

Abstract
A unique feature of Citrobacter koseri is the extremely high propensity to initiate brain abscesses during neonatal meningitis. Previous clinical reports and studies on infant rats have documented many Citrobacter-filled macrophages within the ventricles and brain abscesses. It has been hypothesized that intracellular survival and replication within macrophages may be a mechanism by which C. koseri subverts the host response and elicits chronic infection, resulting in brain abscess formation. In this study, we showed that C. koseri causes meningitis and brain abscesses in the neonatal rat model, and we utilized histology and magnetic resonance imaging technology to visualize brain abscess formation. Histology and electron microscopy (EM) revealed that macrophages (and not fibroblasts, astrocytes, oligodendrocytes, or neurons) were the primary target for long-term C. koseri infection. To better understand C. koseri pathogenesis, we have characterized the interactions of C. koseri with human macrophages. We found that C. koseri survives and replicates within macrophages in vitro and that uptake of C. koseri increases in the presence of human pooled serum in a dose-dependent manner. EM studies lend support to the hypothesis that C. koseri uses morphologically different methods of uptake to enter macrophages. FcγRI blocking experiments show that this receptor primarily facilitates the entry of opsonized C. koseri into macrophages. Further, confocal fluorescence microscopy demonstrates that C. koseri survives phagolysosomal fusion and that more than 90% of intracellular C. koseri organisms are colocalized within phagolysosomes. The ability of C. koseri to survive phagolysosome fusion and replicate within macrophages may contribute to the establishment of chronic central nervous system infection including brain abscesses.
In the United States, 30 to 40% of neonatal meningitis cases are caused by gram-negative bacteria (36, 37). Citrobacter species are gram-negative bacteria responsible for neonatal sepsis, ventriculitis, and meningitis, often causing multiple brain abscesses. Infants with Citrobacter koseri meningitis develop brain abscesses at an alarming frequency of 77%, a number far greater than that with any other pathogen causing neonatal meningitis (19, 47). Transmission to neonates occurs horizontally during nosocomial outbreaks in neonatal hospital wards (49). Also, colonized mothers may facilitate vertical transmission to their children (7, 11, 12). Immunocompromised individuals are also susceptible to Citrobacter infections (41). Symptoms of C. koseri central nervous system (CNS) infection are often nonspecific, and the difficulty of diagnosis is compounded by frequently negative diagnostic tests. Mortality results in 30 to 50% of neonates with meningitis, and 75% of the survivors have severe neurological impairment such as seizures, hearing loss, or mental retardation (36). Of greatest concern is the fact that once a brain abscess is established, it can persist with little response to antibiotic treatment (19, 47). As a result, invasive surgical intervention or multiple aspirations of the abscessed tissue are often prescribed (11, 33).
Often, brain abscesses begin as intense vasculitis, cerebritis, and/or ventriculitis. Local cerebral necrosis serves as the focal point of the abscess. Expansion occurs as infiltrating inflammatory cells accumulate and develop into a vascularized capsule earmarked by peripheral gliosis and/or fibrosis within 10 to 14 days. Kline et al. were the first to use histology to describe C. koseri (previously known as Citrobacter diversus) pathogenesis in the infant rat model (20). Histological examination of infant rat brains infected with C. koseri has shown that this model closely mimics the course of infection observed in humans (20). For example, the infection is age dependent, since 5-day-old infant rats are resistant to C. koseri meningitis, whereas 2-day-old rats are sensitive (20). Furthermore, age dependency is observed even when bacteria are inoculated by intracranial injection (38). As with clinical data, brain abscesses in neonatal rats are para- or periventricular and appear 8 days following infection (20). More recent studies have determined that Citrobacter was able to invade and replicate within human brain microvascular endothelial cells (HBMEC), which constitute the blood-brain barrier (2). Clinical and experimental data suggest that the indolent nature of the infection and the resistance of the bacterium to phagocytic killing may play a role in abscess development (11, 20).
The molecular pathogenesis of C. koseri is poorly understood and widely understudied. Chronic C. koseri infection of the neonatal rat CNS provides a unique opportunity to elucidate bacterial mechanisms necessary for blood-brain barrier penetration and evasion of the host immune response. It has been suggested that the ability of these organisms to survive and replicate within macrophages may account for their persistence and contribute to the establishment of chronic infection resulting in brain abscess formation. In this study, we sought to evaluate and characterize the ability of C. koseri to be taken up, survive, and replicate within macrophages. A better understanding of the role of intracellular survival and replication during C. koseri pathogenesis may lead to novel treatment strategies, thereby considerably reducing the incidence of brain abscess formation, a major cause of morbidity associated with this disease.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
C. koseri strains used in this study are derived from a spontaneous nalidixic acid mutant (SMT319) of strain JLB62 that was isolated from the cerebrospinal fluid (CSF) of an infant with C. koseri meningitis and brain abscess (obtained from Joseph St. Geme III, Washington University School of Medicine). Mutant strain SMT320 constitutively expresses gfp as a result of a random transposition insertion of promoterless mTn10gfp (39) downstream of a constitutive promoter on the C. koseri chromosome (unpublished data). Both SMT319 and SMT320 retain wild-type morphology, growth characteristics, and invasive phenotypes (data not shown). E44, a spontaneous rifampin-resistant mutant of the meningitic Escherichia coli K1 CSF isolate RS218 (O18:K1:H7) (48), and nonpathogenic E. coli K-12 HB101 were used as negative controls. For gentamicin protection assays, bacteria were aerobically grown at 37°C to mid-log phase in brain heart infusion broth with nalidixic acid (15 μg/ml) or rifampin (100 μg/ml) selection. For animal studies, C. koseri strain SMT319 was grown to mid-log phase and washed twice with cold phosphate-buffered saline (PBS). Concentrations of bacterial inoculations were approximated by optical density at 620 nm and confirmed by plating for CFU.
U937 macrophage cultivation and gentamicin protection assay.
U937 macrophages were obtained from the American Type Culture Collection (CRL-1593.2) and seeded into 75-ml tissue culture flasks. Cells were cultivated in RPMI 1640 medium with 2 mM l-glutamine, and the medium was modified to contain 10 mM HEPES, 1 mM sodium pyruvate, 4.5 g of glucose/liter, and 1.5 g of sodium bicarbonate/liter and was supplemented with 10% fetal bovine serum (40). At least 24 h prior to infection, cells were treated with 0.1 μg of phorbol 12-myristate 13-acetate (PMA; Sigma-Aldrich)/ml, placed in 24-well tissue culture plates, and left at 37°C under 5% CO2 to adhere and become activated. Cells were gently washed with RPMI to remove residual PMA, and fresh medium was added prior to infection. U937 human macrophages were infected at a multiplicity of infection of 10 for 45 min at 37°C under 5% CO2. After a 45-min incubation, macrophages were resuspended in U937 medium supplemented with 100 μg of gentamicin/ml and were incubated for an additional 45 min at 37°C under 5% CO2. Macrophages were then washed twice, lysed with 0.5% Triton X, serially diluted, and plated to determine the number of intracellular bacteria at various time points. Results are presented as the percentage of the inoculum that is intracellular. For extended assays (intracellular replication assays), cells were replenished with fresh medium containing 10 μg of gentamicin/ml (above the MIC). Trypan blue exclusion staining indicated that macrophage viability ranged from 80 to 95% and was maintained for at least 72 h. For each indicated time point, results are presented as percent survival of the initial intracellular population recovered at time zero. All assays were performed in triplicate at least three times.
For opsonization studies, macrophage infections occurred in the presence of human pooled serum (Omega, Inc.) at increasing concentrations (2.5, 5, 7.5, and 10%). To denature complement proteins, human pooled serum (HPS) was heat treated for 30 min at 65°C and then used to opsonize C. koseri for 30 min before infection of U937 macrophages. Unopsonized C. koseri and HPS-opsonized C. koseri were used as controls. All assays were performed in duplicate, at least three times.
To block FcγRI-mediated uptake, PMA-stimulated U937 macrophages were treated with 0, 15, or 25 μg of human monomeric anti-CD64 immunoglobulin G1 (IgG1) (clone 10.1; Santa Cruz Biotechnology, Inc.) for 30 min at 37°C under 5% CO2 prior to infection. C. koseri was opsonized with 10% HPS in PBS for 30 min at 37°C with shaking before infection.
Animal infections.
Timed pregnant (embryonic day 14 [E14]) Sprague-Dawley rats (Charles River Laboratories) were obtained and gave birth in our vivarium after a 21-day gestation period. Litters averaged 12 pups and were kept with the mother in an opaque polypropylene cage under a Small Animal Isolator (Forma Scientific, Inc.). Two-day-old rat pups were anesthetized with isoflurane and inoculated with 105 bacteria in PBS via both intraperitoneal (i.p.) (0.1 ml) and intranasal (i.n.) (0.01 ml) routes. i.n. inoculations were administered via a P-20 Pipetman with a round gel loading tip into the left nostril with the head tilted back at a 70° angle. Blood and CSF samples were collected from anesthetized rat pups at the indicated time points. Blood and CSF samples were aseptically collected via intracardiac and cisterna magna punctures, respectively, as previously described (1). CSF and blood samples (10 μl) were inoculated into Luria broth and agar plates with appropriate antibiotic selection. Rats were then euthanized, and whole brains were removed. All animal experiments were performed according to protocols approved by the Childrens Hospital Los Angeles (CHLA) Institutional Animal Care and Use Committee.
Histopathology.
Whole brains of infected neonatal rats were fixed in 10% buffered formalin, routinely processed, and then paraffin embedded. Coronal sections of 4 to 5 μm thickness were cut and stained with hematoxylin and eosin (H&E). Histological analysis was completed by one pathologist (I.G.-G.) who was blinded to the inoculum and time postinfection of each rat at the time of sacrifice. CSF samples were methanol fixed on glass slides and processed for Gram staining (Difco). Animals infected with PBS were analyzed as controls.
Magnetic resonance imaging (MRI).
Brains (fixed in 4% formalin) were imaged with a 1.5T General Electric clinical scanner, by using a custom coil with a diameter of 25 mm. Both gradient echo and three-dimensional sequences were used. The images produced were 256 by 256 by 16 voxels, with a voxel size of 0.260 by 0.260 by 0.400 mm. The brain was oriented so that the highest resolution was obtained in the coronal plane. In all cases, the images were postprocessed using Amira software (Template Graphics Software, version 2.3). The resulting images were examined and reported by a radiologist (H.A.P.).
EM.
Transmission electron microscopy (transmission EM) was used to visualize C. koseri uptake by U937 macrophages. Thirty minutes following infection, the macrophages were washed four times with RPMI and gently removed from a 75-ml tissue culture flask by using a rubber policeman. The macrophages were centrifuged for 10 min at 1,700 × g. The pellet was fixed with 2.5% glutaraldehyde in 0.1 M PBS. C. koseri-infected rat brains and CSF samples were also fixed with 2.5% glutaraldehyde in 0.1 M PBS. Samples were postfixed for 1 h with 2% OsO4, rinsed, dehydrated through graded ethanol solutions, and embedded in polypropylene oxide. Ultrathin sections mounted on collodion one-hole grids were stained with uranyl acetate and lead citrate and were examined by transmission EM with a Philips CM transmission electron microscope.
Lysosomal fusion studies.
PMA-stimulated U937 macrophage monolayers were washed and infected as described above with SMT320. A LysoTracker Red (Molecular Probes, Eugene, Oreg.) 1 mM stock solution was diluted in U937 medium to yield a working concentration of 50 nM. Vital cells were infected and stained for 30 min with LysoTracker Red according to the manufacturer's instructions. In three independent experiments, the total number of intracellular SMT320 organisms was counted and the percentage that colocalized with LysoTracker Red was determined. Confocal micrographs were acquired using a Leica (Wetzlar, Germany) TCS SP confocal DM IRBE inverted microscope (63×/1.4NA Plan-apochromat objective lens; electronic zoom, 3.1×; image width and height, 51 μm; optical slice, approximately 0.4 μm) at the CHLA Congressman Dixon Cellular Imaging Facility. An argon ion 488-nm laser line was used to excite green fluorescent protein (GFP; BD Clontech, Palo Alto, Calif.), and a krypton ion 568-nm laser line was used to excite LysoTracker Red (Molecular Probes). Images were acquired either as Z-series (step size, 0.4 μm; 4-frame average) or as single XY images (16-frame average). Leica LCS 2.968 software was used for all image acquisition and analysis in this study. Contrast adjustment was performed, and scale bars for publication were created, with Adobe (San Jose, Calif.) Photoshop (version 6.0).
RESULTS
Analysis of neonatal rat brain abscesses during C. koseri infection.
To better understand the pathogenesis of chronic C. koseri infection at the cellular level, we utilized the neonatal rat model. As previously described, 2-day-old rats were each inoculated both i.p. and i.n. with 4 × 105 CFU of C. koseri (SMT319) (20). Bacteremia occurred within 24 h (data not shown), and marked ventriculitis developed within the brain by 72 h (Fig. 1A). Brain abscesses were well established between 8 and 10 days (Fig. 1B and 2). These results are consistent with earlier work by Kline et al. establishing this animal model and the kinetics of infection (20). In addition, MRI was employed to visualize infected rat brains. As shown in Fig. 2, at day 8 postinfection, ventriculitis, ventriculomegaly, and paraventricular brain abscesses were readily visualized by use of this technique (Fig. 2).
FIG. 1.

Histological examination of C. koseri CNS infection in the neonatal rat. (A through C) H&E-stained brain sections from neonatal rats infected with C. koseri (SMT319) at the indicated days postinfection. (A) C. koseri infection infiltrating through the olfactory region (O) into the right ventricle (RV). (B) Brain abscess in the third ventricle. Box encloses area of capsule enlarged in panel C. (C) Enlarged section of panel B; abscess wall showing fibroblasts (F), vascular proliferation (V), macrophages (M), neutrophils (N), and red blood cells (R). (D) Tissue section from a brain abscess that was Gram stained by using the MacCullum-Goodpasture technique, showing extracellular C. koseri and intracellular C. koseri within macrophages and neutrophils.
FIG. 2.

MRI image of a neonatal rat brain 8 days after infection with C. koseri (SMT319), showing a severe abscess. The coronal image depicts the walled-abscess on the right side of the brain (thick arrow) and severe bilateral ventriculitis and ventriculomegaly (thin arrow). The resolution of this image is 0.011811 mm.
(i) Histological examinations.
To visualize pathogenesis prior to and during brain abscess formation, brain tissues from neonatal rats infected with C. koseri (SMT319) were processed and examined histologically 2 to 9 days postinfection. Prior to brain abscess formation (i.e., day 6), i.n. inoculation caused local infection of the olfactory region. Direct extension of the infection from the olfactory region to the ventricles resulted in meningitis, ventriculitis with loss of ependymal cells, encephalitis, and necrosis (Fig. 1A to C). At later times, 8 to 10 days postinfection, histological examinations of neonatal rat paraventricular brain abscesses showed well-developed capsules comprising fibroblasts, vascular proliferation, neutrophils, and macrophages (Fig. 1B and C). Furthermore, the presence of free and intracellular bacteria in the brain abscess was demonstrated by Gram staining (Fig. 1D). Also, Gram stains of CSF samples collected from infected rats demonstrated that C. koseri was extracellular in the CSF and within membrane-bound vacuoles of infiltrating macrophages (Fig. 3).
FIG. 3.

Gram stain of neonatal rat CSF collected 6 days after infection with C. koseri (SMT319). Note macrophages (M) and neutrophils (N) containing numerous gram-negative C. koseri organisms within phagosomes.
(ii) EM analysis.
EM analysis of brain abscesses 7 days postinfection demonstrated that C. koseri was primarily intracellular within membrane-bound vacuoles of macrophages, neutrophils, resident macrophages (i.e., microglia), and brain microvascular endothelial cells (Fig. 4). C. koseri was not observed in fibroblasts, astrocytes, oligodendrocytes, or neurons (Fig. 4A through D). Further, there was no evidence of vasculitis in the blood vessels of the brain despite the advanced stage of the disease (Fig. 1A and B). Although macrophages were heavily infected with C. koseri, macrophage necrosis was inconspicuous within the brain tissue and CSF (Fig. 1D, 3, and 4). EM studies also indicated that C. koseri was actively dividing while inside macrophages (Fig. 4C).
FIG. 4.

EM examination of C. koseri CNS infection in the neonatal rat brain. Brain tissues and CSF samples were collected from neonatal rats 7 and 6 days, respectively, after infection with C. koseri (SMT319) and were processed for EM analysis. Arrows indicate single or multiple bacteria within vacuoles. (A) C. koseri within a brain microvascular endothelial cell (E). (B) C. koseri in extracellular space between brain microvascular endothelial cells, a pericyte (P), and an uninfected fibroblast (F). Note infiltrating macrophage (M). (C) C. koseri (arrow indicates dividing bacteria) within several macrophages next to an uninfected fibroblast and uninfected granulocytes (G). (D) C. koseri within a resident macrophage adjacent to an ependymal cell (EP). (E) Infiltrating macrophage harboring four C. koseri bacteria in a neonatal rat CSF sample taken 6 days postinfection. Bars, 1 μm.
Our observations of C. koseri pathogenesis in the neonatal rat model indicate that it closely mimics the human disease and that C. koseri is primarily intimately associated with macrophages, including resident brain macrophages.
Macrophage uptake of C. koseri.
The ability of macrophages to internalize C. koseri in vitro was established by determining the number of intracellular CFU following the gentamicin protection assay. We utilized PMA-stimulated U937 human macrophages, a cell line with markers and characteristics similar to those of the microglia (5, 9). The percentage of the initial C. koseri (SMT319) inoculum found to be intracellular following 45 min of uptake was 3.4% ± 0.04% (Fig. 5). This was three- to sixfold more than that of the E. coli K-12 strain HB101 or the E. coli K1 strain E44, respectively (Fig. 5). To evaluate the effect of opsonization of C. koseri on macrophage uptake, gentamicin protection assays were performed in the presence of HPS. As shown in Fig. 5, C. koseri uptake in a medium containing 10% HPS resulted in 27% ± 2.71% of the inoculum becoming internalized compared to 3.4% ± 0.04% taken up without HPS. In addition, the number of intracellular C. koseri organisms recovered increased in a dose-dependent manner. Interestingly, opsonization of both nonpathogenic and pathogenic E. coli strains led to an increase in uptake, yet there did not appear to be a dose-dependent relationship such as that observed for C. koseri (Fig. 5). In addition, the levels of enhanced uptake for opsonized meningitic E. coli K1 were not nearly up to the level for C. koseri.
FIG. 5.

Macrophage uptake of C. koseri and effect of serum opsonization. The gentamicin protection assay was performed on human U937 macrophages infected with mid-log-phase bacteria. Experiments were performed with media treated with increasing (0 to 10%) concentrations of HPS. Results are presented as the mean percentage of the inoculum that was internalized ± standard error from a representative assay carried out in triplicate. At least three independent experiments were observed with similar results.
EM analysis was used to visualize U937 human macrophage uptake of C. koseri (SMT319) opsonized with 10% HPS. We observed two morphologically distinct processes of bacterial uptake. First, C. koseri appears to “sink” into macrophages and is contained in a tight vacuole; characteristic of uptake via complement receptors (Fig. 6A and B) (44). Second, C. koseri also appears to be taken up by macrophage pseudopodia, resulting in a spacious vacuole (Fig. 6C and D). This mechanism of uptake is characteristic of bacteria being taken up by Fc receptors (44).
FIG. 6.

EM analysis of U937 human macrophages following a 30-min infection with C. koseri. Shown are a macrophage ingesting C. koseri (SMT319) in a manner morphologically similar to complement-coated uptake (A), resulting in tight phagosomes (B), and IgG-opsonized uptake (C), resulting in internalization of bacteria within spacious phagosomes (D). Bars, 1 μm.
With evidence that C. koseri uptake is augmented by serum components (such as complement and IgG), we first sought to determine if enhanced uptake was due to complement protein. Gentamicin protection assays were carried out following opsonization of C. koseri (SMT319) with HPS that was heat treated to denature complement proteins without affecting IgG. Heat killing of HPS before bacterial opsonization did not affect the ability of C. koseri to be taken up by macrophages (Fig. 7). In contrast to our EM studies, these data indicate that complement proteins do not play a significant role during macrophage uptake. Taken together, these findings show that uptake of opsonized C. koseri may occur via IgG or another, unidentified means of phagocytic uptake.
FIG. 7.

Macrophage uptake of C. koseri and effect of opsonization with heat-inactivated serum. The gentamicin protection assay was performed on human U937 macrophages infected with mid-log-phase C. koseri (SMT319). Experiments were performed with C. koseri opsonized with 10% HPS or 10% heat-inactivated HPS (ΔHPS). Results are presented as the mean percentage of the inoculum internalized ± standard error from a representative assay carried out in triplicate. Three independent experiments were conducted with similar results.
To determine if the FcγI receptor (FcγRI) facilitates C. koseri (SMT319) uptake (via IgG opsonization), we blocked the FcγRI on the surface of the macrophage before bacterial infection. We observed a dose-dependent relationship as we pretreated macrophages with increasing concentrations of the blocking antibody. Following blocking with 25 μg of an anti-FcγRI antibody, the number of intracellular bacteria decreased more than 80% (Fig. 8). Taken together, these data show that C. koseri uptake by macrophages is enhanced by serum opsonization and is mediated primarily by FcγRI.
FIG. 8.

Effect of FcγRI blocking on macrophage uptake of C. koseri. The gentamicin protection assay was performed on human U937 macrophages preincubated with increasing concentrations (0 to 25 μg) of a human anti-FcγRI antibody. Macrophages were then infected with mid-log-phase opsonized C. koseri (SMT319). Results are presented as the percentage of the maximum number of C. koseri organisms that were intracellular within unblocked macrophages, arbitrarily set at 100%. Data from three independent experiments (± standard errors) were combined. Experiments were performed at least in duplicate.
C. koseri survival and replication within macrophages.
We next examined if C. koseri (SMT319) is able to survive in the long term or replicate within macrophages. Results from C. koseri gentamicin protection assays during a 72-h time course showed that the number of CFU recovered from infected macrophages increased over time (Fig. 9). In contrast, the nonpathogenic E. coli K-12 strain HB101 was rapidly killed by the macrophages (Fig. 9). In addition, in agreement with our other studies, the meningitic E. coli K1 strain E44 demonstrated long-term survival within macrophages (V. T. Hill et al., submitted for publication). Further, although serum opsonization enhanced uptake (Fig. 5), it did not influence the ability of C. koseri to survive and replicate within macrophages (Fig. 10). These data indicate that C. koseri is steadily maintained and replicates within the macrophages for at least 3 days.
FIG. 9.

Intracellular replication of C. koseri within macrophages. The gentamicin protection assay was performed on human U937 macrophages infected with mid-log-phase bacteria to estimate the number of intracellular bacteria at various time points up to 72 h (T72). For each time point indicated, results are presented as percent survival of the initial intracellular population recovered at time zero. Data are means ± standard errors from a representative assay performed in triplicate. A minimum of three independent experiments were performed with similar results.
FIG. 10.

Intracellular replication of C. koseri within macrophages following opsonized uptake. The gentamicin protection assay was performed on human U937 macrophages infected with HPS-opsonized C. koseri (SMT319) grown to mid-log phase to estimate the number of intracellular bacteria at various time points up to 72 h. For each time point indicated, results are presented as percent survival of the initial intracellular population recovered at time zero. Data are means ± standard errors from a representative assay performed in triplicate. Three independent experiments were conducted with similar results.
C. koseri survives phagolysosomal fusion.
Previous studies with macrophages have demonstrated that late endosomal and lysosomal markers such as Rab7 and Lamp1 colocalize with LysoTracker Red (46). To obtain a better understanding of the potential mechanism(s) of C. koseri intracellular survival, we used confocal fluorescent microscopy with LysoTracker Red staining and a constitutively gfp-expressing wild-type C. koseri strain (SMT320) to localize macrophage lysosomes and intracellular bacteria during infection, respectively (46). We found that 93% ± 0.3% of intracellular gfp-expressing C. koseri organisms colocalized with stained lysosomes (Fig. 11). These findings suggest that C. koseri survives phagolysosomal fusion in macrophages.
FIG. 11.
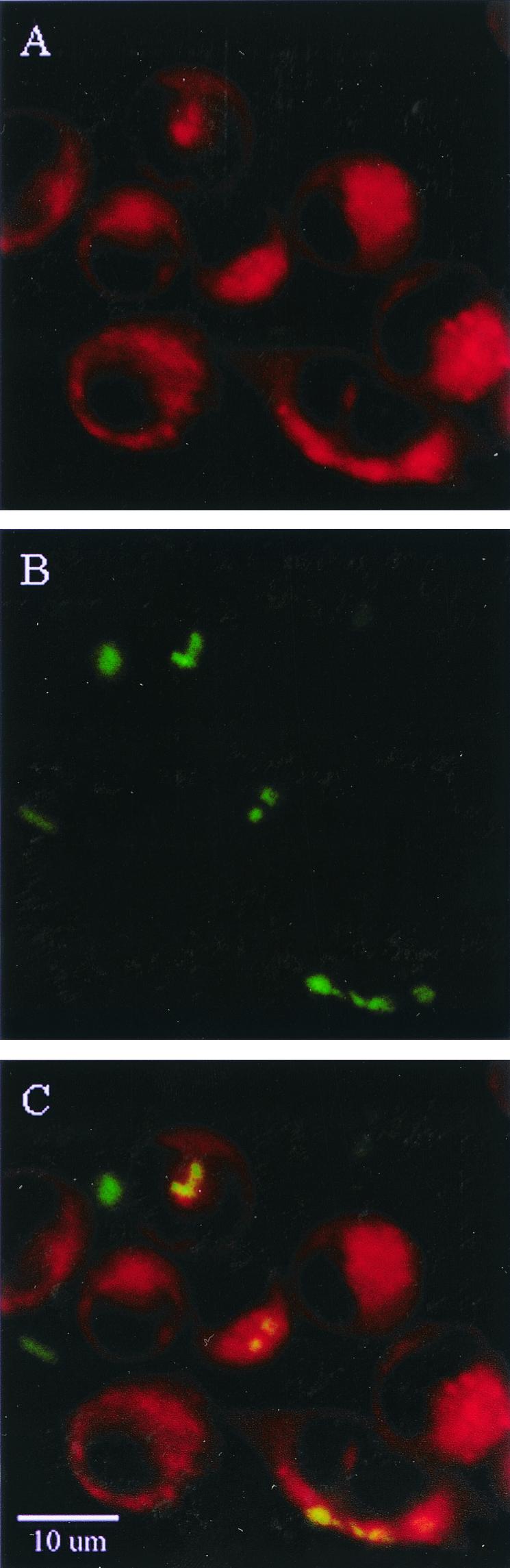
Confocal fluorescence microscopy of C. koseri in macrophages, showing that lysosomes colocalize with intracellular bacteria. Following infection of U937 macrophages with SMT320 (a gfp-expressing C. koseri strain), cells were stained with LysoTracker Red. (A) U937 macrophage with stained lysosomes (red punctate staining). (B) Constitutively gfp-expressing C. koseri (green). (C) Overlay of confocal images depicted in panels A and B. C. koseri organisms (green) that colocalize with stained lysosomes (red) appear yellow. All Z series were analyzed to localize bacteria to an individual plane, of which the single confocal image plane was taken as a representative image.
DISCUSSION
Neonatal C. koseri infection, characterized by meningitis and brain abscesses, frequently results in lifelong debilitating neurological sequelae. Although the ability of meningitic bacteria to invade the CNS has been described by in vitro and in vivo models of infection (2, 3, 14, 15, 18, 28, 31, 32, 35, 45), there is limited information pertaining to the ability of C. koseri to evade host defense mechanisms, establish chronic infection, and cause brain abscesses (2, 4, 20). Our study and earlier work by Kline et al. have shown that the development of Citrobacter meningitis, ventriculitis, and brain abscesses in the neonatal rat can be observed via distinct pathological disease stages (20). Further, our study suggests that MRI could allow noninvasive visualization of progressive brain abscess formation in the neonatal rat. In addition, the goal of the work presented here was to obtain a better understanding of C. koseri CNS infection at the cellular level in vitro and in vivo. We found that during CNS infection in neonatal rats, C. koseri is primarily intracellular within macrophages and neutrophils and is able to survive phagolysosomal fusion and to replicate intracellularly over the long term within macrophages.
EM analysis of abscessed brain tissue offers insight into the host-pathogen interactions occurring at the cellular level. Previous studies by Badger et al. have documented the ability of Citrobacter to invade and replicate within HBMEC in vitro (2). We have supported these findings in vivo by visualizing C. koseri within rat brain microvascular endothelial cells. It should be noted that vasculitis is not evident during the chronic phase of C. koseri infection either clinically or in the neonatal rat (20; this study). This is unlike the scenario in more acute meningitic infections, which cause considerable damage to blood vessels (6, 29). It is also interesting that, although the region of the brain most vulnerable to bacterial penetration is the choroid plexus, C. koseri infection rarely involves these tissues (20; this study). Again, this is in contrast to other hematogenous meningitic bacterial infections (24, 29, 42).
During C. koseri infection of the CNS, accumulation of necrotic tissue and infiltrating phagocytes results in the proportional expansion of the brain abscess (26). Prominent features observed during our EM analysis of infected brain tissue were a strong association of C. koseri with infiltrating macrophages and replication of C. koseri within membrane-enclosed phagosomes of these macrophages. Further, although macrophages were heavily infected, necrosis was inconspicuous. The ability of C. koseri to survive and replicate in these long-lived and abundant immune cells without killing them is clearly an advantageous component of its pathogenic potential, as it is for other intracellularly replicating organisms (8, 21, 23, 34).
EM data visualized C. koseri uptake by U937 macrophages and indicated that uptake occurred by two morphologically distinct processes reminiscent of Fc receptor- and complement-mediated uptake (44). Further, functional assays indicate that FcγRI is primarily mediating C. koseri uptake. However, since C. koseri uptake occurs without serum, other mechanisms could be facilitating internalization (i.e., mechanisms morphologically similar to complement uptake). Different methods of uptake expose internalized bacteria to different bactericidal components of the macrophage. For instance, the oxidative burst and proinflammatory response are triggered when the Fcγ receptor is involved in phagocytosis of opsonized bacteria (13, 16, 22, 25). In contrast, uptake by complement receptors does not induce a respiratory burst (27, 50). In our study, opsonization of C. koseri enhanced macrophage uptake but did not affect intracellular survival or replication. This suggests that the ability of C. koseri to survive and replicate within macrophages is independent of the method of uptake. These data are reminiscent of the intracellular pathogen Brucella abortus in that survival within macrophages was not significantly affected by entering through the Fc receptor as opposed to nonopsonized uptake (10, 17).
Professional phagocytes are the cornerstone of the innate immune response against microbial pathogens. Internalized pathogens are sequestered in phagocytic vacuoles, where they are exposed to an acidic, nutrient-poor environment and must contend with lysosomal fusion. In response to this selective pressure, pathogens have evolved mechanisms enabling survival and even replication under these normally adverse conditions. For example, Brucella suis and Salmonella enterica actively invade host cells and form modified vacuoles to prevent or tolerate lysosomal fusion, respectively (30, 43). Late-endosomal and lysosomal markers such as Rab7 and Lamp1 have been shown to colocalize with LysoTracker Red in macrophages (46), thus supporting its usefulness in this study to identify these compartments in relation to C. koseri phagosomes. Our studies show that following infection, C. koseri colocalized with lysosomes within macrophages. Specifically, more than 90% of intracellular C. koseri bacteria colocalized with lysosomes 30 min following infection. Temporal issues such as a lag time between uptake and phagolysosomal fusion for recently internalized C. koseri may account for the minority of intracellular C. koseri organisms that had not colocalized within lysosomes. It remains to be seen whether the C. koseri mechanism(s) for survival of phagolysosomal fusion is similar to that of S. enterica.
The pathogenesis of C. koseri infection has evolved an elegant balance of host-pathogen interactions, evident in its often-insidious onset and ultimately devastating outcome. It enters and replicates in brain microvascular endothelial cells and in resident and infiltrating macrophages, thus escaping many host defenses. Studies are in progress to determine specific virulence factors necessary for C. koseri survival of phagolysosomal fusion and for C. koseri intracellular replication as well as to determine the precise role of the host inflammatory response in the progress of the disease. Enhanced knowledge of Citrobacter pathogenesis will aid in the development of novel strategies to prevent and treat this devastating disease.
Acknowledgments
We acknowledge the Childrens Hospital of Los Angeles Research Institute Small Animal Imaging Research Center for the development of the MRI methods used in this study. We thank George McNamara for assistance with the fluorescent microscopy experiments performed at the CHLA Research Institute Image Core Facility. We thank Allison Peters and Mary Kenner for technical assistance. We appreciate Kevin Nash and Clark Inderlied for critical review of the manuscript.
This work was generously supported by a CHLA Research Institute Career Development Award and by CHLA Department of Pathology Start-up Funds (to J.L.B.).
Editor: B. B. Finlay
REFERENCES
- 1.Badger, J. L., and K. S. Kim. 1998. Environmental growth conditions influence the ability of Escherichia coli K1 to invade brain microvascular endothelial cells and confer serum resistance. Infect. Immun. 66:5692-5697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Badger, J. L., M. F. Stins, and K. S. Kim. 1999. Citrobacter freundii invades and replicates in human brain microvascular endothelial cells. Infect. Immun. 67:4208-4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Badger, J. L., C. A. Wass, and K. S. Kim. 2000. Identification of Escherichia coli K1 genes contributing to human brain microvascular endothelial cell invasion by differential fluorescence induction. Mol. Microbiol. 36:174-182. [DOI] [PubMed] [Google Scholar]
- 4.Baumeister, F. A., M. Hofer, H. Kuster, and B. H. Belohradsky. 2000. CSF interleukin-6 in neonatal Citrobacter ventriculitis after meningitis. Infection 28:243-245. [DOI] [PubMed] [Google Scholar]
- 5.Chabot, S., D. Charlet, T. L. Wilson, and V. W. Yong. 2001. Cytokine production consequent to T cell-microglia interaction: the PMA/IFN gamma-treated U937 cells display similarities to human microglia. J. Neurosci. Methods 105:111-120. [DOI] [PubMed] [Google Scholar]
- 6.Dodge, P. R., and M. N. Swartz. 1965. Bacterial meningitis: a review of selected aspects. II. Special neurological problems, postmeningitis complications and clinicopathological correlations. N. Engl. J. Med. 272:954-960. [DOI] [PubMed] [Google Scholar]
- 7.Doran, T. I. 1999. The role of Citrobacter in clinical disease of children: review. Clin. Infect. Dis. 28:384-394. [DOI] [PubMed] [Google Scholar]
- 8.Drevets, D. A. 1999. Dissemination of Listeria monocytogenes by infected phagocytes. Infect. Immun. 67:3512-3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eales-Reynolds, L. J., H. Laver, and H. Mojtahedi. 2001. Evidence for the expression of the EGF receptor on human monocytic cells. Cytokine 16:169-172. [DOI] [PubMed] [Google Scholar]
- 10.Eze, M. O., L. Yuan, R. M. Crawford, C. M. Paranavitana, T. L. Hadfield, A. K. Bhattacharjee, R. L. Warren, and D. L. Hoover. 2000. Effects of opsonization and gamma interferon on growth of Brucella melitensis 16M in mouse peritoneal macrophages in vitro. Infect. Immun. 68:257-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feferbaum, R., E. M. Diniz, M. Valente, C. R. Giolo, R. A. Vieira, A. L. Galvani, M. E. Ceccon, M. C. Araujo, V. L. Krebs, and F. A. Vaz. 2000. Brain abscess by Citrobacter diversus in infancy: case report. Arq. Neuropsiquiatr. 58:736-740. [DOI] [PubMed] [Google Scholar]
- 12.Finn, A., G. H. Talbot, E. Anday, M. Skros, M. Provencher, and C. Hoegg. 1988. Vertical transmission of Citrobacter diversus from mother to infant. Pediatr. Infect. Dis. J. 7:293-294. [DOI] [PubMed] [Google Scholar]
- 13.Heller, T., J. E. Gessner, R. E. Schmidt, A. Klos, W. Bautsch, and J. Kohl. 1999. Fc receptor type I for IgG on macrophages and complement mediate the inflammatory response in immune complex peritonitis. J. Immunol. 162:5657-5661. [PubMed] [Google Scholar]
- 14.Hoffman, J. A., C. Wass, M. F. Stins, and K. S. Kim. 1999. The capsule supports survival but not traversal of Escherichia coli K1 across the blood-brain barrier. Infect. Immun. 67:3566-3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang, S. H., C. Wass, Q. Fu, N. V. Prasadarao, M. Stins, and K. S. Kim. 1995. Escherichia coli invasion of brain microvascular endothelial cells in vitro and in vivo: molecular cloning and characterization of invasion gene ibe10. Infect. Immun. 63:4470-4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Indik, Z. K., J. G. Park, S. Hunter, and A. D. Schreiber. 1995. Structure/function relationships of Fc gamma receptors in phagocytosis. Semin. Immunol. 7:45-54. [DOI] [PubMed] [Google Scholar]
- 17.Jones, S. M., and A. J. Winter. 1992. Survival of virulent and attenuated strains of Brucella abortus in normal and gamma interferon-activated murine peritoneal macrophages. Infect. Immun. 60:3011-3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim, K. S., H. Itabashi, P. Gemski, J. Sadoff, R. L. Warren, and A. S. Cross. 1992. The K1 capsule is the critical determinant in the development of Escherichia coli meningitis in the rat. J. Clin. Investig. 90:897-905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kline, M. W. 1988. Citrobacter meningitis and brain abscess in infancy: epidemiology, pathogenesis, and treatment. J. Pediatr. 113:430-434. [DOI] [PubMed] [Google Scholar]
- 20.Kline, M. W., S. L. Kaplan, E. P. Hawkins, and E. O. Mason, Jr. 1988. Pathogenesis of brain abscess formation in an infant rat model of Citrobacter diversus bacteremia and meningitis. J. Infect. Dis. 157:106-112. [DOI] [PubMed] [Google Scholar]
- 21.Kohler, S., F. Porte, V. Jubier-Maurin, S. Ouahrani-Bettache, J. Teyssier, and J. P. Liautard. 2002. The intramacrophagic environment of Brucella suis and bacterial response. Vet. Microbiol. 90:299-309. [DOI] [PubMed] [Google Scholar]
- 22.Kusner, D. J., C. F. Hall, and S. Jackson. 1999. Fc gamma receptor-mediated activation of phospholipase D regulates macrophage phagocytosis of IgG-opsonized particles. J. Immunol. 162:2266-2274. [PubMed] [Google Scholar]
- 23.Leung, K. Y., and B. B. Finlay. 1991. Intracellular replication is essential for the virulence of Salmonella typhimurium. Proc. Natl. Acad. Sci. USA 88:11470-11474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levine, S. 1987. Choroid plexus: target for systemic disease and pathway to the brain. Lab. Investig. 56:231-233. [PubMed] [Google Scholar]
- 25.Looney, R. J. 1993. Structure and function of human and mouse FcγRII. Blood Cells 19:353-359. [PubMed] [Google Scholar]
- 26.Mathisen, G. E., and J. P. Johnson. 1997. Brain abscess. Clin. Infect. Dis. 25:763-779; quiz 780-781. [DOI] [PubMed] [Google Scholar]
- 27.Mosser, D. M., and P. J. Edelson. 1987. The third component of complement (C3) is responsible for the intracellular survival of Leishmania major. Nature 327:329-331. [DOI] [PubMed] [Google Scholar]
- 28.Moxon, E. R., M. P. Glode, A. Sutton, and J. B. Robbins. 1977. The infant rat as a model of bacterial meningitis. J. Infect. Dis. 136(Suppl.):S186-S190. [DOI] [PubMed] [Google Scholar]
- 29.Moxon, E. R., A. L. Smith, D. R. Averill, and D. H. Smith. 1974. Haemophilus influenzae meningitis in infant rats after intranasal inoculation. J. Infect. Dis. 129:154-162. [DOI] [PubMed] [Google Scholar]
- 30.Naroeni, A., N. Jouy, S. Ouahrani-Bettache, J. P. Liautard, and F. Porte. 2001. Brucella suis-impaired specific recognition of phagosomes by lysosomes due to phagosomal membrane modifications. Infect. Immun. 69:486-493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nassif, X. 1999. Interaction mechanisms of encapsulated meningococci with eucaryotic cells: what does this tell us about the crossing of the blood-brain barrier by Neisseria meningitidis? Curr. Opin. Microbiol. 2:71-77. [DOI] [PubMed] [Google Scholar]
- 32.Nizet, V., K. S. Kim, M. Stins, M. Jonas, E. Y. Chi, D. Nguyen, and C. E. Rubens. 1997. Invasion of brain microvascular endothelial cells by group B streptococci. Infect. Immun. 65:5074-5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Renier, D., C. Flandin, E. Hirsch, and J. F. Hirsch. 1988. Brain abscesses in neonates. A study of 30 cases. J. Neurosurg. 69:877-882. [DOI] [PubMed] [Google Scholar]
- 34.Rhoades, E. R., and H. J. Ullrich. 2000. How to establish a lasting relationship with your host: lessons learned from Mycobacterium spp. Immunol. Cell Biol. 78:301-310. [DOI] [PubMed] [Google Scholar]
- 35.Ring, A., J. N. Weiser, and E. I. Tuomanen. 1998. Pneumococcal trafficking across the blood-brain barrier. Molecular analysis of a novel bidirectional pathway. J. Clin. Investig. 102:347-360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saez-Llorens, X., and G. H. McCracken, Jr. 1998. Enterobacter, p. 1279-1282. In R. D. Feigin and J. D. Cherry (ed.), Textbook of pediatric infectious diseases, 4th ed. W. B. Saunders, Philadelphia, Pa.
- 37.Shattuck, K. E., and T. Chonmaitree. 1992. The changing spectrum of neonatal meningitis over a fifteen-year period. Clin. Pediatr. (Philadelphia) 31:130-136. [DOI] [PubMed] [Google Scholar]
- 38.Soriano, A. L., R. G. Russell, D. Johnson, R. Lagos, I. Sechter, and J. G. Morris, Jr. 1991. Pathophysiology of Citrobacter diversus neonatal meningitis: comparative studies in an infant mouse model. Infect. Immun. 59:1352-1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stretton, S., S. Techkarnjanaruk, A. M. McLennan, and A. E. Goodman. 1998. Use of green fluorescent protein to tag and investigate gene expression in marine bacteria. Appl. Environ. Microbiol. 64:2554-2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sundstrom, C., and K. Nilsson. 1976. Establishment and characterization of a human histiocytic lymphoma cell line (U-937). Int. J. Cancer 17:565-577. [DOI] [PubMed] [Google Scholar]
- 41.Tang, L. M., S. T. Chen, and T. N. Lui. 1994. Citrobacter meningitis in adults. Clin. Neurol. Neurosurg. 96:52-57. [DOI] [PubMed] [Google Scholar]
- 42.Tuomanen, E. 1996. Entry of pathogens into the central nervous system. FEMS Microbiol. Rev. 18:289-299. [DOI] [PubMed] [Google Scholar]
- 43.Uchiya, K., M. A. Barbieri, K. Funato, A. H. Shah, P. D. Stahl, and E. A. Groisman. 1999. A Salmonella virulence protein that inhibits cellular trafficking. EMBO J. 18:3924-3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Underhill, D. M., and A. Ozinsky. 2002. Phagocytosis of microbes: complexity in action. Annu. Rev. Immunol. 20:825-852. [DOI] [PubMed] [Google Scholar]
- 45.Unkmeir, A., K. Latsch, G. Dietrich, E. Wintermeyer, B. Schinke, S. Schwender, K. S. Kim, M. Eigenthaler, and M. Frosch. 2002. Fibronectin mediates Opc-dependent internalization of Neisseria meningitidis in human brain microvascular endothelial cells. Mol. Microbiol. 46:933-946. [DOI] [PubMed] [Google Scholar]
- 46.Via, L. E., R. A. Fratti, M. McFalone, E. Pagan-Ramos, D. Deretic, and V. Deretic. 1998. Effects of cytokines on mycobacterial phagosome maturation. J. Cell Sci. 111:897-905. [DOI] [PubMed] [Google Scholar]
- 47.Vogel, L. C., L. Ferguson, and S. P. Gotoff. 1978. Citrobacter infections of the central nervous system in early infancy. J. Pediatr. 93:86-88. [DOI] [PubMed] [Google Scholar]
- 48.Weiser, J. N., and E. C. Gotschlich. 1991. Outer membrane protein A (OmpA) contributes to serum resistance and pathogenicity of Escherichia coli K-1. Infect. Immun. 59:2252-2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Williams, W. W., J. Mariano, M. Spurrier, H. D. Donnell, Jr., R. L. Breckenridge, Jr., R. L. Anderson, I. K. Wachsmuth, C. Thornsberry, D. R. Graham, D. W. Thibeault, et al. 1984. Nosocomial meningitis due to Citrobacter diversus in neonates: new aspects of the epidemiology. J. Infect. Dis. 150:229-235. [DOI] [PubMed] [Google Scholar]
- 50.Wright, S. D., and S. C. Silverstein. 1983. Receptors for C3b and C3bi promote phagocytosis but not the release of toxic oxygen from human phagocytes. J. Exp. Med. 158:2016-2023. [DOI] [PMC free article] [PubMed] [Google Scholar]