side chain. His-148 stabilizes the phenolic oxygen of Tyr-66 by donating a hydrogen bond to it. Water molecule W22 donates a hydrogen to the phenolic oxygen of Tyr-66 and to the main chain oxygen of Thr-203 and accepts the hydrogen from the side chain oxygen of Ser-205. The transition between the two states should be a fast process, since it only involves proton motion.
Further stabilization of the ionized state of the fluorophore occurs through a rearrangement of Thr-203 (Fig. 3, state B). The side chain of Thr-203 occupies a different conformation in which the side chain oxygen stabilizes the ionic state of the fluorophore by donating a hydrogen bond to it. The shift of the Thr-203 main chain causes the loss of the hydrogen bond between its carbonyl oxygen and water W22. However, the water molecule establishes a new hydrogen bond to the carbonyl oxygen of Asn-146. Additionally, there is a loss of the hydrogen bond between Ser-205 and Glu-222. The hydrogen on Glu-222 is proposed to move from the anti to syn conformation, which is much more favorable (35).
A shift from the I to the B state is a major step that requires a change in the conformation of Thr-203, the breakage of the Ser-205—Glu-222 hydrogen bond and probably also the anti–syn isomerization of Glu-222. Therefore, we suggest that this would be a slow or infrequent process, competing with other fast processes.
The conservation of charge and number of hydrogens between the A and B states explains why external pH variation over a wide range is unable to shift the equilibrium between the two conformations (14). However, in the process of the photoisomerization, the equilibrium is shifted toward the minor fraction. UV irradiation does shift the ratio by pumping a proton relay from the chromophore excited state through the water molecule W22 and Ser-205 to Glu-222. The driving force for the proton migrations would be the large increase in acidity of phenols in their excited states (34). The unfavorable syn to anti rearrangement of the hydrogens on Glu-222 necessary for recreating the Ser-205—Glu-222 hydrogen bond may explain the slow return to the equilibrium dark-adapted state of the chromophore.
Evidence Supporting the Proposed Mechanism.
Additional evidence in support of the proposed mechanism is found among the data provided by structural, mutagenesis and spectroscopic work.
The presence of a hydrogen bond between Glu-222 and Ser-205 in the WT structure supports the idea of a hydrogen-bonding network connecting Glu-222 and Tyr-66 through which the proton relocation occurs, as presented in Fig. 3, state A. In the S65T mutant, this hydrogen bond is absent, thus destroying an effective hydrogen bonding pathway, fitting with state B (Fig. 3).
The unusual shape of the electron density for the side chain of Thr-203 pointed to the existence of an additional conformation. An omit 2Fo-Fc map shows clear electron density for two conformations (Fig. 2d). Therefore, the side chain of Thr-203 was modeled in two orientations and refined with appropriate occupancies. It has been found that a major fraction (85%) of the side chain is present in the conformation facing away from the hydroxyl group of Tyr-66 (for a detailed explanation, see Materials and Methods). A minor fraction (15%) of the side chain has the conformation seen in the S65T structure, in which it makes a hydrogen bond to the phenolic hydroxyl of Tyr-66. The ratio of calculated occupancies is in excellent agreement with the calculated 6:1 molar ratio between the 395-nm and 475-nm absorption peaks (33). The mutation changing Thr-203 to Ile results in elimination of the 475-nm absorption peak without altering the 395-nm peak (36). This mutation supports the proposal that Thr-203 is necessary for the formation and stabilization of the ionized chromophore, which in our mechanism is responsible for the 475-nm absorption peak.
The electron density distribution for the side chains of Glu-222 and His-148 implies the presence of additional conformations for both side chains (Fig. 2c). However, due to the relatively poor quality of the electron density map at these positions, it was not possible to determine alternative side chain conformations for Glu-222 and His-148.
Several mutants replacing Ser-65 have been reported: Gly, Ala, Cys, Val, and Thr and in each of these mutants the absorption peak at 395 nm has been suppressed in favor of a peak at 470–490 nm (11, 37). Analysis of the S65T structure (19) suggests that steric hindrance by the extra methyl group forces the OH to rotate to donate a hydrogen bond to the carbonyl of Val-61 rather than to the carboxylate of Glu-222. This change in hydrogen bonding prevents ionization of Glu-222 and permits permanent ionization of the chromophore, so that the only excitation peak is in the blue–green region. Therefore, the S65T structure represents state B in the proposed mechanism. The other substitutions at position 65 would also prevent donation of a hydrogen bond to Glu-222 as necessary for state A. Apparently only Ser has the ability to easily switch from being a donor of the hydrogen bond in state A to being an acceptor of the hydrogen bond from Glu-222 in state B. In state A and state B, the side chain of Ser-65 is stabilized by a water molecule W12.
Interestingly, we see no evidence for an alternative position of Ser-65. Apparently the B state does not require movement of this oxygen to the position seen in the S65T structure. Consequently, Glu-222 probably has a different position in the B state of WT compared with S65T, as implied by the electron density distribution around the Glu-222 side chain (Fig. 2). These changes explain why the longer-wavelength absorption peak in S65T has red-shifted 14 nm (489 nm) compared with the WT GFP (475 nm). In the WT structure, the Ser-65 OH dipole moment is roughly parallel to the long axis of the chromophore and points in the direction of the phenolate (Fig. 4a), which will electrostatically raise the energy required to transfer electron density from phenolate to imidazolidine carbonyl in the excited state. In the S65T structure, however, the dipoles are pointing roughly perpendicular to the long axis of the chromophore or slightly to the right, which should have little electrostatic interaction with the chromophore’s change in the dipole moment (Fig. 4b).
Figure 4.
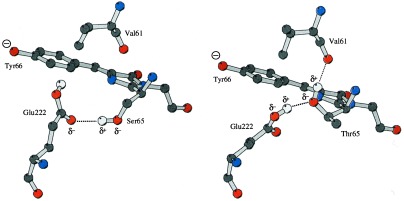
View of the orientation of the hydrogen bonds in (a) the minor form of the WT GFP structure (state B) and (b) the S65T structure. The chromophore and the side chains of Glu-222 and Val-61 are shown as a ball-and-stick model. The nitrogens are shown in blue, oxygens in red, carbons in dark gray, and hydrogens in light gray. The figure was produced by molscript (29).
Another mutation in support of the proposed mechanism is the Glu-222 → Gly mutation which suppresses the hydrogen bonding between Glu-222 and Ser-65 and gives rise only to a 481-nm peak (36). Glu-222 is needed for establishing the hydrogen-bonding network and more importantly for the inhibition of chromophore ionization by its negative charge.
Our model proposes detailed structures to explain recent data from femtosecond-excited state dynamics studies (33) of the fluorescence emission of GFP. These have indicated that there may be three ground states of the chromophore. Two of these states are proposed to be ground states (A and B) that can be slowly interconverted, with a third (I), probably an unrelaxed form of B and intermediate on the interconversion pathway. The A and B states were proposed to be associated with absorption at 395 nm and 475 nm. Irradiation of the 395-nm energy feature resulted in a rapid time-dependent rise of fluorescence at 508 nm that was slowed dramatically upon substitution of exchangeable hydrogens for deuterium, suggesting that proton motion is required for emission from this state (33). Excitation of the 470-nm feature resulted in essentially instantaneous onset of fluorescence, suggesting that proton motion is not required for emission from this state. Irradiation of the 395-nm feature leads to photoisomerization, a loss of absorbance at this wavelength concomitant with a rise in absorption at 470 nm. Chattoraj et al. (33) proposed the following model: the most stable ground state, A, upon excitation to A*, deprotonates in a rapid and efficient process to form species I*, which then fluoresces at 508 nm. According to this model, A can slowly be interconverted to B; however, this interconversion is slow and inefficient. Fluorescence emission from state B* does not involve proton motion. The spectroscopic observations support the identification of the energetic states, A, I, and B (33), with the structural states A, I, and B of Fig. 3, as we have proposed from the comparison of the WT and S65T atomic models.
The photoisomerizability of WT GFP is potentially quite useful in cell biology. The molecules labeled by fusion to GFP can be marked by localized UV (<400 nm) exposure, which enhances the fluorescence excitable at longer wavelengths such as 475 nm. The brightened GFPs can be imaged over time to reveal the subsequent spatial locations of the chimeric molecules. Protein diffusion, exchange, and active targeting are thus quantitatively revealed (15). Such experiments are analogous to the use of caged fluorophores. In the case of GFP, the great advantage is that its photochromic fluorophore could be synthesized and targeted by molecular biology in situ rather than by in vitro chemical conjugation followed by microinjection back into cells as is necessary at present. However, in WT GFP the quantum yield for photoisomerization and the optical contrast between the virgin and UV-exposed proteins leave much to be desired. Upon UV exposure the blue excitation amplitude increases, but the maximum increase found to date is a factor of three (15) and a larger enhancement would be preferable. Now that the molecular basis for photoisomerization in WT GFP is understood in some detail, it may be possible to design mutants. The most desirable mutants would start out almost entirely in state A, with negligible excitation amplitude at 475–490 nm, but could then be photoisomerized almost entirely to state B, with high amplitude at wavelengths similar to Ser-65 mutants. Spontaneous reversion back to state A in minutes to hours might be allowable or even desirable in many cell biological experiments, because the same GFP-tagged proteins might be then reconverted and viewed multiple times.
Acknowledgments
We thank to R. Huber for initial support (K.B.) and A. Perrakis for help with the calculations (K.B. and T.K.S.). This work was supported by Netherlands Organization for Scientific Research/Netherlands Foundation for Chemical Research (K.B. and T.K.S.) and the National Science Foundation (S.J.R.).
ABBREVIATIONS
- GFP
green fluorescent protein
- WT
wild type
Footnotes
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, Chemistry Department, Brookhaven National Laboratory, Upton, NY 11973 (reference 1EMB).
References
- 1.Prendergast F G, Mann K G. Biochemistry. 1978;17:3448–3453. doi: 10.1021/bi00610a004. [DOI] [PubMed] [Google Scholar]
- 2.Prasher D C, Eckenrode V K, Ward W W, Prendergast F G, Cormier M J. Gene. 1992;111:229–233. doi: 10.1016/0378-1119(92)90691-h. [DOI] [PubMed] [Google Scholar]
- 3.Cubitt A B, Heim R, Adams S R, Boyd A E, Gross L A, Tsien R Y. Trends Biochem Sci. 1995;20:448–455. doi: 10.1016/s0968-0004(00)89099-4. [DOI] [PubMed] [Google Scholar]
- 4.Prasher D C. Trends Genet. 1995;11:320–323. doi: 10.1016/s0168-9525(00)89090-3. [DOI] [PubMed] [Google Scholar]
- 5.Gerder H-H, Kaether C. FEBS Lett. 1996;389:44–47. doi: 10.1016/0014-5793(96)00586-8. [DOI] [PubMed] [Google Scholar]
- 6.Bokman S H, Ward W W. Biochem Biophys Res Commun. 1981;101:1372–1380. doi: 10.1016/0006-291x(81)91599-0. [DOI] [PubMed] [Google Scholar]
- 7.Ward W W, Bokman S H. Biochemistry. 1982;21:4535–4540. doi: 10.1021/bi00262a003. [DOI] [PubMed] [Google Scholar]
- 8.Shimomura O. FEBS Lett. 1979;104:220–222. [Google Scholar]
- 9.Cody C W, Prasher D C, Westler W M, Prendergast F G, Ward W W. Biochemistry. 1993;32:1212–1218. doi: 10.1021/bi00056a003. [DOI] [PubMed] [Google Scholar]
- 10.Heim R, Prasher D C, Tsien R Y. Proc Natl Acad Sci USA. 1994;91:12501–12504. doi: 10.1073/pnas.91.26.12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heim R, Cubitt A B, Tsien R Y. Nature (London) 1995;373:663–664. doi: 10.1038/373663b0. [DOI] [PubMed] [Google Scholar]
- 12.Morise H, Shimomura O, Johnson F H, Winant J. Biochemistry. 1974;13:2656–2662. doi: 10.1021/bi00709a028. [DOI] [PubMed] [Google Scholar]
- 13.Ward W W, Cody C W, Hart R C, Cormier M J. Photochem Photobiol. 1980;31:611–615. [Google Scholar]
- 14.Ward W W, Prentice H J, Roth A F, Cody C W, Reeves S C. Photochem Photobiol. 1982;35:803–808. [Google Scholar]
- 15.Yokoe H, Meyer T. Nat Biotech. 1996;14:1252–1256. doi: 10.1038/nbt1096-1252. [DOI] [PubMed] [Google Scholar]
- 16.Cormack B P, Valdivia R H, Falkow S. Gene. 1996;173:33–38. doi: 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- 17.Kahana J, Silver P. In: Current Protocols in Molecular Biology. Ausubel F M, Brent R, Kingston R, Moore D, Seidman J, Smith J, Struhl K, editors. New York: Wiley; 1996. pp. 9.7.22–9.7.28. [Google Scholar]
- 18.Levy J P, Muldoon R R, Zolotukhin S, Link C J J. Nat Biotech. 1996;14:610–614. doi: 10.1038/nbt0596-610. [DOI] [PubMed] [Google Scholar]
- 19.Ormö M, Cubitt A B, Kallio K, Gross L A, Tsien R Y, Remington S J. Science. 1996;273:1392–1395. doi: 10.1126/science.273.5280.1392. [DOI] [PubMed] [Google Scholar]
- 20.Yang F, Moss L G, Phillips G N. Nat Biotech. 1996;14:1246–1252. doi: 10.1038/nbt1096-1246. [DOI] [PubMed] [Google Scholar]
- 21.Chalfie M, Tu Y, Euskirchen G, Ward W W, Prasher D C. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- 22.McPherson A. Eur J Biochem. 1989;189:1–23. doi: 10.1111/j.1432-1033.1990.tb15454.x. [DOI] [PubMed] [Google Scholar]
- 23.Matthews B W. J Mol Biol. 1968;33:491–497. doi: 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- 24.Leslie A G W. In: Crystallographic Computing. Moras D, Podjarni A D, Thierry J C, editors. Oxford: Oxford Univ. Press; 1991. pp. 50–61. [Google Scholar]
- 25.Collaborative Computational Project, Number 4. Acta Crystallogr D. 1994;50:760–763. [Google Scholar]
- 26.Tronrud D E, Ten Eyck L F, Matthews B W. Acta Crystallogr A. 1987;43:489–501. [Google Scholar]
- 27.Jones T A. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 28.Laskowski R A, MacArthur M W, Moss D S, Thornton J M. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 29.Kraulis P. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 30.Bacon D J, Anderson W F. J Mol Graphics. 1988;6:219–220. [Google Scholar]
- 31.Merritt E A, Murphy M E P. Acta Crystallogr D. 1994;50:869–873. doi: 10.1107/S0907444994006396. [DOI] [PubMed] [Google Scholar]
- 32.Nicholls A, Sharp K, Honig B. Proteins Struct Funct Genet. 1991;11:281–285. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 33.Chattoraj M, King B A, Bublitz G U, Boxer S G. Proc Natl Acad Sci USA. 1996;93:8362–8367. doi: 10.1073/pnas.93.16.8362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stewart R. The Proton: Applications to Organic Chemistry. Orlando, FL: Academic; 1985. pp. 228–234. [Google Scholar]
- 35.Gandour R. Bioorg Chem. 1981;10:169–176. [Google Scholar]
- 36.Ehrig T, O’Kane D J, Prendergast F G. FEBS Lett. 1995;367:163–166. doi: 10.1016/0014-5793(95)00557-p. [DOI] [PubMed] [Google Scholar]
- 37.Delagrave S, Hawtin R E, Silva C M, Yang M M, Youvan D C. Bio/Technology. 1995;13:151–154. doi: 10.1038/nbt0295-151. [DOI] [PubMed] [Google Scholar]