

Abstract
Flowers sensu lato are short, specialized axes bearing closely aggregated sporophylls. They are typical for seed plants (spermatophytes) and are prominent in flowering plants sensu stricto (angiosperms), where they often comprise an attractive perianth. There is evidence that spermatophytes evolved from gymnosperm-like plants with a fern-like mode of reproduction called progymnosperms. It seems plausible, therefore, that the stamens/carpels and pollen sacs/nucelli of spermatophytes are homologous to fern sporophylls and sporangia, respectively. However, the exact mode and molecular basis of early seed and flower evolution is not yet known. Comparing flower developmental control genes to their homologs from lower plants that do not flower may help to clarify the issue. We have isolated and characterized MADS-box genes expressed in gametophytes and sporophytes of the fern Ceratopteris. The data indicate that at least two different MADS-box genes homologous to floral homeotic genes existed in the last common ancestor of contemporary vascular plants, some descendants of which underwent multiple duplications and diversifications and were recruited into novel developmental networks during the evolution of floral organs.
MADS-box genes encode a family of highly conserved transcription factors that play important roles in signal transduction and developmental control in plants, animals, and fungi (1–4). For example, homeotic organ identity genes belonging to the MADS-box gene family are components of complex networks of genes that sculpt the structure of dicotyledonous flowers (1, 3–7). The development of monocotyledonous flowers and of gymnospermous reproductive axes is probably under the control of orthologs of these genes (3, 4, 8–10).
MADS domain proteins, like many other eukaryotic transcription factors, have a modular structural organization (2). It is very similar in almost all known seed plant MADS domain proteins, including MADS (M-), intervening (I-), keratin-like (K-), and C-terminal (C-) domains (4, 11, 12).
The M-domain is by far the most highly conserved region of the proteins (11). In most cases, it is found at the N terminus of the putative proteins although some plant proteins contain additional residues N-terminal to the M-domain. The M-domain is the major determinant of DNA binding, but it also performs dimerization and accessory factor binding functions (2). The I-domain, directly downstream of the M-domain, comprises ≈30 amino acids but is somewhat variable in length (12). It is only relatively weakly conserved among plant M-domain proteins (11). In some Arabidopsis M-domain proteins, it has been shown that the I-domain constitutes a key molecular determinant for the selective formation of DNA-binding dimers (13). The K-domain, which is not present in any of the animal or fungal M-domain proteins known so far (3, 4), is characterized by a conserved, regular spacing of hydrophobic residues (Fig. 1), which is proposed to allow for the formation of an amphipathic helix. It is assumed that such an amphipathic helix interacts with that of another K-domain-containing protein, thus promoting dimerization (2, 7). The most variable region, both in sequence and length, is the C-domain at the C terminus of the M-domain proteins. The function of that domain is unknown, and it has turned out to be dispensable for DNA binding and protein dimerization in the case of some floral, homeotic M-domain proteins (e.g., see ref. 15).
Figure 1.

Sequence comparison among M-domain proteins from plants, indicating the conserved domain structures. Conceptual amino acid sequences of MADS-box genes from Ceratopteris (CRM) were aligned to one representative of each of the different subfamilies of M-domain proteins from plants that have been identified up to now (4). A “<” sign at the beginning of some sequences indicates that they are incomplete at the N terminus because of the cloning procedures used. The M-, I-, K-, and C-domains are indicated. In the region of the K-domain, hydrophobic amino acids (L, I, V, and M) are shown in bold, and positions in which more than 75% of the sequences have a hydrophobic residue are marked by a star. The complete names of the fern genes are CRM1;Ce.ric, CRM3;Ce.ric, CRM2;Ce.pte, CRM4;Ce.pte, CRM5;Ce.pte, CRM6;Ce.pte, and CRM7;Ce.pte, which indicate their origin from different Ceratopteris species (richardii and pteroides, respectively) in accordance with recent guidelines to naming sequenced plant genes (14).
It is a corollary of evolutionary developmental biology that the same genes that control the development of organs also played important roles during the evolution of these organs (3). Thus, the establishment of floral homeotic genes and changes in their regulation or function may have contributed to the establishment and structural evolution of flowers (3). Therefore, the phylogeny of MADS-box genes is of central evolutionary interest. Previous reconstructions of the phylogeny of the MADS-box gene family revealed the existence of defined gene subfamilies, indicated by the appearance of distinct subtrees (clades) of phylogenetic trees (4, 11, 16). Most subfamily members share highly related functions, e.g. as floral meristem or organ identity genes, suggesting that the emergence of the respective gene subfamilies may have played a pivotal role in the evolution of several complex eukaryotic body structures, such as flowers (3, 4, 16). Thus, the question arises as to when these plant-specific subfamilies arose during evolution and what functions they exerted before they became the “molecular architects of the flower.”
Clarifying the origin of the genes controlling flower development has been hampered by the fact that relatives of these genes from nonseed plants have not been reported up to now. For the first isolation of such genes, we chose the homosporous fern Ceratopteris because of its advantages as a plant model system (17, 18) and the relative close relationship between pteridophytes and seed plants among land plants. Like all other ferns, Ceratopteris does not form ovules or seeds and does not aggregate its sporophylls into flower-like structures. These properties are primitive characteristics with respect to vascular plants as a whole (19). Here we show that ferns contain a considerable number of MADS-box genes. The conceptual proteins of the genes that have been analyzed so far have domain structures that are very similar to those of seed plant M-domain proteins. However, the respective genes are expressed in the haploid as well as the diploid phase of the fern life cycle, which is an unusual feature compared with seed plant MADS-box genes. In line with this, phylogeny reconstructions revealed that these genes are first representatives of new subfamilies rather than members of any of the MADS-box gene subfamilies known from seed plants. The phylogenetic implications of these findings are discussed.
MATERIALS AND METHODS
Isolation of cDNA.
CRM1 and CRM3 cDNA were isolated by heterologous screening of a cDNA phage library representing gametophytic mRNA of the Ceratopteris richardii diploid, wild-type, inbred strain Hnn (20). Radioactively labeled MADS-box gene cDNA of the Antirrhinum majus homeotic genes SQUAMOSA, DEFICIENS, GLOBOSA, and PLENA (1, 4) were used as hybridization probes. CRM2, CRM4–7, and OPM1 partial cDNA were isolated by 3′-rapid amplification of cDNA ends (RACE) (21), using different oligonucleotides (sequences available upon request) as MADS-box-specific primers and polyA+–RNA isolated from juvenile as well as fertile fronds of either Ceratopteris pteroides or Ophioglossum pedunculosum sporophytes (obtained from C. Neinhuis, Botanical Garden of the University of Bonn, Bonn, Germany) as template. In some cases, second rounds of PCR with nested MADS-box primers were carried out with primary amplification products. RACE products were cloned into a standard plasmid vector, pGEM-T (Promega).
Southern and Northern Analyses.
Gene-specific hybridization probes for Southern and Northern analyses mainly were obtained from the region downstream of the MADS-box to avoid cross-hybridization with other gene family members even under stringent hybridization conditions. For the synthesis of probes, linear PCR was used essentially as described (22), but PCR products of CRM gene cDNA were used as templates, and different gene-specific oligonucleotides were used as primers. A MADS-box probe was obtained by amplifying the MADS-box regions of CRM1 and CRM3 by PCR.
Southern blots were prepared by standard methods (23) with 15 μg of DNA per lane, isolated from C. richardii strain Hnn, and digested with different restriction enzymes. The filters were hybridized at 68°C (high stringency) or at 60°C (moderate stringency) for 16 h in 5× SSPE [standard saline phosphate/EDTA (0.18 M NaCl/10 mM phosphate, pH 7.4/1 mM EDTA)], 5× Denhardt’s solution, 0.5% SDS, and 20 μg/ml herring sperm DNA and washed at 68°C in 0.1× SSPE/0.1% SDS (high stringency) or at 60°C in 1× SSPE/0.1% SDS (moderate stringency).
For Northern analysis, the probes were hybridized to RNA blots containing 20 μg per lane of total RNA isolated from C. richardii strain Hnn by the guanidinium chloride method (23). RNA sources were gametophytes grown on solid medium with Parker–Thompson salts (18) and harvested 11 days after spore soaking or fertile fronds of sporophytes that had been cultivated as described (18). The filters were hybridized at 42°C for 16 h in 5× SSPE, 50% formamide, 10× Denhardt’s solution, 0.5% SDS, and 100 μg/ml herring sperm DNA and washed at 68°C in 0.2× SSC/0.1% SDS (23). The blots were rehybridized using a nuclear-encoded Ceratopteris glyceraldehyde-3-phosphate dehydrogenase (GAPA) gene (J. P., W. M., unpublished data) as a control for RNA loading.
Construction of Phylogenetic Trees.
Alignment of conceptual amino acid sequences was done by the Genetics Computer Group (Madison, WI) program pileup. Phylogenetic trees were constructed as described (4), using the neighbor-joining algorithm as provided by the program neighbor of the phylogeny inference package (phylip). As data, derived amino acid sequences of all available MIKC-type genes [i.e., all known plant MADS-box genes except some transposon-like elements carrying a MADS-box (3, 4, 22); for a definition of MIKC, see Results) were used. Non-MIKC-type M-domain protein sequences were omitted in these analyses because they have no sequences outside the M-domain that can reliably be aligned to MIKC-type proteins. Sources of sequences that had not been determined in our laboratory have been described elsewhere (4), except for PRMADS1, PRMADS2, SAMADSA, and SAMADSB (GenBank database accession nos. U42399U42399, U42400U42400, U25696U25696, and U25695U25695). A 170-amino acid sequence [comprising the M-domain (60 amino acids) plus the 110 amino acids downstream of the M-domain including the I- and K-domains] was used in the analyses, or fragments of that sequence were used if the complete sequence was not available. Distances were calculated by the phylip program protdist, using the percentage accepted mutation model of amino acid transition. To test the statistical significance of the phylogenetic trees, 100 bootstrap samples were generated from each data set using the seqboot program of the phylip package. Except for minor changes at the tips and in deep branching, the tree presented in Fig. 4 is concordant with trees derived from parsimony analysis and with trees based on M-domain sequences only [constructed as described by Theissen et al. (4)], comprising also all known non-MIKC-type M-domain protein sequences (data not shown).
Figure 4.

Phylogenetic tree showing the relationships among all MIKC-type M-domain proteins known. Genus names of species from which the respective genes were isolated are given in parentheses behind the protein names. Genes of gymnosperms are indicated by shaded boxes, and those of ferns are indicated by inverted boxes. All other genes have been isolated from angiosperms (4). The numbers next to some nodes give bootstrap percentages, shown only for relevant nodes (mainly those defining gene subfamilies). Subfamilies (4) are labeled by brackets at the right margin. They generally represent monophyletic gene clades (4), except for the subfamily of TM3-like genes, which appears here, but not in other tree constructions (4), as paraphyletic to TM8.
RESULTS
Cloning and Structural Evaluation of MADS-Box Gene cDNA from Ferns.
By screening a cDNA library with heterologous hybridization probes derived from Antirrhinum MADS-box genes and by 3′-RACE cloning, seven different cDNA of Ceratopteris MADS-box genes were isolated. The proteins encoded by them were designated CRM1–CRM7 (for Ceratopteris MADS1–7; see Fig. 1). Sequence comparisons with representatives of each of the M-domain protein subfamilies known to date (4) indicated no obvious similarity of CRM gene products to animal or fungal proteins outside the M-domain (data not shown) but revealed that the products encoded by the fern genes generally have the same domain structure as M-domain proteins of seed plants (Fig. 1), including M-, I-, K-, and C-domains (4, 11, 12) (Fig. 1). The high conservation of the M-domain is also obvious for the sequences from Ceratopteris (Fig. 1), suggesting that the fern M-domains are under similar general functional constraints as those of seed plants. The presence of K-domains in all CRM proteins makes it conceivable that they interact with other K-domain-containing proteins via these regions, as has been suggested for seed plant M-domain proteins. Although the I-domain is only relatively weakly conserved among plant M-domain proteins in general (11, 12), CRM1, CRM2, CRM4, and CRM5 show high sequence similarity also in that region (Fig. 1). In the case of some Arabidopsis M-domain proteins, the I-domain constitutes a key determinant for the selective formation of DNA-binding dimers (13), so it seems likely that the similarity of CRM1, CRM2, CRM4, and CRM5 in that region reflects similarities in the dimerization specificity of these proteins. CRM1, CRM2, CRM4, and CRM5 also show considerable sequence similarity in the C-domain (Fig. 1). Unfortunately, a function has not yet been attributed to the C-domain of M-domain proteins, so the functional relevance of that sequence similarity is unknown at the moment.
The similarity between the CRM genes and most seed plant MADS-box genes with respect to M-domain sequence and overall domain structure clearly indicates that they share a common ancestor from which they were derived by gene duplications, sequence diversification, and fixation. The identified fern genes are thus clearly homologs of the MADS-type floral homeotic genes known from angiosperms. There is no indication that domain shuffling occurred within the genealogy of these genes. Genes of this type will henceforth be called MIKC-type MADS-box genes according to the four regions (M, I, K, and C) present in each of them. Using 3′-RACE, we also isolated a cDNA encoding a MIKC-type gene from Ophioglossum (sequence not shown but available under database accession no. Y08244Y08244), thereby demonstrating that MIKC-type genes are not only present in leptosporangiate ferns like Ceratopteris but also in eusporangiate ferns.
The CRM cDNA Represent Seven Different Genomic Loci.
Hybridization of Southern blots containing genomic DNA of the Hnn inbred strain of C. richardii with different probes specific for each of the CRM genes under stringent conditions gave single bands at different lengths in each case, indicating that CRM1–7 represent seven different genomic loci (genes) (Fig. 2). Using a MADS-box probe for hybridization under conditions of moderate stringency revealed the presence of more than 15 different MADS-box genes in the Ceratopteris genome (data not shown).
Figure 2.

Southern blot analysis of CRM genes in C. richardii. DNA from the inbred strain Hnn was digested with HindIII (H) or EcoRV (E) as indicated above the lanes, electrophoresed, blotted onto nylon membranes, and hybridized under stringent conditions with probes specific for the different CRM genes as depicted.
CRM1 and CRM3 Are Expressed in Both Major Phases of the Fern Life Cycle.
Northern blots revealed that CRM1 and CRM3 are transcribed in the sporophytic (diploid) as well as the gametophytic (haploid) phase of the fern life cycle (Fig. 3). This is in remarkable contrast to the situation in seed plants, in which expression of a MIKC-type gene in gametophytes has not been clearly demonstrated to date, even when gene expression was restricted to stamens, carpels, or ovules. Expression in sporophytes and gametophytes suggests a more ubiquitous function of the fern genes in the control of development or cell differentiation than the temporally and spatially quite restricted functions of the homeotic genes determining floral organ identity of angiosperms (1–7).
Figure 3.
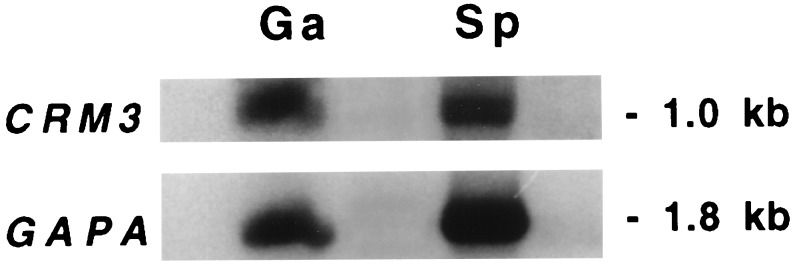
Northern blot analysis indicates that CRM3 is expressed during sporophytic and gametophytic phases of the fern life cycle. Note that the autoradiography showing CRM3 expression was exposed ≈5 times longer than the GAPA loading control. RNA sources were gametophytes (Ga) harvested 11 days after spore soaking or fertile fronds of the sporophyte (Sp) as indicated. CRM1 expression was found to be very similar to that of CRM3, albeit weaker (data not shown).
Phylogeny Reconstructions.
To determine the evolutionary relationship between the fern genes and the other known MADS-box genes, we carried out phylogeny reconstructions with all available MIKC-type genes. It turned out that the CRM genes clearly were not members of any of the distinct gene clades previously identified from seed plants (ref. 4; Fig. 4). Rather, they are first representatives of new gene subfamilies.
The gene phylogeny in Fig. 4 reveals a complex but intriguing pattern of lineage-specific increases in the number of MADS-box genes during tracheophyte evolution. With the exception of AGL12-, AGL17-, TM3-, and TM8-like genes, all angiosperm clades of MIKC-type genes characterized to date contain at least two members encoded by a single plant species, documenting continued diversification and fixation of MIKC-type genes during angiosperm evolution (4). The fact that CRM1- and CRM6-like proteins form two clades that comprise only fern proteins (Fig. 4) suggests that the gene duplications that led to the respective genes were restricted to the lineage leading to extant pteridophytes and are thus clearly distinct from those that gave rise to floral homeotic genes of angiosperms. Isolation of a CRM6-like gene (OPM1) from an eusporangiate fern (Ophioglossum), which is quite distantly related to the leptosporangiate Ceratopteris, reveals that CRM6-like genes must have been established before the separation of the lineages that led to Ophioglossaceae and leptosporangiate ferns (Fig. 4).
The three subfamilies of MIKC-type proteins (CRM1-, CRM6-, and CRM3-like sequences) from ferns are interspersed among spermatophyte sequence clades in Fig. 4; no pair out of these three subfamilies forms sister clades. However, phylogeny reconstructions based on input data that are slightly different from those used for Fig. 4 sometimes yield trees in which the CRM1- and CRM6-like proteins form sister clades (data not shown). As in earlier studies (4, 11, 16), therefore, the relationships among some of the major gene clades (i.e., the “deep branching” of the MADS-box gene tree) remain undetermined. However, in all tree reconstructions, CRM3 was never grouped together with the other fern proteins within a single clade. A conservative interpretation of all available data thus leads to the conclusion that at least two different MIKC-type genes existed in the last common ancestor of ferns and seed plants.
The evidence available to date is compatible with the assumption that few MIKC-type genes (compared with the high number of MADS-box genes in present day angiosperms and ferns) were present in the last common ancestor of ferns and seed plants because the reconstructed gene phylogeny reveals that many of the gene duplications that led to the high number of present day MIKC-type genes occurred independently in the lineages leading to ferns and seed plants.
DISCUSSION
Fig. 4 shows that there are four different clades containing MADS-box gene members from both gymnosperms and angiosperms (AG-, AGL2-, AGL6-, and TM3-like genes), indicating that at least four different MIKC-type genes existed in the last common ancestor of contemporary seed plants. Analysis of seed plant MIKC-type genes has revealed a tight linkage between the evolutionary and functional relationships of MIKC-type transcription factors, suggesting that the establishment of a number of seed plant gene clades was tightly correlated with the establishment of some spermatophytic reproductive characters (3, 4, 11, 16). For example, the members of at least three clades common to seed plants (AG-, AGL2-, and AGL6-like genes) are expressed during flower development, with AG-like genes known to determine the identity of reproductive organs, i.e., stamens and carpels (e.g., AG), or ovules (e.g., FBP11) (4, 24). The fact that, from ferns, no genes have been isolated that are within the gene clades known from seed plants might thus be correlated to the absence of seed plant-specific structures, like ovules, in these ferns.
Our results suggest that there was a relatively small pool of MIKC-type genes in the common ancestor of vascular plants, which is assumed to have existed ≈395 million years ago (MYA) (25). Descendants of that pool of genes have been recruited by gene duplication and diversification in the lineage leading to spermatophytes to give rise to floral homeotic genes. According to different estimations based on molecular data, the last common ancestor of extant seed plants existed ≈285–350 MYA (ref. 26 and W.M., V. Goremykin, and S. Hansmann, unpublished data). It is conceivable that, in the time interval before the radiation of extant seed plants but subsequent to their divergence from pteridophytes, some, if not all, clades of floral homeotic genes had been established. It was in that time interval [from Middle Devonian to Early Carboniferous (Tournaisian)] that progymnosperms existed, i.e. plants that already had gymnospermous wood but still had a pteridophytic, free-sporing mode of reproduction (19). It is intriguing to think, therefore, that, during that time, the establishment of some seed plant-specific MADS-box gene clades (e.g., AG-like genes) in progymnosperms might have been an important aspect to confer seed plant reproductive structures [like integumented, indehiscent megasporangia (i.e., ovules)] to plants that still had a pteridophytic mode of reproduction but otherwise were already gymnosperm-like. Thus, the progenitor of extant seed plants may have been established. In line with this, the oldest known seed plant (Elkinsia) has been preserved in the fossil record of that time (Late Devonian, ≈365 MYA), and different intermediate stages in the evolution of the ovule have been found in the fossil record of the Lower Carboniferous, ≈350 MYA (27).
A “crude molecular clock” estimation, although based only on the analysis of flowering plant MADS-box gene sequences, suggested that the floral homeotic genes might have been established fairly rapidly ≈340 MYA in the lineage leading to seed plants (11). Our findings clearly are compatible with that notion. During extensive cloning efforts comprising the screening of cDNA libraries of Ceratopteris gametophytes and sporophytes as well as RACE cloning and using probes and primers derived from Antirrhinum MADS-box genes, we have found no evidence to date for the existence of orthologs and functional equivalents of floral homeotic genes in Ceratopteris (data not shown). Therefore, there is currently no support for alternative estimations (11) of the age of floral homeotic genes, suggesting that orthologs of them should be present in all land plant species and even in the chlorophyte algal groups. Cloning all MADS-box genes from Ceratopteris will further help to clarify that issue. Additional work will reveal whether the absence of pteridophyte orthologs of spermatophyte floral homeotic genes causally correlates to the lack of angiosperm-specific organs in ferns.
Acknowledgments
We thank C. Neinhuis for leaf material from Ceratopteris pteroides and Ophioglossum pedunculosum, Leslie G. Hickok for spores of C. richardii strain Hnn, and Zsuzsanna Schwarz–Sommer and Hans Sommer for cDNA of Antirrhinum MADS-box genes. We also thank the Automatic DNA Isolation and Sequencing team for sequencing some of the cDNA clones.
ABBREVIATIONS
- M-domain
MADS domain
- I-domain
intervening domain
- K-domain
keratin-like domain
- C-domain
C-terminal domain
- RACE
rapid amplification of cDNA ends
- MYA
million years ago
Footnotes
Data deposition: The nucleotide sequence data of the cDNA corresponding to the protein sequences reported in this paper have been deposited in the European Molecular Biology Laboratory, GenBank, and DNA Data Base in Japan Nucleotide Sequence databases (accession nos. Y08014Y08014, Y08015Y08015, and Y08239–Y08244Y8239Y8240Y8241Y8242Y8243Y8244).
References
- 1.Schwarz-Sommer Z, Huijser P, Nacken W, Saedler H, Sommer H. Science. 1990;250:931–936. doi: 10.1126/science.250.4983.931. [DOI] [PubMed] [Google Scholar]
- 2.Shore P, Sharrocks A D. Eur J Biochem. 1995;229:1–13. doi: 10.1111/j.1432-1033.1995.tb20430.x. [DOI] [PubMed] [Google Scholar]
- 3.Theissen G, Saedler H. Curr Opin Genet Dev. 1995;5:628–639. doi: 10.1016/0959-437x(95)80032-8. [DOI] [PubMed] [Google Scholar]
- 4.Theissen G, Kim J T, Saedler H. J Mol Evol. 1996;43:484–516. doi: 10.1007/BF02337521. [DOI] [PubMed] [Google Scholar]
- 5.Coen E S, Meyerowitz E M. Nature (London) 1991;353:31–37. doi: 10.1038/353031a0. [DOI] [PubMed] [Google Scholar]
- 6.Weigel D, Meyerowitz E M. Cell. 1994;78:203–209. doi: 10.1016/0092-8674(94)90291-7. [DOI] [PubMed] [Google Scholar]
- 7.Ma H. Genes Dev. 1994;8:745–756. doi: 10.1101/gad.8.7.745. [DOI] [PubMed] [Google Scholar]
- 8.Schmidt R J, Veit B, Mandel M A, Mena M, Hake S, Yanofsky M F. Plant Cell. 1993;5:729–737. doi: 10.1105/tpc.5.7.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Theissen G, Strater T, Fischer A, Saedler H. Gene. 1995;156:155–166. doi: 10.1016/0378-1119(95)00020-7. [DOI] [PubMed] [Google Scholar]
- 10.Tandre K, Albert V A, Sundas A, Engström P. Plant Mol Biol. 1995;27:69–78. doi: 10.1007/BF00019179. [DOI] [PubMed] [Google Scholar]
- 11.Purugganan M D, Rounsley S D, Schmidt R J, Yanofsky M F. Genetics. 1995;140:345–356. doi: 10.1093/genetics/140.1.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma H, Yanofsky M F, Meyerowitz E M. Genes Dev. 1991;5:484–495. doi: 10.1101/gad.5.3.484. [DOI] [PubMed] [Google Scholar]
- 13.Riechmann J L, Krizek B A, Meyerowitz E M. Proc Natl Acad Sci USA. 1996;93:4793–4798. doi: 10.1073/pnas.93.10.4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Price C A, Reardon E M, Lonsdale D M. Plant Mol Biol. 1996;30:225–227. doi: 10.1007/BF00020109. [DOI] [PubMed] [Google Scholar]
- 15.Zachgo S, de Andrade Silva E, Motte P, Tröbner W, Saedler H, Schwarz-Sommer Z. Development. 1995;121:2861–2875. doi: 10.1242/dev.121.9.2861. [DOI] [PubMed] [Google Scholar]
- 16.Doyle J J. Syst Biol. 1994;43:307–328. [Google Scholar]
- 17.Chasan R. Plant Cell. 1992;4:113–115. doi: 10.1105/tpc.4.2.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hickok L G, Warne T R, Fribourg R S. Int J Plant Sci. 1995;156:332–345. [Google Scholar]
- 19.Beck C B, editor. Origin and Evolution of Gymnosperms. New York: Columbia Univ. Press; 1988. [Google Scholar]
- 20.Scott R, Hickok L. Am J Bot. 1987;74:1872–1877. [Google Scholar]
- 21.Frohman M A, Dush M K, Martin G R. Proc Natl Acad Sci USA. 1988;85:8998–9002. doi: 10.1073/pnas.85.23.8998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fischer A, Baum N, Saedler H, Theissen G. Nucleic Acids Res. 1995;23:1901–1911. doi: 10.1093/nar/23.11.1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 24.Colombo L, Franken J, Koetje E, van Went J, Dons H J M, Angenent G C, van Tunen A J. Plant Cell. 1995;7:1859–1868. doi: 10.1105/tpc.7.11.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stewart W N, Rothwell G W. Paleobotany and the Evolution of Plants. 2nd Ed. Cambridge, U.K.: Cambridge Univ. Press; 1993. [Google Scholar]
- 26.Savard L, Li P, Strauss S H, Chase M W, Michaud M, Bousquet J. Proc Natl Acad Sci USA. 1994;91:5163–5167. doi: 10.1073/pnas.91.11.5163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taylor T N, Taylor E L. The Biology and Evolution of Fossil Plants. Englewood Cliffs, NJ: Prentice–Hall; 1993. [Google Scholar]