

Abstract
The structure of mitogen-activated protein (MAP) kinase p38 has been solved at 2.1-Å to an R factor of 21.0%, making p38 the second low activity MAP kinase solved to date. Although p38 is topologically similar to the MAP kinase ERK2, the phosphorylation Lip (a regulatory loop near the active site) adopts a different fold in p38. The peptide substrate binding site and the ATP binding site are also different from those of ERK2. The results explain why MAP kinases are specific for different activating enzymes, substrates, and inhibitors. A model presented for substrate and activator interactions has implications for the evolution of protein kinase cascades.
Mitogen-activated protein (MAP) kinases are ubiquitous signaling molecules responsive to hormones, cytokines, environmental stresses, and other extracellular stimuli. The activation of these protein kinases leads to diverse regulatory events, particularly proliferation and differentiation. MAP kinase p38 (1–4) is activated by proinflammatory cytokines and environmental stresses such as osmotic shock and UV light, and is essential for the lipopolysaccharide-induced translation of tumor necrosis factor in monocytes. p38 is one of nearly a dozen mammalian MAP kinases that have been cloned (2, 5–11). Each MAP kinase is characterized by the substrates it phosphorylates and by the distinct MAP/ERK kinases (MEKs) by which it is preferentially activated.
MAP kinase p38 phosphorylates and activates both nuclear transcription factors ATF2 (12) and GADD153 (13), and protein kinase targets such as MAPKAP kinases 2 and 3 (3, 4, 14). p38 is phosphorylated and activated specifically by MEK3 (15, 16) and MEK6 (16–19), and together with c-Jun N-terminal kinases 1 and 2 (JNK1 and JNK2) by MEK4 (15, 20). A pyrimidine imidazole compound found to inhibit the synthesis of tumor necrosis factor has been shown to bind tightly to p38 (2), suggesting that pharmacological inhibitors can bind to individual members of this kinase family.
While MAP kinases are selective in their interactions with activators and substrates, they share a common regulatory mechanism, being activated 100- to 1000-fold or more (21–23) by dual phosphorylation on a conserved threonine and a conserved tyrosine residue in the phosphorylation Lip (residues Leu-171–Val-183) near the active site (24). MAP kinases also share a common specificity for substrates containing proline in the P+1 position (25, 26).
Here we describe the structure of unphosphorylated, low activity p38 at 2.1-Å resolution. The structure is compared with that of ERK2, which we solved previously (27) to understand how the common mechanism of activation by dual phosphorylation and the common specificity for proline is integrated with the unique specificity of these kinases for their activating enzymes, substrates, and inhibitors.
METHODS
Expression, Purification, and Crystallization.
A vector encoding murine p38 in pET-14b (Novagen) has 19 extra amino acids at the N terminus, (M)GSSHHHHHHSSGLVPRGSHMSQERPT, encoding a His6 Ni2+-nitrilotriacetic acid agarose affinity tag, a thrombin cleavage site (underlined), and eight other residues (1). The bacterial strain BL21(DE3) was used for expression. Cell cultures were grown for ≈12 hr at 30°C without induction. Cells were pelleted by centrifugation and resuspended in 50 mM Tris, pH 7.9/0.5 M NaCl/5 mM imidazole. Flash-frozen cells were thawed and lysed by adding 1 mg/ml lysozyme and 0.3% Nonidet P-40 detergent followed by gentle sonication. The supernatant was loaded onto Ni2+-nitrilotriacetic acid agarose (Qiagen), and p38 was eluted with 50–250 mM imidazole in lysis buffer. The fractions were further purified on a Mono Q column (HR 10/10, Pharmacia) using a gradient of 0.1–0.5 M NaCl in 50 mM Tris·HCl, pH 7.4/2 mM DTT. In a third step, hydrophobic interaction chromatography on a Phenyl Superose column (HR 10/10, Pharmacia) was carried out in a gradient from 2M to 20 mM NaCl in 50 mM Tris·HCl, pH 7.4/2 mM DTT. Finally, p38 was purified on a Mono S column (HR 5/5, Pharmacia) using a gradient of 50–250 mM NaCl in 50 mM Hepes, pH 6.8/4 mM DTT. After the above four purification steps, the protein was homogeneous, as assessed by silver staining of gels that underwent SDS/PAGE and isoelectric focusing gel electrophoresis.
Crystallization conditions were identified in a crystal screening (28) kit from Hampton Research (Riverside, CA). Optimization of the best conditions led to crystals, with use of 18% polyethylene glycol (PEG) 8000/0.2 M Mg(OAc)2/0.1 M Hepes, pH 7.0. The protein concentration was ≈10 mg/ml in a buffer of 50 mM NaCl/1 mM EDTA/10 mM DTT/1 mM benzamidine/1 μM pepstatin/10 μg/ml leupeptin/25 mM Hepes, pH 7.4. Microseeding was used to produce larger crystals. The average size of the p38 crystals was 0.5 × 0.25 × 0.08 mm. The largest reached 1.0 × 0.5 × 0.15 mm. Crystals were cryoprotected at a final concentration of 35% PEG 400 added to mother liquor. Seven transfers to successively more concentrated PEG 400 solutions were used. Crystals were then flash-frozen in liquid nitrogen in a nylon loop.
Data Collection and Structure Solution.
Data were collected both at Brookhaven National Laboratory with a synchrotron beam line X12C using 1.0-Å radiation at −175°C and at the University of Texas Southwestern Medical Center on an RAXIS IIC with a rotating anode generator (Rigaku, Tokyo; model RU-300) using 1.54-Å radiation. The synchrotron data collected on Mar image plates had a diffraction limit of 2.1-Å. Images were indexed and scaled using denzo and scalepack (29). The statistics for the data and structure refinement are given in Tables 1 and 2. Starting phases for the p38 structure were taken from the molecular replacement solution found using the program amore (30) and the coordinates for the entirety of the low activity form of ERK2 (27). While the first maps showed buildable density in the C-terminal domain, initially density for the smaller N-terminal domain was broken. Density for the N-terminal domain appeared after several cycles of model building and refinement. Density interpretation and model building were done in o (31) using 2|Fobs| − |Fcalc| and |Fobs| − |Fcalc| sigmaA weighted (32) maps calculated by refmac in ccp4 (33). Subsequent to each round of model building, cycles of refinement were carried out in x-plor (34). Simulated annealing omit maps were used as needed for residues 33–36, 55–57, 119–122, 170–189, and 320–330. In the final model of p38, residues 33–36, 119–122, and 171–178 have weak electron density and are disordered. Almost all of the peptide torsion angles (98.5%) fall within the most favored regions or generally allowed regions of the Ramachandran (φ,ψ) diagram as defined in procheck (35). The present model includes 132 water molecules.
Table 1.
X-ray diffraction data
| Native 1 (20°C/RAXIS) | Native 2 (−175°C/X12C) | |
|---|---|---|
| Space group | P212121 | P212121 |
| Unit cell, Å | a = 46.10 | a = 45.76 |
| b = 86.92 | b = 84.93 | |
| c = 126.54 | c = 123.91 | |
| Resolution, Å | 2.5 | 2.1 |
| No. of measurements | 29,966 | 206,687 |
| Unique reflections | 17,246 | 25,393 |
| Completeness, % (overall/outer shell) | 93.5/65.9 | 86.0/65.6 |
| I/σ (overall/outer shell) | 14/3.2 | 20/5.3 |
| Rmerge,* % | 7.0 | 4.2 |
Rmerge = 100 × ∑|Ii − 〈I〉|/∑Ii, where Ii is the intensity of an individual measurement, and 〈I〉 is the mean intensity of this reflection.
Table 2.
Molecular replacement data and refinement statistics
| Molecular replacement | |
| Method | AMoRe* |
| Search model | ERK2 |
| Resolution range, Å | 9.0–3.5 |
| Rotation function | 6.5σ |
| Translation function | 6.7σ |
| Rigid body | |
| Correlation coefficient | 26.4 |
| R factor | 51.7 |
| Refinement | |
| Resolution, Å | 6.0–2.1 |
| No. of reflections (F > 2σ) | 23,950 |
| Total no. of atoms | 2834 |
| Working R factor,† % | 21.0 |
| Free R factor,‡ % | 24.1 |
| rms deviation | |
| Bond length, Å | 0.010 |
| Bond angles | 1.58° |
| B values,§ Å2 | |
| Main chain | 28.4 |
| Side chain | 30.0 |
| No. of water molecules | 132 |
AMoRe, Automatic Molecular Replacement (30).
R factor = 100 × ∑ ||Fobs| − |Fcal||/∑|Fobs|, where |Fobs| and |Fcal| are the observed and calculated structure factor amplitudes, respectively.
Free R factor was calculated using 10% of the data which was excluded from the refinement.
B values, Averaged B values for all nonhydrogen atoms in the current model. For the N-terminal domain, B(main chain) = 32.0, and for the C-terminal domain, excluding the felixible Lip region of residues 170–178, B(main chain) = 23.8.
RESULTS
Overall Structure.
The present model of p38 contains a single 351-aa polypeptide chain and 132 water molecules, and has been refined to an R factor of 21.0% (Rfree = 24.1%, Table 2) to 2.1-Å resolution. The phosphorylation site residues are well resolved in the electron density (Fig. 1). At the N terminus, three residues derived from the native sequence and 19 residues from the vector are not visible in electron density; at the C terminus, last six residues are not observed. Like other protein kinases, p38 is composed of two domains, a 135-residue N-terminal domain and a 225-residue C-terminal domain. The N-terminal domain is composed largely of β-sheets, whereas the C-terminal domain is largely helical. The catalytic site is at the junction between the two domains (36).
Figure 1.

Electron density, around the phosphorylation sites Thr-180 and Tyr-182 and around the P+1 specificity pocket, contoured at 1.25σ and drawn in o (31). The map was calculated with the program refmac in ccp4 (33) with sigmaA weighted coefficients (32) and model-derived phases. Dashed yellow lines denote hydrogen bonds between Arg-186 and the backbone carbonyls of Met-179 and Gly-181. The dashed red line denotes stacking interaction between Gly-181 and the backbone carbonyl of Thr-226 (distance of 3.1 Å).
Topologically, the structure of p38 is similar to that of ERK2, with which it shares approximately 40% sequence identity. Of the 351 aa present in the model, 247 have the same secondary structure and packing interactions as those in ERK2. A sequence alignment based on the structure is presented in Fig. 2. Cα atoms of corresponding residues from both domains superimpose with an rms deviation of 1.7 Å. The individual domains, however, are more closely related; the C-terminal domains superimpose with an rms deviation of 0.8 Å, and the N-terminal domains with a deviation of 1.2 Å. The discrepancy between the whole molecule and domain superpositions is a consequence of a difference in domain separation in the two molecules (Fig. 3), discussed below. The comparisons below are based on a superposition of Cα atoms of residues between Ala-111[Thr-108] and Ala-320[Ala-323]¶ that occupy corresponding positions.
Figure 2.

Sequence alignment of mouse p38, rat ERK2, human JNK1, and mouse cAPK, based on structural comparisons where available. Subdomains are labeled according to ref. 37. Phosphorylation sites are denoted by asterisks. Brackets denote the secondary structure elements in other kinases not present in p38.
Figure 3.
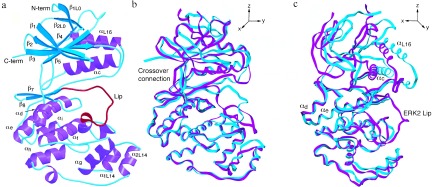
(a) Ribbon diagram of p38 drawn using molscript (38). Helices are purple, β-strands are cyan, and the Lip is red. Note the turn of helix formed by the Lip. (b and c) Superposition of p38 (cyan) and ERK2 (magenta) drawn with setor (39) in two views, approximately 90° apart. Molecules were superimposed using corresponding Cα atoms within the C-terminal domain.
Phosphorylation Lip and P+1 Specificity Pocket.
The phosphorylation Lip (Figs. 1, 3, and 4a) is the locus of major conformational differences between p38 and ERK2. The Lip in p38 consists of 13 residues, Leu-171[168]–Val-183[186], six residues shorter than the Lip in ERK2. Both p38 and ERK2 are activated by phosphorylation on a single threonine and a single tyrosine residue, Thr-180[183] and Tyr-182[185], in the Lip. The architecture of the Lip residues is completely different in p38 compared with ERK2 (Fig. 4). The differences in conformation commence at Gly-170[167], in the conserved DFG of subdomain VII (Fig. 2), and end with Thr-185[188]. Leu-171[168]–Asp-177[Pro-174] assume a nearly extended conformation, traversing the top of the C-terminal domain in the active site along a direct path. Leu-171 is in contact with Arg-70 in helix C, but the remainder of this segment makes few contacts and is partially disordered.
Figure 4.

The P+1 specificity pocket and phosphorylation Lip region of p38 (a) and ERK2 (b) drawn in setor. In p38, the shorter Lip is formed into a helical turn near the P+1 site, and the phosphorylation site tyrosine is on the surface. In ERK2, the Lip takes a wide excursion around the side chains of a neighboring surface loop, and the phosphorylation site tyrosine is buried. p38 superimposed on ERK2 in the neighborhood of the ATP binding site identified in ERK2 (c) and putative substrate binding site (d). Dark gray denotes p38; light gray and italic labeling denote ERK2.
The phosphorylation site sequence Thr-180–Gly-181–Tyr-182 makes a surface turn (Figs. 1 and 4a). The phosphorylation site residue Tyr-182 forms an edge-to-face interaction with Trp-187 and is also associated with a water molecule. Thr-180, like Tyr-182, is on the surface. By comparison, Tyr-185 of ERK2 is buried (Fig. 4b). The backbone carbonyl of Gly-181 forms an unusual π interaction with the backbone carbonyl of Thr-226 in L13 [the loop following helix F (27)] in p38. The surface turn buries Arg-186[189], and the carbonyl oxygen atoms of Met-179 and Gly-181 accept hydrogen bonds from the guanidinium moiety of Arg-186. Residues Val-183–Arg-186 are formed into a distorted helical turn and make a short helix with residues Arg-186–Arg-189. Thr-185[188], a semiconserved catalytic residue, is displaced relative to ERK2.
The position of the P+1 specificity pocket is known from the structure of cAPK (ref. 40; marked in Fig. 4b). This site is occupied by Arg-192 in ERK2 but otherwise is open for substrate interactions. In p38, however, this site is abolished because it is blocked by the turn of helix Val-183–Arg-186 (Figs. 3a and 4a). Thus it is anticipated that the structure changes on phosphorylation and activation to form the P+1 binding site.
The conformation of the Lip region in p38 resembles CDK2 in some respects. First, residues Ala-172–Glu-178 at the beginning of the Lip assume positions corresponding to Ala-150–Val-156 in low activity CDK2 (ref. 41; data not shown). Second, Tyr-200 (Fig. 4a) is hydrogen bonded to the Arg-149Nɛ, similar to the corresponding side chain interactions observed in CDK2 (42) as well as cAPK (36). Interestingly, ERK2/Lys-201–Tyr-203, corresponding to p38/Met-198–Tyr-200, adopts a completely different fold (Fig. 4b), forming very tight interactions with the Lip.
The Lip in ERK2 has been shown to be involved in MEK interactions. Robinson et al. (43) showed that reducing the length of the Lip modestly influenced the ability of ERK2 to be phosphorylated by MEK1 and MEK2. In contrast, it has been found that lengthening the Lip did not alter the level of phosphorylation of p38 by MEK3 or MEK4 (Y. Jiang, Z. Li, E. M. Schwartz, K. Guan, R. J. Ulevitch and J. Han, unpublished work). Apparently, adding residues does not affect activation of p38. Both studies showed that mutating the intervening residue between the threonine and tyrosine had no effect on the activation by MEKs, in contrast to what had been suggested (12). Robinson and Cobb (unpublished data) have shown that an ERK3/ERK2 chimeric kinase joined between subdomains V and VI is phosphorylated strongly by MEK1 and MEK2, the natural activator of ERK2 (45). Thus, sequences in the C-terminal domain drive recognition toward MEK (Fig. 2) in this chimeric kinase. Brunet and Pouysségur (46) show that a p38/ERK2 chimeric with the junction between subdomains III and V (helix C, the crossover connection, and helix D; Fig. 2) is activated by both p38 and ERK2 regulators. This suggests that MEKs interact with the N-terminal domain of the MAP kinase. However, because this experiment involved expression and stimulation of transfected cells, the results may be influenced by interactions with scaffolding proteins or other molecules.
Putative Phosphate Binding Ligands.
In cAPK, covalently bound phosphate is ligated by charged residues around the phosphorylation Lip (36, 47). As in low activity ERK2, the phosphorylation site residues Thr-180 and Tyr-182 in p38 are not aligned with any of these positively charged residues. The binding site that is conserved with other kinases consists of residues Arg-149[146] in the catalytic loop, Arg-173[170] in the Lip region, and Arg-67[65] and Arg-70[68] in helix C (36, 47, 48). This site has been shown recently to form the phosphate binding site in phosphorylated CDK2 (42).
Two other arginine residues near the phosphorylation Lip, Arg-186[189] and Arg-189[192], are conserved only among the MAP kinases (although CDK2 has an arginine residue corresponding to Arg-189). In low activity ERK2 (49), these arginines are not aligned with the phosphorylation site residues but are found ligated by a sulfate of crystallization, suggesting that the arginines may bind to phosphothreonine or phosphotyrosine in the active structure. In p38, these arginines are also not aligned with the phosphorylation sites, and, in fact, Arg-186 is buried (Fig. 4a). Thus, activation of p38 may involve rearrangement of the Lip, and other residues, to form these interactions.
Domain Structure.
p38 has a wider domain separation in the low activity structure than does ERK2. The domain structure is coupled to the conformation of the Lip through interactions between the Lip and helix C (Fig. 3). Helix C straddles the Lip, with Arg-67[65] facing into the active site and Arg-70[68] facing the surface of the molecule, in contact with Leu-171 and Glu-328 in the C-terminal domain (data not shown). The fourth and fifth turns of helix C, however, are tethered to the C-terminal domain by packing interactions of Leu-74[Ile-72] and Met-78[Phe-76] between helix E and β7 in the C-terminal domain. The packing interactions of these residues are very similar to those in ERK2, but the axis of helix C is rotated, contributing to the wider domain separation (Fig. 3c). The rotation axis that relates the N-terminal domains of p38 and ERK2 lies approximately along helix C (about the y axis in Fig. 3c), similar to that noted for casein kinase Iδ in comparison to cAPK (50). The axes of the domain rotations observed in cAPK (51) and twitchin (52) are 90° away (approximately about the x axis in Fig. 3b) from that in p38.
The N terminus of helix C is shifted 6 Å in p38 relative to ERK2, as result of this rotation and the interactions of helix C with the Lip. αL16 is even further extended, shifted by 7 Å relative to ERK2 (Fig. 3c). Although αL16 is a signature of the MAP kinase family (Fig. 2), it is missing in an alternatively spliced form of p38, Mxi2 (53). We observe a less intimate interaction of αL16 in p38, largely due to the greater hydrophilicity of residues forming contacts with the body of the N-terminal domain Thr-341[Val-345] and Tyr-342[Phe-346] (data not shown). Also, p38 lacks an intimate interaction present in ERK2 between Phe-346 and Arg-89 (p38 has Tyr-342 and Thr-91 in corresponding positions that are not in contact).
Differences in the crossover connection (His-107–Asp-112) between the two domains also affect the domain separation (Fig. 4c). Met-109[106], a conserved residue in the crossover connection, occupies a different position in p38. Met-109 makes contact with Ala-157[Leu-154] in β7 and the ATP site; the smaller alanine residue in p38 creates a pocket in which the Met-109 packs. Apparently carried along with this conformational difference is Leu-108[105]. Leu-108 makes similar contacts with the N-terminal domain in p38 and ERK2, but because of the difference in the backbone position of Leu-108 and Met-109, Leu-108 is farther away from the C-terminal domain in p38, contributing to a wider domain separation.
The ATP Binding Site.
ATP does not bind to crystals of low activity p38, as judged by diffusion experiments. The structure reveals that the ATP site is malformed in p38 (Fig. 4c). The crossover connection, His-107–Asp-112[Asp-104–Asp-109], discussed above, is at the back of the ATP binding site in ERK2, but in p38, residues His-107–Asp-112 are shifted on average ≈2.5 Å. Met-109[106] is shifted even more, 3.5 Å relative to ERK2, in toward the ATP binding site. As a consequence, the backbone of His-107 blocks the ATP binding site observed in ERK2 (27). The conformational differences may be related in part to the difference in the pocket for Met-109 discussed above. The wider domain separation in p38 appears to be responsible for a significant increase in volume of the ATP binding site. Val-38[37] and Lys-53[52], residues in the N-terminal domain that contact ATP in ERK2, are shifted by 4 Å, increasing the separation between these ATP binding residues in the two domains by nearly that amount.
In addition to these global differences in the ATP binding site, amino acid replacements are present in residues known to contact ATP in ERK2. First, Lys-112 is hydrogen bonded to the ribose ring of ATP in ERK2 and is replaced by Asn-115 in p38. Modeling suggests that the shorter asparagine residue would not be able to contact ATP in p38. Second, Leu-154 contacts the adenine ring of ATP in ERK2 and is replaced by Ala-157 in p38. The effect of this replacement on the domain structure was discussed above. Finally, Cys-164 is in the ATP binding pocket in ERK2, although not in contact with ATP. It is replaced by Leu-167 in p38, making the pocket smaller. Each of these replacements contributes to the size, shape, hydrophobicity, and charge of the ATP binding site. It is interesting that two of these positions are also different in the JNKs (Fig. 2).
Substrate Specificity Determinants.
A surface groove formed by helices D, F, and G, and L13 (Figs. 3a and 4d) within the C-terminal domain has been shown to contact substrates, substrate analogs, or pseudosubstrates in the protein kinases for which structural data are available, the cAMP-dependent protein kinase (40) and twitchin (52). Four amino acid replacements in p38 relative to ERK2 in this region could affect specificity, assuming that MAP kinases also bind substrates in this region: residues Asn-114[Tyr-111], Cys-119[Thr-116], and Lys-121[His-118] in helix D and Gly-219[Asn-222] in helix F. JNK1 and JNK2 (11) have different sequences at these sites as well (Fig. 2). The backbone trace of Gln-120[117] and Lys-121[His-118] is also distinct in p38.
DISCUSSION
The structure of p38, together with that of ERK2, shows how unique specificity for activators is achieved within the MAP kinase family, despite the common regulatory mechanism by dual phosphorylation. The Lip must bind into the active site of the activating MEK, and thus may be involved in directing specificity toward MEK. The structure of p38 reveals that the Lip is unique in both conformation and sequence in the low activity form, thus modulating interactions with MEK. This idea is supported by mutational analysis in ERK2, as discussed above. The data of Brunet and Pouysségur (46) suggest that sequences in the N-terminal domain are also involved in pathway specificity.
However, the Lip regions of p38 and ERK2 are likely to be more similar to one another in the active conformation. Among all of the MAP kinases, the phosphorylation sites (Thr-180 and Tyr-182), and the putative phosphate binding ligands (Arg-67, Arg-70, Arg-149, Arg-173, Arg-186, and Arg-189) are conserved in homologous positions, and thus may interact similarly in different active MAP kinases. Also, each MAP kinase is specific for proline in the P+1 site. The position of the P+1 site is known to be adjacent to the Lip (Fig. 4b), and the residues that form the P+1 pocket (residues Val-183–Arg-189) are all conserved among the MAP kinases. The P+1 site is blocked completely in low activity p38, and thus large conformational changes in this region must occur.
Substrate Specificity.
Each member of the MAP kinase family has distinct substrates, despite a common specificity for proline-containing peptides. Substrates of the JNK MAP kinases have been studied by deletion analysis, showing that residues N-terminal to the phosphorylation site are involved in binding to kinase. In ATF2, 20 residues N-terminal to the phosphorylation site have been shown to be essential for tight binding to JNK1 (54, 55). Similarly, the δ domain of c-Jun, which is directly N-terminal to the phosphorylation site of c-Jun, is known to be essential for JNK interactions (56). In the ERK2 substrate c-Myc, the ERK2 binding site has been narrowed to 100 residues N-terminal to the phosphorylation site (57).
The regions of the kinase that interact with substrates also have been identified in JNKs. The Karin laboratory (11) showed that JNK2-JNK1 chimeras containing N-terminal sequences through L13 and helix G bind c-Jun with high affinity, characteristic of wild-type JNK2. From these data, an overall model for the interaction of substrates is that sequences N-terminal to the phosphorylation site of substrates binds in a surface groove near helices D, F, and G of the kinase, similar to the interactions observed in cAPK and twitchin.
We observe several amino acid differences in this region between p38 and ERK2 that could contribute to their specificity. It is interesting that the backbone of p38 and ERK2 superimpose closely in the substrate binding region, with an rms deviation of only 0.8 Å, so that modeling of the specificity surface of one MAP kinase from the coordinates of another should be relatively accurate.
Model for the Evolution of Protein Kinase Cascades.
In summary, interactions with activators are mediated by the Lip, and residues in the N-terminal domain, and interactions that discriminate among MAP kinase substrates are mediated near helices D, F, and G (Fig. 4d). Data available for other protein kinases also implicate the Lip in activating phosphorylations (41, 58, 59) and helices D and G in substrate interactions (36, 52). Thus, the sites at which protein kinases bind to activating protein kinases are distinct from those that recognize substrates. The spatial separation allows these interactions with activators and substrates to evolve independently.
Protein Kinase Inhibitor Specificity.
It might be anticipated that ATP-analog inhibitors would bind similarly to various protein kinases. Surprisingly, it is possible to obtain low molecular weight inhibitors that are directed toward single protein kinases, even single MAP kinases (60, 61). This is so in spite of the available structural data which show that most small molecule inhibitors of protein kinases bind in the ATP binding pocket (62, 63). The structure of p38 reveals differences in the ATP binding site, relative to ERK2 and other protein kinases, that can account for inhibitor specificity. First, p38 has sequence variations in residues in the known ATP binding site (residues Asn-115, Ala-157, and Leu-167). Second, there are differences in the backbone conformation in the crossover connection (His-107–Asp-112) that forms one wall of the site (27). Third, there are very large differences in the domain structure, due in part to the differences in the phosphorylation Lip, and differences in the position of Met-109, discussed above. Fourth, the phosphorylation Lip itself binds closer into the active site and can interact with inhibitors directly. It is apparent that protein kinases are different from other families of enzymes such as serine proteases, in which the active sites of multiple enzymes are highly similar (44). Thus, the problem of making specific inhibitors to these proteins may not be as difficult as it is for serine proteases. It will be interesting to determine to what extent the structural differences observed in the low activity structures are maintained in the active conformations of the MAP kinases.
Acknowledgments
We are greatly indebted to the crystallographic group of the University of Texas Southwestern Medical Center at Dallas for technical help. This research was supported by National Institutes of Health Grant DK46993, State of Texas Technology Transfer Grant 18596, and Welch Foundation Grant I1128.
ABBREVIATIONS
- MAP
mitogen-activated protein
- MEK
MAP/ERK kinase
- JNK
c-Jun N-terminal kinase
Footnotes
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, Chemistry Department, Brookhaven National Laboratory, Upton, NY 11973 (reference 1P38).
The residue number appearing in brackets after the p38 residue refers to the corresponding ERK2 residue number.
References
- 1.Han J, Lee J-D, Bibbs L, Ulevitch R J. Science. 1994;265:808–811. doi: 10.1126/science.7914033. [DOI] [PubMed] [Google Scholar]
- 2.Lee J C, Laydon J T, McDonnell P C, Gallagher T F, Kumar S, Green D, McNulty D, Blumenthal M J, Heys J R, Landvatter S W, Strickler J E, McLaughlin M M, Siemens I R, Fisher S M, Livi G P, White J R, Adams J L, Young P R. Nature (London) 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- 3.Rouse J, Cohen P, Trigon S, Morange M, Alonso-Llamazares A, Zamanillo D, Hunt T, Nebreda A R. Cell. 1994;78:1027–1037. doi: 10.1016/0092-8674(94)90277-1. [DOI] [PubMed] [Google Scholar]
- 4.Freshney N W, Rawlinson L, Guesdon F, Jones E, Cowley S, Hsuan J, Saklatvala J. Cell. 1994;78:1039–1049. doi: 10.1016/0092-8674(94)90278-x. [DOI] [PubMed] [Google Scholar]
- 5.Boulton T G, Yancopoulos G D, Gregory J S, Slaughter C, Moomaw C, Hsu J, Cobb M H. Science. 1990;249:64–67. doi: 10.1126/science.2164259. [DOI] [PubMed] [Google Scholar]
- 6.Cheng M, Boulton T G, Cobb M H. J Biol Chem. 1996;271:8951–8958. doi: 10.1074/jbc.271.15.8951. [DOI] [PubMed] [Google Scholar]
- 7.Lee J-D, Ulevitch R J, Han J. Biochem Biophys Res Commun. 1995;213:715–724. doi: 10.1006/bbrc.1995.2189. [DOI] [PubMed] [Google Scholar]
- 8.Zhou G, Bao Z Q, Dixon J E. J Biol Chem. 1995;270:12665–12669. doi: 10.1074/jbc.270.21.12665. [DOI] [PubMed] [Google Scholar]
- 9.Lechner C, Zahalka M A, Giot J-F, Møller N P H, Ullrich A. Proc Natl Acad Sci USA. 1996;93:4355–4359. doi: 10.1073/pnas.93.9.4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dérijard B, Hibi M, Wu I-H, Barrett T, Su B, Deng T, Karin M, Davis R J. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 11.Kallunki T, Su B, Tsigelny I, Sluss H K, Dérijard B, Moore G, Davis R, Karin M. Genes Dev. 1994;8:2996–3007. doi: 10.1101/gad.8.24.2996. [DOI] [PubMed] [Google Scholar]
- 12.Raingeaud J, Gupta S, Rogers J S, Dickens M, Han J, Ulevitch R J, Davis R J. J Biol Chem. 1995;270:7420–7426. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- 13.Wang X Z, Ron D. Science. 1996;272:1347–1349. doi: 10.1126/science.272.5266.1347. [DOI] [PubMed] [Google Scholar]
- 14.McLaughlin M M, Kumar S, McDonnell P C, Van Horn S, Lee J C, Livi G P, Young P R. J Biol Chem. 1996;271:8488–8492. doi: 10.1074/jbc.271.14.8488. [DOI] [PubMed] [Google Scholar]
- 15.Dérijard B, Raingeaud J, Barrett T, Wu I-H, Han J, Ulevitch R J, Davis R J. Science. 1995;267:682–685. doi: 10.1126/science.7839144. [DOI] [PubMed] [Google Scholar]
- 16.Raingeaud J, Whitmarsh A J, Barrett T, Dérijard B, Davis R J. Mol Cell Biol. 1996;16:1247–1255. doi: 10.1128/mcb.16.3.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han J, Lee J-D, Jiang Y, Li Z, Feng L, Ulevitch R J. J Biol Chem. 1996;271:2886–2891. doi: 10.1074/jbc.271.6.2886. [DOI] [PubMed] [Google Scholar]
- 18.Stein B, Brady H, Yang M X, Young D B, Barbosa M S. J Biol Chem. 1996;271:11427–11433. doi: 10.1074/jbc.271.19.11427. [DOI] [PubMed] [Google Scholar]
- 19.Moriguchi T, Kuroyanagi N, Yamaguchi K, Gotoh Y, Irie K, Kano T, Shirakabe K, Muro Y, Shibuya H, Matsumoto K, Nishida E, Hagiwara M. J Biol Chem. 1996;271:13675–13679. doi: 10.1074/jbc.271.23.13675. [DOI] [PubMed] [Google Scholar]
- 20.Lin A, Minden A, Martinetto H, Claret F-X, Lange-Carter C, Mercurio F, Johnson G L, Karin M. Science. 1995;268:286–290. doi: 10.1126/science.7716521. [DOI] [PubMed] [Google Scholar]
- 21.Payne D M, Rossomando A J, Martino P, Erickson A K, Her J-H, Shabanowitz J, Hunt D F, Weber M J, Sturgill T W. EMBO J. 1991;10:885–892. doi: 10.1002/j.1460-2075.1991.tb08021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robbins D J, Cobb M H. Mol Biol Cell. 1992;3:299–308. doi: 10.1091/mbc.3.3.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robbins D J, Zhen E, Owaki H, Vanderbilt C A, Ebert D, Geppert T D, Cobb M H. J Biol Chem. 1993;268:5097–5106. [PubMed] [Google Scholar]
- 24.Anderson N G, Maller J L, Tonks N K, Sturgill T W. Nature (London) 1990;343:651–653. doi: 10.1038/343651a0. [DOI] [PubMed] [Google Scholar]
- 25.Alvarez E, Northwood I C, Gonzalez F A, Latour D A, Seth A, Abate C, Curran T, Davis R J. J Biol Chem. 1991;266:15277–15285. [PubMed] [Google Scholar]
- 26.Clark-Lewis I, Sanghera J S, Pelech S L. J Biol Chem. 1991;266:15180–15184. [PubMed] [Google Scholar]
- 27.Zhang F, Strand A, Robbins D, Cobb M H, Goldsmith E J. Nature (London) 1994;367:704–711. doi: 10.1038/367704a0. [DOI] [PubMed] [Google Scholar]
- 28.Jancarik J, Kim S-H. J Appl Crystallogr. 1991;24:409–411. [Google Scholar]
- 29.Otwinowski Z. In: Data Collection and Processing. Sawyer L, Isaacs N, Bailey S W, editors. Warrington, U.K.: Science and Engineering Council/Daresbury Laboratory; 1993. pp. 56–62. [Google Scholar]
- 30.Navaza J. Acta Crystallogr A. 1994;50:157–163. [Google Scholar]
- 31.Jones, T. A. & Kjeldgaard, M. (1993) o: The Manual (Uppsala University, Uppsala), Version 5.9.
- 32.Read R J. Acta Crystallogr A. 1986;42:140–149. [Google Scholar]
- 33.Collaborative Computing Project Number 4. Acta Crystallogr D. 1994;50:760–763. [Google Scholar]
- 34.Brünger, A. T. (1992) x-plor: A System for X-Ray Crystallography and NMR (Yale University, Department of Molecular Biophysics, New Haven, CT), Version 3.1.
- 35.Laskowski R A, MacArthur M W, Moss D S, Thornton J M. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 36.Knighton D R, Zheng J, Ten Eyck L F, Ashford V A, Xuong N-H, Taylor S S, Sowadski J M. Science. 1991;253:407–413. doi: 10.1126/science.1862342. [DOI] [PubMed] [Google Scholar]
- 37.Hanks S K, Quinn A M, Hunter T. Science. 1988;241:42–52. doi: 10.1126/science.3291115. [DOI] [PubMed] [Google Scholar]
- 38.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 39.Evans S V. J Mol Graphics. 1993;11:134–138. doi: 10.1016/0263-7855(93)87009-t. [DOI] [PubMed] [Google Scholar]
- 40.Knighton D R, Zheng J, Ten Eyck L F, Xuong N-H, Taylor S S, Sowadski J M. Science. 1991;253:414–420. doi: 10.1126/science.1862343. [DOI] [PubMed] [Google Scholar]
- 41.De Bondt H L, Rosenblatt J, Jancarik J, Jones H D, Morgan D O, Kim S-H. Nature (London) 1993;363:595–602. doi: 10.1038/363595a0. [DOI] [PubMed] [Google Scholar]
- 42.Russo A A, Jeffrey P D, Pavletich N P. Nat Struct Biol. 1996;3:696–700. doi: 10.1038/nsb0896-696. [DOI] [PubMed] [Google Scholar]
- 43.Robinson M J, Cheng M, Khokhlatchev A, Ebert D, Ahn N, Guan K-L, Stein B, Goldsmith E, Cobb M H. J Biol Chem. 1996;271:29734–29739. doi: 10.1074/jbc.271.47.29734. [DOI] [PubMed] [Google Scholar]
- 44.Greer J. Proteins. 1990;7:317–334. doi: 10.1002/prot.340070404. [DOI] [PubMed] [Google Scholar]
- 45.Zheng C-F, Guan K-L. J Biol Chem. 1993;268:11435–11439. [PubMed] [Google Scholar]
- 46.Brunet A, Pouysségur J. Science. 1996;272:1652–1655. doi: 10.1126/science.272.5268.1652. [DOI] [PubMed] [Google Scholar]
- 47.Johnson L N, Noble M E M, Owen D J. Cell. 1996;85:149–158. doi: 10.1016/s0092-8674(00)81092-2. [DOI] [PubMed] [Google Scholar]
- 48.Goldsmith E J, Cobb M H. Curr Opin Struct Biol. 1994;4:833–840. doi: 10.1016/0959-440x(94)90264-x. [DOI] [PubMed] [Google Scholar]
- 49.Zhang J, Zhang F, Ebert D, Cobb M H, Goldsmith E J. Structure. 1995;3:299–307. doi: 10.1016/s0969-2126(01)00160-5. [DOI] [PubMed] [Google Scholar]
- 50.Longenecker K L, Roach P J, Hurley T D. J Mol Biol. 1996;257:618–631. doi: 10.1006/jmbi.1996.0189. [DOI] [PubMed] [Google Scholar]
- 51.Cox S, Radzio-andzelm E, Taylor S S. Curr Opin Struct Biol. 1994;4:893–901. doi: 10.1016/0959-440x(94)90272-0. [DOI] [PubMed] [Google Scholar]
- 52.Hu S-H, Parker M W, Lei J Y, Wilce M C J, Benian G M, Kemp B E. Nature (London) 1994;369:581–584. doi: 10.1038/369581a0. [DOI] [PubMed] [Google Scholar]
- 53.Zervos A S, Faccio L, Gatto J P, Kyriakis J M, Brent R. Proc Natl Acad Sci USA. 1995;92:10531–10534. doi: 10.1073/pnas.92.23.10531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Livingstone G, Patel G, Jones N. EMBO J. 1995;14:1785–1797. doi: 10.1002/j.1460-2075.1995.tb07167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gupta S, Campbell D, Dérijard B, Davis R J. Science. 1995;267:389–402. doi: 10.1126/science.7824938. [DOI] [PubMed] [Google Scholar]
- 56.Dai T, Rubie E, Franklin C C, Kraft A, Gillespie D A F, Avruch J, Kyriakis J M, Woodgett J R. Oncogene. 1995;10:849–855. [PubMed] [Google Scholar]
- 57.Gupta S, Davis R J. FEBS Lett. 1994;353:281–285. doi: 10.1016/0014-5793(94)01052-8. [DOI] [PubMed] [Google Scholar]
- 58.Hubbard S R, Wei L, Ellis L, Hendrickson W A. Nature (London) 1994;372:746–754. doi: 10.1038/372746a0. [DOI] [PubMed] [Google Scholar]
- 59.Mansour S J, Matten W T, Hermann A S, Candia J M, Rong S, Fukasawa K, Vande Woude G F, Ahn N G. Science. 1994;265:966–970. doi: 10.1126/science.8052857. [DOI] [PubMed] [Google Scholar]
- 60.Lee J C, Adams J L. Curr Opin Biotechnol. 1995;6:657–661. doi: 10.1016/0958-1669(95)80108-1. [DOI] [PubMed] [Google Scholar]
- 61.Fry D W, Bridges A J. Curr Opin Biotechnol. 1995;6:662–667. doi: 10.1016/0958-1669(95)80109-x. [DOI] [PubMed] [Google Scholar]
- 62.Schulze-Gahmen U, Brandsen J, Jones H D, Morgan D O, Meijer L, Vesely J, Kim S-H. Proteins. 1995;22:378–391. doi: 10.1002/prot.340220408. [DOI] [PubMed] [Google Scholar]
- 63.De Azevedo W F, Jr, Mueller-Dieckmann H-J, Schulze-Gahmen U, Worland P J, Sausville E, Kim S-H. Proc Natl Acad Sci USA. 1996;93:2735–2740. doi: 10.1073/pnas.93.7.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]