

Abstract
Background
A potential benefit of profiling of tissue samples using microarrays is the generation of molecular fingerprints that will define subtypes of disease. Hierarchical clustering has been the primary analytical tool used to define disease subtypes from microarray experiments in cancer settings. Assessing cluster reliability poses a major complication in analyzing output from clustering procedures. While most work has focused on estimating the number of clusters in a dataset, the question of stability of individual-level clusters has not been addressed.
Results
We address this problem by developing cluster stability scores using subsampling techniques. These scores exploit the redundancy in biologically discriminatory information on the chip. Our approach is generic and can be used with any clustering method. We propose procedures for calculating cluster stability scores for situations involving both known and unknown numbers of clusters. We also develop cluster-size adjusted stability scores. The method is illustrated by application to data three cancer studies; one involving childhood cancers, the second involving B-cell lymphoma, and the final is from a malignant melanoma study.
Availability
Code implementing the proposed analytic method can be obtained at the second author's website.
Background
Due to the advent of high-throughput microarray technology, scientists have conducted global molecular profiling studies in cancer [1-3]. One of the scientific goals of these experiments is the discovery of disease subtypes defined by the gene expression data that are more predictive of clinical outcomes (disease recurrence, survival, disease-free survival, etc.) than usual clinical correlates. Development of such a molecular classification system can potentially lead to more tailored therapies for patients as well as better diagnostic procedures.
Hierarchical clustering has been an important tool in the discovery of disease subtypes in microarray data [4]. Such procedures typically output a dendrogram that groups samples. Determining the reliability of clustering procedures poses a major problem in the interpretation and analysis of microarray data.
One important related question is estimating the true number of clusters in a dataset so that clusters which arise due to random chance can be separated from those which represent "true" clusters. The null hypothesis that is being tested here is that of no structure in the data. This is often referred to as a global hypothesis of clustering. Several methods have addressed this issue: these include the proposals of Hartigan [5], Krzanowski and Lai [6], Tibshirani et al. [7], Ben-Hur et al. [8] and Dudoit and Fridlyand [9]. In addition, there have been alternative clustering methodologies developed for microarray data [10,11]. Still more work has been done on assessing the validity of a clustering procedure based on the jackknife [12] and bootstrap methods [13].
A second hypothesis of interest in clustering problems is testing to determine if particular clusters found represent reliable clusters. In contrast to the global test of clustering described in the previous paragraph, inference regarding particular clusters is local in nature. There has been some recent work focused on assessing the local reliability of clusters [14,15]. While the global and local hypotheses involve clustering are different, it is obvious that the particular clusters found depend on the number of clusters one determines to be in the dataset.
In most microarray studies, the number of samples profiled is much smaller than the number of genes and ESTs represented on the chip. Due to the number of elements spotted on the microarray, it is reasonable to assume that there is redundant information available on them. Consequently, if we cluster samples based on a subset of the spots on the microarray, stable clusters should be replicated on average. This statement heuristically describes our approach to assessing the reliability of clustering analyses of microarray data. It involves performing sensitivity analyses using random subspace methods. The approach is relatively generic and can be applied to any clustering algorithm. We will focus primarily on hierarchical clustering since that is the technique used most often in the analysis of microarray data. While we are primarily interested in clustering samples, these methods can be utilized for clustering genes as well. These techniques have been examined for supervised learning problems [16]; their application to clustering techniques appears to be novel. The issue addressed in this paper is separate from estimating the number of clusters in a dataset. However, the two problems are related; in particular, the sensitivity measures we develop depend on the number of clusters. In System and Methods, we describe the data used, outline hierarchical clustering and summarize the procedure of Ben-Hur et al. [8] for estimating the number of clusters. Two approaches are taken in this paper. For the first, we assume that the number of clusters is known; sensitivity measures using random subspace methods are calculated. In the second situation, the number of clusters is unknown. We address this problem by proposing a two-stage procedure in which the number of clusters is estimated at the first stage and sensitivity measures are calculated at the second. These techniques are described in Systems and methods and compared with the methods of McShane et al. [14] and Tibshirani et al. [15]. We have programmed our procedures in the R language; in Implementation, we discuss the software. We use these methods to re-analyze three publicly available datasets in the literature: a childhood cancer study [3], a B-lymphoma study [2], and a cutaneous melanoma study [1]. These analyses are summarized in Results. Finally, in Discussion, we make some concluding remarks.
Systems and methods
Data and clustering procedures
We will let x1, ..., xn denote the p-dimensional vectors of gene expression profiles; n is the number of samples profiled. In what follows, we assume that the data have been preprocessed and normalized. Thus, our procedures work with both oligonucleotide and cDNA microarrays.
Since we will be primarily applying our methods to hierarchical clustering procedures, we briefly summarize the method here.
Hierarchical clustering
To implement the standard method for the analysis of gene expression data from microarray experiments, one first constructs a similarity measure for each pair of objects. Some examples are given in Table 1. Clustering is based on a pairwise distance matrix between objects, where distance is defined to be one minus the association measure.
Table 1.
Distance measures used for hierarchical cluster analysis
| Name | d(xi,xj) |
| Euclidean | |
| Manhattan | |
| Canberra | |
| Maximum | max1≤k≤p|xik-xjk| |
Hierarchical clustering methods fall into two classes: agglomerative nesting methods and divisive analysis methods [17]. Agglomerative nesting algorithms proceed in the same general manner: begin with n singleton clusters; the closest pair of distinct clusters is found and merged, leaving (n - 1) singleton clusters and one cluster with two distinct objects; the dissimilarity matrix is updated to take into account the merging that has occurred; based on the new dissimilarity matrix, the two closest distinct clusters are found and merged; iterate until one cluster consisting of all n objects remains.
The opposite to agglomerative nesting is a divisive analysis approach. Heuristically, the algorithm begins with one cluster of n objects. The object in the cluster that has the greatest dissimilarity to the other elements (the seed) is then separated to form a so-called splinter group and the remaining elements in the original cluster are examined to see whether or not additional elements should be added to the splinter group. Two clusters result. The diameter of each cluster (the largest distance between observations in the same cluster) is then computed to see which one is greater. The steps above are repeated with the cluster that has the greater diameter. Iterate until there are n singleton clusters. The distance for separate clusters can be defined based on average linkage or one of the other methods described above.
The algorithms described are a fraction of the available methods for clustering gene expression data. Other techniques that have been used include self-organizing maps, minimal spanning trees, spectral analysis and k-means clustering. While the methods described in this paper can be used with any of these clustering procedures, we focus on hierarchical clustering due to its popularity and to facilitate comparisons with other proposals.
Estimating number of clusters
In the Algorithm section, we discuss a two-stage procedure for performing sensitivity analysis of clustering output when the number of clusters is not fixed a priori. The method involves estimating the number of clusters at the first stage and then computing random subspace-based sensitivity measures at the second stage. We looked at the literature for the various proposals of estimating the number of clusters. Based on our experience with real datasets, the best performance seemed to be given by the method of Ben-Hur et al. [8]. We now briefly describe their procedure. It should be pointed out that our approach is relatively generic and that any method for estimating the number of clusters can be used in the first stage.
In the approach of Ben-Hur et al. [8], the samples are partitioned into k clusters. We then rerun the clustering algorithm based on the subsampling a fraction of the samples and group the subsamples into k clusters. We then compute a similarity index of the subsamples, the correlation coefficient between the clusters for the resampled data with those for the original data based on the definition given by Fowlkes and Mallows [18]. We repeat this several times to get a histogram of correlation coefficient values. We then vary k and redo the procedure.
Algorithm
Random subspace methods for known number of clusters
In this section, we assume that the number of clusters is known to be some number, say K. Thus, the samples {1,2, ..., n} are partitioned into K sets A1, ..., AK. To apply the random subspace, we randomly choose a subset D of the indices {1,2, ..., p}, where the cardinality of D is d; We the choice of d is discussed later. We then create a new dataset 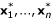 , where
, where 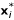 is the d-dimensional subvector of xi (i = 1, ..., n). We create a new dissimilarity matrix based on the , i = 1, ..., n and rerun the hierarchical clustering procedure. The resulting dendrogram is cut into K clusters,
is the d-dimensional subvector of xi (i = 1, ..., n). We create a new dissimilarity matrix based on the , i = 1, ..., n and rerun the hierarchical clustering procedure. The resulting dendrogram is cut into K clusters, 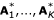 . We then check to see if Ai ⊂
. We then check to see if Ai ⊂ 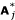 for i, j = 1, ..., K. The random subspace selection is repeated B times. For each of the original sets A1, ..., AK, we compute the proportion of B samples in which the set appears. This is our sensitivity measure. If the value is close to 1, then this evidence that the cluster is stable. On the other hand, if the proportion is small, then this provides less evidence of the stability of the cluster.
for i, j = 1, ..., K. The random subspace selection is repeated B times. For each of the original sets A1, ..., AK, we compute the proportion of B samples in which the set appears. This is our sensitivity measure. If the value is close to 1, then this evidence that the cluster is stable. On the other hand, if the proportion is small, then this provides less evidence of the stability of the cluster.
These sensitivity measures will depend on the choice of d. Larger values of d tend to yield larger sensitivity measures while the converse holds for small d. Our experience has been to choose d to be within between .75 and .85 times p.
While we have presented the procedure from a purely algorithmic point of view, there is some theoretical justification for our procedure. Since we are computing proportions based on B random subsets of (1, ..., p), the sensitivity measure can be thought as a probabilistic quantity that is averaged over B models. This provides an analogue of stacking or combining models [19,20] for unsupervised learning. It might be also possible to calculate sensitivity measures that average both over d as well as over subsets of (1, ..., p), but we will not pursue that here.
It is obvious that the criteria Ai ⊂ (i, j = 1, ..., K) will favor smaller clusters. We will also calculate a size-adjusted cluster stability score. If Pi represents cluster stability score for the ith cluster (i = 1, ..., K), then the size-adjusted score is 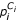 , where Ci = 1/(ln|Ai| + 1), |Ai| is the size of the ith cluster, and ln(x) represents the natural logarithm of x. For two given clusters that have the same unadjusted cluster stability score, the adjusted cluster stability score will be greater for that with the larger number of clusters.
, where Ci = 1/(ln|Ai| + 1), |Ai| is the size of the ith cluster, and ln(x) represents the natural logarithm of x. For two given clusters that have the same unadjusted cluster stability score, the adjusted cluster stability score will be greater for that with the larger number of clusters.
Random subspace methods for unknown number of clusters
Having developed a method for using random subspace techniques in, we can summarize our method when the number of clusters is not known a priori by the following two-stage method. First, we estimate the number of clusters at the first stage using the method of Ben-Hur et al. (2002). Next, conditional on the number of clusters estimated at the first stage, use the random subspace method developed in the previous section for calculating the sensitivity measures of the K* clusters.
Comparisons with other proposals
Two other techniques for assessing the reliability of individual clusters are R-index procedure of McShane et al. [14] and the cluster scoring method of Tibshirani et al. [15]. We now compare and contrast our method with these two works.
A recent paper by McShane et al. [14] describes the application of the R-index [18] for inference regarding the local hypothesis of clustering. Note that we use the R-index for addressing the problem of number of clusters, which is the global hypothesis of clustering. The authors create new datasets based on adding independent normal random errors to the original dataset and then determine the proportion of pairs of specimens within the original cluster for which the members of pairs stay together in the re-clustered perturbed dataset. While the method bears some relationships with ours, there are several operational differences. First, their method requires adding independent noise to the original data. By contrast, our method involves subsampling genes from the expression profiles. Second, while they use the overall experimental variance for data perturbation, this choice is relatively ad hoc. Our method requires no specification of added error variance. Furthermore, the added noise in their procedure is independent across genes, which is not a biologically plausible assumption. Our procedure avoids such independence assumptions.
In the method of Tibshirani et al. [15], a hierarchical clustering is performed on the genes. The average gene expression profile in each cluster is associated with a clinical response. A set of winning clusters is then found, and permutation methods are used to assess the reliability of the winning clusters. One fundamental difference between our method and that of Tibshirani et al. [15] is that their method requires a clinical outcome. Their goals is to correlate gene expression patterns with a response, and a by-product of their procedure is a score associating each cluster with the clinical response. Our procedure, by contrast, does not require a clinical response and can be used on the gene expression data only.
Implementation
We have written macros in R for implementing the methods we have proposed for genes and samples. They are obtainable from the second author's website at the following URL: http://www.sph.umich.edu/~ghoshd/COMPBIO/.
Results
We now discuss the application of the proposed methodology to three microarray datasets: one from a childhood cancer study [3], one from a lymphoma study [1] and the final is from a cutaneous melanoma study [2].
For each dataset, the Ben-Hur et al. [8] algorithm was applied to hierarchical cluster solutions obtained using average and complete-linkage upon standardization of gene expression values. At each iteration of the algorithm, two data subsets were created by randomly selecting 65% of the available samples. Correlations between the cluster designations for the members of each subset pair were calculated using the Jaccard co-efficient. For each cluster number, k, considered, 100 correlations were computed and the distribution of correlation coefficients was mapped. The distributions obtained for various cluster numbers were compared to determine the best estimate of the true number of clusters. In instances for which the true number of clusters was not obvious, both visual inspection of the original dendrogram and examination of the result obtained using the other linkage method for that dataset were considered.
After estimating the true number of clusters, we then calculated cluster stability scores using d = 0.85 p, 0.75 p, 0.5 p and 0.25 p, where p is the number of genes. For each setting, B = 100 cluster trials were performed. Both unadjusted and cluster size-adjusted scores were calculated.
In the Khan dataset, gene expression values were measured for p = 2308 genes on a total of n = 89 subjects. For these data, application of the Ben-Hur et al. [8] algorithm in addition to other methods described above resulted in estimates of five and seven for the number of clusters in the average- and complete-linkage solutions, respectively. To account for the presence of three singletons, the dendrogram for the average-linkage solution was cut at k = 8 to ensure five non-singleton clusters. The results of random gene subset clustering using average-linkage are shown in Table 2. Similarly, results from the analysis of the complete-linkage solution are presented in Table 3. The average linkage clustering method finds a cluster of 66 childhood cancers which contains cancers of different sites of origins, so it is not very meaningful clinically. Similarly, the clustering results from the complete linkage analyses do not suggest the presence of any meaningful clusters, although the cluster of seven samples with a high stability score are from the same tumor type (Ewing sarcoma).
Table 2.
Cluster stability scores for Khan et al. [3] data
| Cluster | |||||
| Gene % | 1 | 2 | 3 | 4 | 5 |
| 85 | 0.12 (0.66) | 0.63 (0.82) | 0.29 (0.66) | 0.87 (0.95) | 1.00 (1.00) |
| 75 | 0.07 (0.59) | 0.56 (0.78) | 0.23 (0.61) | 0.86 (0.95) | 1.00 (1.00) |
| 50 | 0.03 (0.51) | 0.31 (0.61) | 0.07 (0.41) | 0.85 (0.95) | 0.97 (0.98) |
| 25 | 0.00 (0.00) | 0.10 (0.38) | 0.03 (0.30) | 0.58 (0.83) | 0.88 (0.93) |
Note: Average linkage hierarchical clustering used here. The sizes of clusters 1–5 are 66,4,7,7 and 2, respectively. Gene % represents percentage (out of 100) of p = 2308 genes used for calculating cluster stability scores. Numbers in parentheses represent cluster size-adjusted stability scores.
Table 3.
Cluster stability scores for Khan et al. [3] data
| Cluster | |||||||
| Gene % | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
| 85 | 0.63 (0.89) | 0.53 (0.83) | 0.04 (0.43) | 0.79 (0.92) | 0.15 (0.64) | 0.67 (0.87) | 0.62 (0.7) |
| 75 | 0.61 (0.88) | 0.42 (0.77) | 0.02 (0.37) | 0.71 (0.88) | 0.04 (0.47) | 0.64 (0.86) | 0.60 (0.7) |
| 50 | 0.17 (0.64) | 0.05 (0.41) | 0.00 (0.00) | 0.31 (0.66) | 0.01 (0.33) | 0.36 (0.71) | 0.69 (0.8) |
| 25 | 0.06 (0.49) | 0.01 (0.26) | 0.00 (0.00) | 0.14 (0.49) | 0.00 (0.00) | 0.21 (0.59) | 0.47 (0.6) |
Note: Complete linkage hierarchical clustering used here. The sizes of clusters 1–7 are 19, 11, 18, 6, 26, 7 and 2, respectively. Gene % represents percentage of p = 2308 genes used for calculating cluster stability scores. See note to Table 2.
In the Alizadeh dataset, data were available on n = 96 samples for whom gene expression values on p = 4026 different genes were measured. Application of the Ben-Hur et al. [8] methodology to the average-linkage solution suggested the presence of eight true clusters in the data. A similar estimate was assumed for the complete-linkage solution since no conclusive result was obtained. In both instances, the dendrograms were cut at larger values of k to account for the presence of singletons (eight and five for the average and complete-linkage results, respectively). Tables 4 and 5 display the cluster stability scores for the average and complete linkage analyses, respectively. The cluster 2 found in both analyses is the same or contains the cluster of diffuse large B-cell lymphoma identified by Alizadeh et al. [1].
Table 4.
Cluster stability scores for Alizadeh et al. [1] data
| Cluster | ||||||||
| Gene % | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| 85 | 1.00 (1.00) | 0.19 (0.70) | 1.00 (1.00) | 0.39 (0.64) | 0.42 (0.75) | 1.00 (1.00) | 0.99 (0.99) | 1.00 (1.00) |
| 75 | 1.00 (1.00) | 0.12 (0.70) | 0.99 (1.00) | 0.35 (0.61) | 0.44 (0.76) | 1.00 (1.00) | 0.92 (0.95) | 1.00 (1.00) |
| 50 | 0.97 (0.99) | 0.11 (0.62) | 0.95 (0.99) | 0.28 (0.55) | 0.34 (0.69) | 1.00 (1.00) | 0.73 (0.83) | 0.84 (0.90) |
| 25 | 0.90 (0.95) | 0.02 (0.43) | 0.77 (0.94) | 0.08 (0.30) | 0.37 (0.71) | 1.00 (1.00) | 0.41 (0.59) | 0.63 (0.76) |
Note: Average linkage hierarchical clustering used here. The sizes of clusters 1–8 are 3, 40, 26, 3, 7, 5, 2 and 2, respectively. Gene % represents percentage of p = 4026 genes used for calculating cluster stability scores. See note to Table 2.
Table 5.
Cluster stability scores for Alizadeh et al. [1] data
| Cluster | ||||||||
| Gene % | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| 85 | 0.98 (0.99) | 0.19 (0.70) | 0.98 (0.99) | 0.72 (0.87) | 0.99 (1.00) | 1.00 (1.00) | 1.00 (1.00) | 1.00 (1.00) |
| 75 | 0.89 (0.96) | 0.10 (0.61) | 0.95 (0.99) | 0.57 (0.79) | 0.92 (0.96) | 0.98 (0.99) | 1.00 (1.00) | 0.98 (0.99) |
| 50 | 0.62 (0.86) | 0.08 (0.59) | 0.71 (0.90) | 0.36 (0.65) | 0.75 (0.87) | 0.82 (0.93) | 0.97 (0.99) | 0.88 (0.97) |
| 25 | 0.35 (0.71) | 0.03 (0.48) | 0.49 (0.81) | 0.13 (0.43) | 0.53 (0.74) | 0.66 (0.86) | 0.82 (0.91) | 0.72 (0.92) |
Note: Complete linkage hierarchical clustering used here. The sizes of clusters 1–8 are 8, 41, 11, 4, 3, 6, 3 and 15, respectively. Gene % represents percentage of p = 4026 genes used for calculating cluster stability scores. See note to Table 2.
The Bittner dataset contained data on n = 31 samples for whom gene expression measurements on p = 3613 genes were used. Application of the Ben-Hur et al. (2002) method in conjunction with visual inspection yielded an estimate of four true clusters for both the complete-linkage clustering solution. This estimate was also used for the average-linkage solution since no conclusive result was obtained. To account for the presence of singletons, dendrograms were cut at k = 5 and k = 8, respectively. Results of random gene subset clustering for both solutions are presented in Tables 6 and 7. While the average linkage results suggest that the melanoma cluster of Bittner et al. [2] should be expanded to include two samples (Cluster 1 in Table 7), this cluster is not found using complete linkage. In addition, the stability of the cluster drops off with decreasing numbers of genes.
Table 6.
Cluster stability scores for Bittner et al. [2] data
| Cluster | ||||
| Gene % | 1 | 2 | 3 | 4 |
| 85 | 0.09 (0.48) | 0.98 (0.99) | 0.09 (0.49) | 0.52 (0.73) |
| 75 | 0.03 (0.35) | 0.90 (0.96) | 0.04 (0.39) | 0.47 (0.70) |
| 50 | 0.03 (0.35) | 0.71 (0.88) | 0.03 (0.36) | 0.34 (0.60) |
| 25 | 0.00 (0.00) | 0.48 (0.77) | 0.01 (0.26) | 0.28 (0.55) |
Note: Complete linkage hierarchical clustering used here. The sizes of clusters 1–4 are 10, 6, 11, and 3, respectively. Gene % represents percentage of p = 3613 genes used for calculating cluster stability scores. See note to Table 2.
Table 7.
Cluster stability scores for Bittner et al. [2] data
| Cluster | ||||
| Gene % | 1 | 2 | 3 | 4 |
| 85 | 0.47 (0.83) | 1.00 (1.00) | 0.29 (0.28) | 0.16 (0.34) |
| 75 | 0.36 (0.78) | 1.00 (1.00) | 0.34 (0.53) | 0.09 (0.24) |
| 50 | 0.14 (0.62) | 0.98 (0.99) | 0.44 (0.62) | 0.06 (0.19) |
| 25 | 0.07 (0.52) | 0.87 (0.90) | 0.33 (0.52) | 0.05 (0.17) |
Note: Average linkage hierarchical clustering used here. The sizes of clusters 1–4 are 21, 2, 2, and 2, respectively. Gene % represents percentage of p = 3613 genes used for calculating cluster stability scores. See note to Table 2.
Discussion
In this paper, we have developed an approach to statistical validation of clustering results based on subsampling methods. One of the advantages of this approach is that it exploits the fact that in microarray experiments, the number of spots on the chip is greater than the number of samples profiled. By subsampling the spots on the chip, we are able to determine which clusters are relatively stable on average. It is important to note that an assumption being made is that there is sufficient correlation on the spots with respect to discriminating between clustered samples. For example, if only one gene on a 10 K chip discriminates two cancer subtypes, then the approach described here might give misleading results. However, given the fact that cancer is a complex trait, it is highly unlikely that all discriminatory information will be available in one gene.
Based on the cluster stability score method, we revisited several datasets from cancer studies to explore the stability of clustered samples. The main point of the analyses was to demonstrate the ability of our method to provide a measure of stability for the clusters that were found. In certain cases, the analyses helped confirm was what found in the previous analyses, while in other cases, they led to clinically nonmeaningful results. These results demonstrate the potential pitfalls of clustering analyses [21].
In many cancer studies, there are additional clinical covariates (e.g., survival time, PSA recurrence) available. One potential method of more formal biological validation is to combine the clustering methodology with correlation of the subsequent output to these covariates. Such an approach was taken in Tibshirani et al. [15]. Due to the variability in gene expression data, it may be potentially desirable to incorporate clinical knowledge into such clustering analyses.
Authors' Contributions
MS carried out the statistical analysis and computer implementation of the proposed study; DG conceived of the study, and participated in its design and coordination. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgments
This work has been supported by a MUNN Idea Grant and Prostate SPORE Seed Grant from the University of Michigan, as well as a Bioinformatics Pilot Award from the University of Michigan and Pfizer.
Contributor Information
Mark Smolkin, Email: Marksmolkin@hotmail.com.
Debashis Ghosh, Email: ghoshd@umich.edu.
References
- Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, Powell JI, Yang L, Marti GE, Moore T, Hudson J, Lu L, Lewis DB, Tibshirani R, Sherlock G, Chan WC, Greiner TC, Weisenburger DD, Armitage JO, Warnke R, Levy R, Wilson W, Grever MR, Byrd JC, Botstein D, Brown PO, Staudt LM. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- Bittner M, Meltzer P, Chen Y, Jiang Y, Seftor E, Hendrix M, Radmacher M, Simon R, Yakhini Z, Ben-Dor A, Sampas N, Dougherty E, Wang E, Marincola F, Gooden C, Lueders J, Glatfelter A, Pollock P, Carpten J, Gillanders E, Leja D, Dietrich K, Beaudry C, Berens M, Alberts D, Sondak V. Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature. 2000;406:536–540. doi: 10.1038/35020115. [DOI] [PubMed] [Google Scholar]
- Khan J, Wei JS, Ringner M, Saal LH, Ladanyi M, Westermann F, Berthold F, Schwab M, Antonescu CR, Peterson C, Meltzer PS. Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nat Med. 2001;7:673–679. doi: 10.1038/89044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartigan J. Clustering Algorithms. New York: Wiley. 1975.
- Krzanowski WJ, Lai YT. A criterion for determining the number of groups in a data set using sum of squares clustering. Biometrics. 1985;44:23–34. [Google Scholar]
- Tibshirani R, Walter G, Hastie T. Estimating the number of clusters in a data set via the gap statistic. J R Stat Soc B. 2001;63:411–423. doi: 10.1111/1467-9868.00293. [DOI] [Google Scholar]
- Ben-Hur A, Elisseeff A, Guyon I. A stability-based method for discovering structure in clustered data. Pac Symp Biocomput. 2002:6–17. [PubMed] [Google Scholar]
- Dudoit S, Fridlyand J. A prediction-based resampling method to estimate the number of clusters in a dataset. Genome Biology. 2002;3:RESEARCH0036. doi: 10.1186/gb-2002-3-7-research0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getz G, Levine E, Domany E. Coupled two-way clustering analysis of gene expression data. Proc Natl Acad Sci. 2000;97:12079–12084. doi: 10.1073/pnas.210134797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Dor A, Shamir R, Yakhini Z. Clustering gene expression patterns. J Comput Biol. 1999;6:281–297. doi: 10.1089/106652799318274. [DOI] [PubMed] [Google Scholar]
- Yeung KY, Haynor DR, Ruzzo WL. Validating clustering for gene expression data. Bioinformatics. 2001;17:309–318. doi: 10.1093/bioinformatics/17.4.309. [DOI] [PubMed] [Google Scholar]
- Kerr MK, Churchill GA. Bootstrapping cluster analysis: assessing the reliability of conclusions from microarray experiments. Proc Natl Acad Sci. 2001;98:8961–8965. doi: 10.1073/pnas.161273698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McShane LM, Radmacher MD, Friedlin B, Yu R, Li MC, Simon R. Methods for assessing reproducibility of clustering patterns observed in analyses of microarray data. Bioinformatics. 2002;18:1462–1469. doi: 10.1093/bioinformatics/18.11.1462. [DOI] [PubMed] [Google Scholar]
- Tibshirani R, Hastie T, Narasimhan B, Eisen M, Sherlock G, Brown P, Botstein D. Exploratory screening of genes and clusters from microarray experiments. Stat Sinica. 2002;12:47–59. [Google Scholar]
- Ho TK. The random subspace method for constructing decision forests. IEEE Trans Pattern Anal Mach Intell. 1998;20:832–844. doi: 10.1109/34.709601. [DOI] [Google Scholar]
- Kaufman L, Rousseeuw P. Finding Groups in Data. New York: John Wiley and Sons. 1990.
- Fowlkes EB, Mallows CL. A method for comparing two hierarchical clusterings. J Am Statist Assoc. 1983;78:553–569. [Google Scholar]
- Wolpert DH. Stacked Generalization. Neural Networks. 1992;5:241–259. [Google Scholar]
- Leblanc M, Tibshirani R. Combining estimates in regression and classification. J Am Statist Assoc. 1996;91:1641–1650. [Google Scholar]
- Goldstein D, Ghosh D, Conlon E. Statistical issues in the clustering of gene expression data. Stat Sinica. 2002;12:219–241. [Google Scholar]