

Abstract
Background
Microarray technology allows the monitoring of expression levels for thousands of genes simultaneously. This novel technique helps us to understand gene regulation as well as gene by gene interactions more systematically. In the microarray experiment, however, many undesirable systematic variations are observed. Even in replicated experiment, some variations are commonly observed. Normalization is the process of removing some sources of variation which affect the measured gene expression levels. Although a number of normalization methods have been proposed, it has been difficult to decide which methods perform best. Normalization plays an important role in the earlier stage of microarray data analysis. The subsequent analysis results are highly dependent on normalization.
Results
In this paper, we use the variability among the replicated slides to compare performance of normalization methods. We also compare normalization methods with regard to bias and mean square error using simulated data.
Conclusions
Our results show that intensity-dependent normalization often performs better than global normalization methods, and that linear and nonlinear normalization methods perform similarly. These conclusions are based on analysis of 36 cDNA microarrays of 3,840 genes obtained in an experiment to search for changes in gene expression profiles during neuronal differentiation of cortical stem cells. Simulation studies confirm our findings.
Background
Biological processes depend on complex interactions between many genes and gene products. To understand the role of a single gene or gene product in this network, many different types of information, such as genome-wide knowledge of gene expression, will be needed. Microarray technology is a useful tool to understand gene regulation and interactions [1-3]. For example, cDNA microarray technology allows the monitoring of expression levels for thousands of genes simultaneously. cDNA microarrays consist of thousands of individual DNA sequences printed in a high density array on a glass slide. After being reverse-transcribed into cDNA and labelled using red (Cy5) and green (Cy3) fluorescent dyes, two target mRNA samples are hybridized with the arrayed DNA sequences or probes. Then, the relative abundance of these spotted DNA sequences can be measured. After image analysis, for each gene the data consist of two fluorescence intensity measurements, (R, G), showing the expression level of the gene in the red and green labelled mRNA samples. The ratio of the fluorescence intensity for each spot represents the relative abundance of the corresponding DNA sequence. cDNA microarray technology has important applications in pharmaceutical and clinical research. By comparing gene expression in normal and tumor tissues, for example, microarrays may be used to identify tumor-related genes and targets for therapeutic drugs [4].
In microarray experiments, there are many sources of systematic variation. Normalization attempts to remove such variation which affects the measured gene expression levels. Yang et al. [5] and Yang et al. [6] summarized a number of normalization methods for dual labelled microarrays such as global normalization and locally weighted scatterplot smoothing (LOWESS [7]). Quackenbush [8] and Bilban et al. [9] provided good reviews on normalization methods. There have been some extensions for global and intensity-dependent normalizations. For example, Kepler et al. [10] considered a local regression to estimate a normalized intensities as well as intensity dependent error variance. Wang et al. [11] proposed a iterative normalization of cDNA microarray data for estimating a normalized coefficients and identifying control genes. Workman et al. [12] proposed a roust non-linear method for normalization using array signal distribution analysis and cubic splines. Chen et al. [13] proposed a subset normalization to adjust for location biases combined with global normalization for intensity biases. Edwards [14] considered a non-linear LOWESS normalization in one channel cDNA microarrays mainly for correcting spatial heterogeneity. The main idea of normalization for dual labelled arrays is to adjust for artifactual differences in intensity of the two labels. Such differences result from differences in affinity of the two labels for DNA, differences in amounts of sample and label used, differences in photomultiplier tube and laser voltage settings and differences in photon emission response to laser excitation. Although normalization alone cannot control all systematic variations, normalization plays an important role in the earlier stage of microarray data analysis because expression data can significantly vary from different normalization procedures. Subsequent analyses, such as differential expression testing would be more important such as clustering, and gene networks, though they are quite dependent on a choice of a normalization procedure [1,3].
Several normalization methods have been proposed using statistical models (Kerr et al. [15]; Wolfinger et al. [16]). However, these approaches assume additive effects of random errors, which needs to be validated. Because they are less frequently used, we have not evaluated them here. Although several normalization methods have been proposed, no systematic comparison has been made for the performance of these methods. In this paper, we use the variability among the replicated slides to compare performance of several normalization methods.
We focus on comparing the normalization methods for cDNA microarrays. A detailed description on normalization methods considered in our study is given in the next section. Complex methods do not necessarily perform better than simpler methods. Complex methods may add noise to the normalized adjustment and may even add bias if the assumptions are incorrect. The fact that a non-linear method linearizes a graph of red intensity versus green intensity or a plot of log(R/G) versus log(RG) is not in itself evidence that the method is not just fitting noise. Consequently, proposed normalization methods require validation.
We first compare the methods using data from cDNA microarrays of 3,840 genes obtained in an experiment to search for changes in gene expression profiles during neuronal differentiation of cortical stem cells. Then, we perform simulation studies to compare these normalization methods systematically. Bolstad et al. [17] compare normalization methods for high density oligonucleotide array data using variance and bias.
The paper is organized as follows. Section 2 describes normalization methods. Section 3 describes the measures for variability to compare normalization methods. Section 4 shows the comparison results for cDNA microarrays obtained from a cortical stem cells experiment along with some simulation results. Finally, Section 5 summarizes the concluding remarks.
Normalization methods
As pointed out by Yang et al. [5], the purpose of normalization is to remove systematic variation in a microarray experiment which affects the measured gene expression levels. They summarized a number of normalization methods: (i) within-slide normalization, (ii) paired-slide normalization for dye-swap experiments, and (iii) multiple slide normalization.
As a first step, one needs to decide which set of genes to use for normalization. Yang et al. [5] suggested three approaches: all genes on the array, constantly expressed genes, and controls. Recently, Tseng et al. [18] suggested using the rank invariant genes.
Let (logG, logR) be the green and red background-corrected intensities. Then, (M, A) are defined by M = log(R/G) and 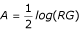 . Once the genes for normalization are selected, the following normalization approaches are available based on (M, A), as described by Yang et al. [5]:
. Once the genes for normalization are selected, the following normalization approaches are available based on (M, A), as described by Yang et al. [5]:
(1) Global normalization (G) using the global median of log intensity ratios
(2) Intensity dependent linear normalization (L)
(3) Intensity dependent nonlinear normalization (N) using a LOWESS curve.
Under ideal experimental conditions, we expect M = 0 for the genes used for normalization. However, quite different patterns may be observed in real microarray experiments. Note that all three normalization methods consider regression models in the form of

For global normalization α0 is estimated by the median of M. For intensity dependent normalization (β0, β1) by least squares estimation, and c(A) by using the robust scatter plot smoother LOWESS. Thus, all three normalization methods are based on regression models of M in terms of A. Let 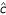 (A) be the fitted value of c(A). The normalization process can be described in terms of (A). For gene j, the normalized ratio
(A) be the fitted value of c(A). The normalization process can be described in terms of (A). For gene j, the normalized ratio 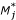 is
is
(Aj) = Mj - (Aj). (1)
Thus, the normalization process transforms (logG, logR) to (M, A), and then we obtain M* using regression models. The statistical analysis can then be performed using the normalized M*. The above normalization methods are mainly for controlling location shifts of logarithmically transformed intensities. They may be applied separately to each grid on the microarray because each grid is robotically printed by a different print-tip. Yang et al. [5] also proposed scale normalization methods. The main purpose of scale normalization is to control between slide variability and it may also be performed separately for each print-tip. Figure 1 shows a flowchart for these normalization methods.
Figure 1.

Flowchart of normalization
The detailed descriptions for each normalization approach considered in this study are given in Tables 1 and 2. We denote global normalization, intensity dependent linear normalization, and intensity dependent nonlinear normalization by G, L, and N, respectively. They all represent location normalization and can be carried out globally or separately over print-tips. Similarly, scale normalization can be carried out globally or separately over print-tips. Furthermore, the global scale normalization can also be performed after print-tip scale normalization. All possible combinations of location and scale normalization are summarized in Tables 1 and 2. For example, GPS.s denotes between-slide scale normalization (.s) after global (G) median location normalization and scale (S) normalization on each print-tip (P).
Table 1.
Abbreviation for Normalization Methods
| Abbreviation | Description |
| O | Original data |
| G | Global median normalization |
| L | Intensity dependent linear normalization |
| N | Intensity dependent nonlinear normalization (LOWESS) |
| P | Print-tip normalization |
| S | Print-tip scale normalization |
| .s | Between-slide scale normalization |
Table 2.
List of Normalization Methods
| List of normalization methods including print-tip scale normalization | ||
| Method | Notation | Description |
| O | Original data | |
| Global | G | Global median normalization |
| GP | Global median normalization on each print-tip | |
| GPS | Global median normalization on each print-tip with scale normalization | |
| G.s | Global median normalization and between-slide scale normalization | |
| GP.s | Global median normalization on each print-tip and between-slide scale normalization | |
| GPS.s | Global median normalization on print-tip with scale normalization and between-slide scale normalization | |
| Linear | L | Intensity dependent linear regression normalization |
| LP | Intensity dependent linear regression normalization on each print-tip | |
| LPS | Intensity dependent linear regression normalization on each print-tip with scale normalization | |
| L.s | Intensity dependent linear regression normalization and between-slide scale normalization | |
| LP.s | Intensity dependent linear regression normalization on each print-tip and between-slide scale normalization | |
| LPS.s | Intensity dependent linear regression normalization on each print-tip with scale normalization and between-slide scale normalization | |
| Nonlinear | N | Intensity dependent nonlinear regression normalization (LOWESS) |
| NP | Intensity dependent nonlinear regression normalization (LOWESS) on each print-tip | |
| NPS | Intensity dependent nonlinear regression normalization (LOWESS) on each print-tip with scale normalization | |
| N.s | Intensity dependent nonlinear regression normalization (LOWESS) and between-slide scale normalization | |
| NP.s | Intensity dependent nonlinear regression normalization (LOWESS) on each print-tip and between-slide scale normalization | |
| NPS.s | Intensity dependent nonlinear regression normalization (LOWESS) on each print-tip with scale normalization and between-slide scale normalization | |
Measures of variation
In order to derive measures of variation, we now use y instead of M to represent the logarithm of the ratio of red and green background-corrected intensities. We introduce subscripts i,j,k and I for describing the cDNA microarray data introduced in the next section. Suppose there are I experimental groups denoted by i, J time points denoted by j, and K replications denoted by k.
Also suppose that there are N genes in each slide. For the simple case when there are I experimental groups but no time sequences, we can simply let J = 1. For the case when there are only time sequences, we can let I = 1. Thus, these subscripts represent general cases.
Let yijkl be the logarithm of the red to green intensity ratio from group i(= 1, ..., I), time j(= 1, ... , J), replication k(= 1, ..., K), and gene l. Using the replicated observations, we want to derive the variability measures of yijkl. It is expected that the better the normalization method, the smaller the variation among the replicated observations. Let σl be the variability measure for the lth gene. We introduce two methods for estimating 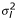 , l = 1, ... , N. Either density plots or box plots for the variability measures can be used for visually comparing different normalization methods.
, l = 1, ... , N. Either density plots or box plots for the variability measures can be used for visually comparing different normalization methods.
Method 1. Pooled variance estimators
For gene l, a simple variance estimator for can be obtained by pooling variance estimators for each i and j. That is,
![]()
where 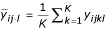 , for i = 1, ..., I; j = 1, ..., J; l = 1, ..., N. Then, we compare the distributions of
, for i = 1, ..., I; j = 1, ..., J; l = 1, ..., N. Then, we compare the distributions of 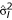 . The better the normalization method, the smaller the variance estimates.
. The better the normalization method, the smaller the variance estimates.
Method 2. Variance estimator using analysis of variance models
Consider the following two-way analysis of variance(ANOVA) model with interactions for each gene.
yijkl = μl + αil + βjl + (αβ)ijl + εijkl, (3)
where i = 1, ..., I, j = 1, ..., J, k = 1, ..., K, and l = 1, ..., N. The gene effects μl capture the overall mean intensity in fluorescent signals for genes across the arrays, groups, and time points. The αil terms account for gene specific group effects representing overall differences between two groups. The βjl account for time effects that capture differences in the overall concentration of mRNA in the samples from the different time points. The terms (αβ)ijl account for the interaction effects between group and time representing the signal contribution due to the combination of group and time.
From this ANOVA model, an unbiased estimate of can be obtained which is the error sum of squares divided by an appropriate degrees of freedom. Let 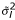 be the variance estimate for the lth gene. Note that for Model (3) the values of are the same with those of (2), if there are no missing observations. However, this measure of variability is more flexible to use in the sense that it can be used with any ANOVA model which fits the intensity data well.
be the variance estimate for the lth gene. Note that for Model (3) the values of are the same with those of (2), if there are no missing observations. However, this measure of variability is more flexible to use in the sense that it can be used with any ANOVA model which fits the intensity data well.
Results
Data
The data studied here are from a study of cortical stem rat cells. The goal of the experiment is to identify genes that are associated with neuronal differentiation of cortical stem cells. A detailed description of data is given by Park et al. [19]. From a developing fetal rat brain, 3,840 genes including novel genes were immobilized on a glass chip and fluorescence-labelled target cDNA were hybridized. After expansion, differentiation to neuronal cells was induced by removing bFGF with or without ciliary neurotrophic factor (CNTF) at six time points (12 hrs, 1 day, 2 days, 3 days, 4 days, 5 days). To get more reliable data, all the hybridization analyses were carried out three times against same RNA, and the scanned images were analyzed using an edge detection mode [20]. As an illustration of normalization methods, we chose one slide among 36 slides to see the effect of normalization. In Figure 2, the original slide is the graph of (logG, logR) before normalization which shows a nonlinear spatial pattern. In this case, three normalization methods yielded quite different results. On the other hand, for the slides with a linear pattern three normalization methods yielded similar results in making the slope 1.
Figure 2.

(A) The original slide with a non-linear pattern. (B-D) Three normalized slides (global median, intensity dependent linear regression, intensity dependent non-linear regression.
For this dataset, Park et al. [19] presented clustering analysis results after nonlinear LOWESS normalization. We applied three normalization methods to all 36 slides and compared the performance of normalization methods using the measures of variation among the replicated observations described in Section 3. This data set is a special case of the one described in Section 3 with two experimental groups, I = 2, six time points, J = 6, and three replications K = 3. For these microarray data, we derived the distribution of variance estimates by the methods given in Section 3. Since the two methods provided quite similar results, we only present the results from the ANOVA model.
Figure 3 shows the dot plots of the mean values of log-transformed variances, say, log(), for the original data, global normalization, intensity dependent linear normalization, and intensity dependent nonlinear normalization, denoted by O, G, L, and N, respectively. Figure 3a is a dot plot showing the mean values of log() from O, G, L, N as well as those corresponding to the scale normalized approaches. Figures 3b to 3d focus on the effect of scale normalization and print-tip stratification.
Figure 3.

Dot Plots of Log-transformed Variance Estimates for Cortical Stem Cells Data. The Y-axis represents normalization methods and the X-axis represents the mean values of log-transformed variance estimates. (A) Dot plots for O, G, G.s, L, L.s, N, and N.s, (B) Dot plots for Global Normalization Methods, (C) Dot plots for Intensity-dependent Linear Normalization Methods, (D) Dot plots for Intensity-dependent Nonlinear Normalization Methods.
As shown in Figure 3a, the three normalization methods G, L, N reduce the variability greatly. The two intensity dependent normalization methods denoted by L and N perform equally well and somewhat better than global normalization.
Figure 3b shows the results of global normalization with different scale normalization approaches. Great differences are not found among the six approaches. Hence global normalization does not appear to be improved by normalizing separately over the grids of spots defined by the print-tips or by scale adjustments.
Figure 3c focuses on intensity dependent linear normalization with different scale normalization approaches. Figure 3d is for intensity dependent non-linear LOWESS normalization with different scale normalization approaches. Neither linear nor non-linear intensity dependent normalization appear to benefit greatly by scale adjustments or by performing normalization by print-tip grids for this set of data.
Based on these figures, we conclude that intensity dependent normalization methods perform better than global normalization methods. Small differences are observed between the linear and nonlinear normalization methods. In addition, small effects are observed for scale normalization methods.
Simulation studies
In order to compare the normalization methods more systematically, we performed a simulation study by generating typical patterns of microarray data. Using simulated data we could compare the normalization methods with regard to bias and mean squared error as well as variance. In general, mean square error (MSE) is defined by the sum of variance and bias2. MSE has been used as a criterion to compare normalization methods for the cases when they can be computed. Bolstad et al. [17] used variance and bias to compare normalization methods for high density oligonucleotide array data. Since we do not know the true values for real datasets, however, we cannot find out whether the normalization methods reduce biases or not, while we can get estimates of variance. For the simulated datasets, on the other hand, we know the true values and thus we can compute biases. From biases and variances we can derive mean square errors. Simulation studies provide some useful information that the public datasets do not provide. After a careful examination of all 36 slides in our study, we considered four typical types of microarray data. These four types are shown in Figure 4. Although they were selected from our study, we think that they represent various linear and nonlinear types of artifacts commonly observed in microarray studies. For simplicity, we generated the replicated microarray data from the same distribution, with 5,000 spots in one microarray.
Figure 4.

Four types of (logG, logR) plots for the simulated microarray data: Type I (A), Type II (B), Type III (C), Type IV (D).
Type I
Figure 4a shows the plot of (logG, logR) for Type I which shows a clear linear pattern. Most observations are scattered around the y = x line showing a high correlation between the two intensity values.
Type II
Figure 4b shows the second type. It has a specific non-linear pattern with many spots distant from the y = x line.
Type III
Figure 4c shows the third type which has the same linear pattern as Type I. However, the lower intensity values have higher variabilities than those of higher intensity values.
Type IV
Figure 4d shows the fourth type. It is a combination of Type II and Type III. That is, it has a non-linear pattern with higher variability at the lower intensities. In order to generate (logGI, logRI), we assume that they are observed from the bivariate normal distribution with mean vector β and covariance matrix Σ, where
![]()
For simplicity, let the random vector (YG, YR)T represent the log-transformed intensity values, say (logGI, logRI). To generate bivariate normal random variables, it is convenient to generate YG first and then generate YR from the conditional distribution of YR| YG = Y. Note that the conditional distribution is given by
![]()
where μR|G = μR - ρσR/σG(Y - μG) and 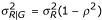 . For Type II, we use the same values of YG generated from Type I. For YR, however, we use the transformed mean and variance to add a pattern like that shown in Figure 4b. That is,
. For Type II, we use the same values of YG generated from Type I. For YR, however, we use the transformed mean and variance to add a pattern like that shown in Figure 4b. That is, 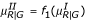 and
and 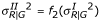 . We first tried generating Type II data by directly transforming the data generated from Type I but could not get a graph with the desired pattern. The functions f1 and f2 are found by trial and error. For Type III, we use a similar approach. That is, we use the same values of YG generated from Type I. For YR, we use the transformed variance to allow larger variability for the lower intensity observations. Thus,
. We first tried generating Type II data by directly transforming the data generated from Type I but could not get a graph with the desired pattern. The functions f1 and f2 are found by trial and error. For Type III, we use a similar approach. That is, we use the same values of YG generated from Type I. For YR, we use the transformed variance to allow larger variability for the lower intensity observations. Thus, 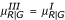 and
and 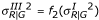 . For simplicity, we use the same function f2 used for Type II. Similarly, for Type IV we use the same values of YG generated from Type I. For YR, we use the transformed mean and variance to add a specific pattern. That is,
. For simplicity, we use the same function f2 used for Type II. Similarly, for Type IV we use the same values of YG generated from Type I. For YR, we use the transformed mean and variance to add a specific pattern. That is, 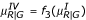 and
and 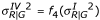 . The functions f3 and f4 are also found by trial and error. For each type, we generate ten replicated samples. For these replicated samples, we apply the normalization methods and draw the distributions for the variation measures. For simplicity, we do not consider the print-tip variation in this simulation. We compare four graphs: original data(O), globally normalized data(G), linear normalized data(L), and non-linear normalized data(N) using LOWESS.
. The functions f3 and f4 are also found by trial and error. For each type, we generate ten replicated samples. For these replicated samples, we apply the normalization methods and draw the distributions for the variation measures. For simplicity, we do not consider the print-tip variation in this simulation. We compare four graphs: original data(O), globally normalized data(G), linear normalized data(L), and non-linear normalized data(N) using LOWESS.
In order to compare these normalization methods, we compute the mean square error(MSE). For each gene, the MSE is computed as the average of the distances between normalized data and true expected value. Figure 5 shows the dot plots for the means of log-transformed MSEs. Figure 5a shows the dot plot for Type I. As expected, the four dots look exactly the same. These plots imply that these normalization approaches are equivalent when normalization is unnecessary. On the other hand, Figure 5b shows quite different patterns for Type II. Note that Type II contains a specific non-linear pattern on the original data. The original data have the largest MSEs. Large differences in MSEs are observed between original data and globally normalized data. Furthermore, there is substantial additional reduction in MSEs for the intensity dependent linear and non-linear normalization approaches, though slight differences are observed between them. Figure 5c shows the results for Type III. Surprisingly, no clear differences are observed among four box plots. This implies that these normalization methods do not reduce the MSEs of data when no specific patterns are observed but variability differs greatly depending on the intensity levels. Finally, Figure 5d shows the results for Type IV which contains a specific non-linear pattern as well as higher variability for lower intensity observations. Only small differences are observed between the original data and globally normalized data. In addition, the reduction of MSEs by linear and non-linear normalization approaches are not as large as those of Type II. One possible explanation is that Type IV appears to have a more similar pattern to Type III than to Type II. Though not reported here, we also examined variances and squared biases. For Types I, III, and IV, three normalization approaches show similar distributions as the original data with regard to these measures. For Type II, however, quite different patterns are observed for variances and biases. For variances, globally normalized data have similar distributions as the original data, while the intensity dependent linear and non-linear normalization approaches have much reduced variances. For biases, similar graphs are obtained. That is, globally normalized data have smaller biases, and intensity dependent normalized data have much smaller biases than original data. Only slight differences are observed between two intensity dependent normalized data. The graphs of biases and variances show that the global normalization method has the effect of reducing biases, while intensity dependent normalization methods have effects on reducing both biases and variation.
Figure 5.

Dot Plots of Log-transformed Variance Estimates for Simulated Data. The Y-axis represents normalization methods and the X-axis represents the mean values of log-transformed variance estimates.
We summarize our findings from the simulation studies. First, the normalization methods performed similarly when the original data has a linear pattern such as Type I, and when there are high variabilities for observations with lower intensities such as Types III. Second, when there is a specific pattern such as Type II, all normalization methods tend to reduce MSEs. Third, in this case, MSEs are dominated by biases. Fourth, only small differences are observed between the intensity-dependent linear and non-linear normalization methods.
Discussion
In this article, we compare normalization methods commonly used to analyze microarray data. The comparison is based on the variability measures derived from the replicated microarray samples. These variability measures can be easily derived from any replicated microarray experiment. As pointed out by many researchers, many undesirable systematic variations are observed in the microarray experiment. Normalization becomes a standard process for removing some of the variation which affects the measured gene expression levels. Although a number of normalization methods have been proposed, it has been difficult to decide which method performs better than the others. Thus, the evaluation of normalization methods in microarray data analysis is indeed an important issue. In this article, we show that the intensity dependent normalization method performs better than the simpler global normalization methods in many cases. We have not been sure about whether apparent nonlinearity of an M-A scatter plot or a scatter plot of red vs green is sufficient basis for feeling confident that a non-linear normalization is useful. Although we have studied only a limited number of data sets, our findings can provide some guidance on the selection of normalization methods. There are clearly cases where intensity dependent normalization performs substantially better than global normalization methods. In most of the cases that we considered, the non-linear intensity dependent procedures did not perform substantially better than a linear intensity dependent method, although there may be datasets where this is not the case. For the cases we considered, we did not see large benefits to separate normalization by print-tip grids or for scale normalization. None of the normalization methods effectively addressing the dependence of the variance of measurements on intensity level. Recently, some other non-linear normalization methods have been employed such as B-splines and Gaussian-kernel fitting [12,21]. These non-linear normalization methods seem to be comparable to the LOWESS normalization. We think the basic idea of non-linear normalization is the same whether they use LOWESS curve, splines, or kernels. They are quite effective in controlling specific non-linear variations. Some methods may have more computational efficiency. We believe that more sophisticated non-linear normalization methods are under development. We recommend that experimentalists examine their data carefully and consider applying intensity-dependent normalization methods routinely. Software for intensity dependent normalization is available on desktop packages such as BRB-ArrayTools http://linus.nci.nih.gov/BRB-ArrayTools and via the internet in SNOMAD http://pevsnerlab.kennedykrieger.org/snomad.htm tools which provide any DNA array researcher with the tools to apply a variety of non-linear intensity-dependent normalization methods to their data [22].
Acknowledgments
Acknowledgement
The authors wish to thank two anonymous referees whose comments were extremely helpful. The work was supported by the IMT2000 contribution from Korea Ministry of Health and Welfare, and Ministry of Information and Communication (01-PJ11-PG9-01BT-00A-0045).
Contributor Information
Taesung Park, Email: tspark@stats.snu.ac.kr.
Sung-Gon Yi, Email: skon@kr.FreeBSD.org.
Sung-Hyun Kang, Email: gadin@statcom.snu.ac.kr.
SeungYeoun Lee, Email: leesy@sejong.ac.kr.
Yong-Sung Lee, Email: yongsung@hanyang.ac.kr.
Richard Simon, Email: RSimon@nih.gov.
References
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. In Proc Natl Acad Sci. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spellman PT, Sherlock G, Zhang MQ, Iyer VR. Comprehensive identification of cell cycle-regulated genes of the yeast saccharomyces cerevisiae by microarray hybridization. Molecular Biology of the Cell. 1998;9:3273–3297. doi: 10.1091/mbc.9.12.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA, Bloomfield CD, Lander ES. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, Powell JI, Yang L, Marti GE, Moore T, Hudson J, Jr, Lu L, Lewis DB, Tibshirani R, Sherlock G, Chan WC, Greiner TC, Weisenburger DD, Armitage JO, Warnke R, Levy R, Wilson W, Grever MR, Byrd JC, Botstein D, Brown PO, Staudt LM. Different type of diffuse large b-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- Yang YH, Dudoit SD, Luu P, Speed TP. Normalization for cDNA Microarray Data. In SPIE BioE. 2001.
- Yang YH, Dudoit S, Luu P, Lin DM, Peng V, Ngai J, Speed TP. Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Research. 2002;30:e15. doi: 10.1093/nar/30.4.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleveland WS. Robust locally weighted regression and smoothing scatterplots. Jounral of the American Statistical Association. 1974;74:829–836. [Google Scholar]
- Quackenbush J. Microarray data normalization and transformation. Natuture Genetics. 2002;Suppl 32:496–501. doi: 10.1038/ng1032. [DOI] [PubMed] [Google Scholar]
- Bilban M, Buehler LK, Head S, Desoye G, Quaranta V. Normalizing DNA microarray data. Curr Issues Mol Biology. 2002;4:57–64. [PubMed] [Google Scholar]
- Kepler TB, Crosby L, Morgan KT. Normalization and analysis of DNA microarray data by self-consistency and local regression. Genome Biology. 2002;3:RESEARCH0037. doi: 10.1186/gb-2002-3-7-research0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Lu J, Lee R, Gu Z, Clarke R. Iterative normalization of cDNA microarray data. IEEE Trans Inf Technol Biomed. 2002;6:29–37. doi: 10.1109/4233.992159. [DOI] [PubMed] [Google Scholar]
- Workman C, Jensen LJ, Jarmer H, Berka R, Gautier L, Nielsen HB, Saxild HH, Nielsen C, Brunak S, Knudsen S. A new non-linear normalization method for reducing variability in DNA microarray experiments. Genome Biology. 2002;3:1–16. doi: 10.1186/gb-2002-3-9-research0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YJ, Kodell R, Sistare F, Thompson KL, Morris S, Chen JJ. Normalization methods for analysis of microarray gene-expression data. Journal of Biopharmaceutical Statistics. 2003;13:57–74. doi: 10.1081/BIP-120017726. [DOI] [PubMed] [Google Scholar]
- Edwards D. Non-linear normalization and background correction in one-channel cDNA microarray studies. Bioinformatics. 2003;19:825–833. doi: 10.1093/bioinformatics/btg083. [DOI] [PubMed] [Google Scholar]
- Kerr MK, Martin M, Churchill GA. Analysis of variance for gene expression microarray data. Journal of Computational Biology. 2000;7:819–837. doi: 10.1089/10665270050514954. [DOI] [PubMed] [Google Scholar]
- Wolfinger RD, Gibson G, Wolfinger ED, Bennett L, Hamadeh H, Bushel P, Afshari C, Paules RS. Assessing gene significance from cDNA microarray expression data via mixed models. Journal of Computational Biology. 2001;8:625–637. doi: 10.1089/106652701753307520. [DOI] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP. Variance and bias to compare normalization methods for high density oligonucleotide array data. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- Tseng GC, Oh MK, Rohlin L, Liao JC, Wong WH. in cDNA microarray analysis: quality filtering, channel normalization, models of variations and assessment of gene effects. Nucleic Acids Research. 2001;29:2549–2557. doi: 10.1093/nar/29.12.2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park T, Yi SG, Lee S, Lee SY, Yoo DH, Ahn JI, Lee YS. Statistical tests for identifying differentially expressed genes in time-course microarray experiments. Bioinformatics. 2003;19:694–703. doi: 10.1093/bioinformatics/btg068. [DOI] [PubMed] [Google Scholar]
- Kim JH, Kim HY, Lee YS. A novel method using edge detection for signal extraction from cDNA microarray image analysis. Experimental and Molecular Medicine. 2001;33:83–88. doi: 10.1038/emm.2001.15. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Gwadry FG, Reinhold WC, Miller LD, Smith LH, Scherf U, Liu ET, Kohn KW, Pommier Y, Weinstein JN. Transcriptional Regulation of Mitotic Genes by Camptothecin-induced DNA Damage: Microarray Analysis of Dose and Time-dependent Effects. CANCER RESEARCH. 2002;62:1688–1695. [PubMed] [Google Scholar]
- Colantuoni C, Henry G, Zeger S, Pevsner J. SNOMAD (Standardization and Normalization of MicroArray Data): Web-Accessible Tools for Gene Expression Data Analysis. Bioinformatics. 2002;18:1540–1541. doi: 10.1093/bioinformatics/18.11.1540. [DOI] [PubMed] [Google Scholar]