

Abstract
The binding of heterotrimeric lymphotoxin, LTα1β2, to the LTβ receptor (LTβR), a member of the tumor necrosis factor receptor (TNFR) superfamily, induces nuclear factor κB (NF-κB) activation and cell death in HT29 adenocarcinoma cells. We now show that treatment with LTα1β2 or agonistic LTβR antibodies causes rapid recruitment of TNFR-associated factor 3 (TRAF3) to the LTβR cytoplasmic domain. Further, stable overexpression of a TRAF3 mutant that lacks the RING and zinc finger domains inhibits LTβR-mediated cell death. The inhibition is specific for LTβR cell death signaling, since NF-κB activation by LTα1β2 and Fas-mediated apoptosis are not inhibited in the same cells. The mutant and endogenous TRAF3s are both recruited at equimolar amounts to the LTβR, suggesting that the mutant disrupts the function of the signaling complex. These results implicate TRAF3 as a critical component of the LTβR death signaling complex and indicate that at least two independent signaling pathways are initiated by LTβR ligation.
Keywords: apoptosis, cytokines, signal transduction, tumor necrosis factor
The tumor necrosis factor (TNF) superfamily of cytokines and receptors can activate nuclear factor κB (NF-κB) and effect cell growth, differentiation, or death. Mutations can result in developmental abnormalities, immunodeficiency, or autoimmune-like disorders. Lymphotoxin (LT)α and LTβ (for review see ref. 1), two members of the TNF cytokine family, are implicated in the embryonic development of secondary lymph organs (2, 3) and in the formation of germinal centers during immune responses in the adult (4–7). LTα can be secreted as a homotrimer or can be anchored to the surface of activated T lymphocytes as a heterotrimer with LTβ, a type II transmembrane glycoprotein (8–10). Secreted LTα homotrimers can bind to the 75- to 80-kDa TNF receptor (TNFR) (TNFR80; type 2 or CD120b) or to the 55- to 60-kDa TNFR (TNFR60; also known as type 1 or CD120a) (11–13), while surface LTα1β2 binds to the LTβ receptor (LTβR) (14). LTβR and the TNFRs have similar cysteine-rich, glycosylated, extracellular domains, and largely nonhomologous transmembrane and cytoplasmic domains (reviewed in ref. 15).
The TNFR cytoplasmic domains lack enzymatic activity, and thus signaling appears to be mediated by interactive cytoplasmic proteins. The cytoplasmic domain of TNFR60 and Fas (Apo-1 or CD95) have homologous death domains which can mediate apoptosis (16–20). Death domains can interact with similar motifs in TRADD (21), MORT1/FADD (22, 23), and a serine kinase, RIP (24). FADD can in turn interact with MACH1/FLICE (25, 26), a cysteine protease that can initiate a cascade of interleukin 1β convertase-related aspartate-specific proteases and effect apoptosis. Although LTβR, CD40, TNFR80, and CD30 do not have death domains, activation of these receptors can also induce cell death in specific cellular contexts (27–30). The cytoplasmic mediators of cell death from these receptors are uncertain.
The TNFR-associated factor 3 (TRAF3) protein is a candidate signaling molecule for the non-death-domain TNFR family members. TRAF3 was originally described as a CD40-binding protein and as a protein associated with LMP1, a dominant oncogene product of Epstein–Barr virus (31–34). TRAF3 also interacts with the cytoplasmic regions of LTβR, TNFR80, and CD30, but not significantly with TNFR60 or Fas (34–36). Like most other TRAFs, TRAF3 has an N-terminal RING finger and several zinc finger domains, a coiled-coil region, and a C-terminal receptor-binding domain that is homologous to other TRAFs (37). Other members of the TRAF family interact with LTβR, TNFR80, CD40, and CD30 (34–40). Overexpression of TRAF2 or TRAF5 can activate NF-κB, a transcription factor controlling expression of genes involved in immune and inflammatory responses (41, 42). TRAF2 also interacts with other cytosolic proteins, such as the cellular homologs of baculovirus inhibitor of apoptosis 1 and 2 (43), I-TRAF/TANK (44, 45), and TRADD, the latter providing an indirect link to TNFR60 (46). TRAF3 does not activate NF-κB, and its functions are uncertain (37, 47). The N-terminal putative zinc-binding domains of TRAF1, TRAF2, TRAF3, and TRAF5 are implicated as effectors in NF-κB or CD23 activation (31, 39, 40, 47, 48).
Although initiating events in signal transduction frequently involve ligand-induced receptor aggregation and clustering of cytoplasmic domains, TRAF3 interaction with TNFRs has not been previously demonstrated to be ligand dependent. We have therefore examined the effect of ligands on the association of the LTβR with TRAF3 in HT29, an adenocarcinoma cell line. We found that LTα1β2 induces TRAF3 association with LTβR in a time- and dose-dependent process. Having established TRAF3 as a component of the LTβR signaling complex, we investigated whether overexpression of a TRAF3 N-terminal deletion mutant in HT29 cells could abrogate the cell death response. We found that the TRAF3 N-terminal deletion mutant inhibits the LTβR-mediated cell death response, but not LTβR-mediated NF-κB activation or Fas-mediated cell death. LTβR signaling appears to bifurcate, with TRAF3 being in the pathway leading to cell death and possibly other TRAFs mediating the pathway to NF-κB activation.
METHODS
Antibodies and Cytokines.
Recombinant LTα, TNF (49), and soluble LTα1β2 (50) produced with a truncated version of LTβ lacking the cytosolic and transmembrane domains were provided by Jeffrey Browning (Biogen). The methods for production and characterization of antibodies to receptor-Fc fusion proteins have been described (51). The goat anti-LTβR does not crossreact with TNFR60, TNFR80, or Fas as measured by immunoprecipitation of proteins overexpressed in COS7 cells, nor does it inhibit the binding of radioiodinated TNF or LTα to TNFR60-Fc or TNFR80-Fc. The presence of the ligand does not block the reactivity of the goat anti-LTβR with the receptor, although the antiserum when prebound is competitive for binding of LTα1β2. An IgG fraction, prepared by ammonium sulfate precipitation and ion-exchange chromatography, was exhaustively dialyzed against Hanks’ balanced salt solution, pH 7.4, and filter sterilized for use in tissue culture. Preimmune goat serum or IgG was not toxic to HT29.14S cells. Rabbit anti-TRAF3 was produced by immunization with a synthetic N-terminal peptide (residues 5–25) of TRAF3, coupled to keyhole limpet hemocyanin as a carrier. The antiserum was used at 1:1000 dilution and showed no crossreactivity in Western blots with TRAF1, -2, or -5 made by in vitro translation. Immune complexes were detected with donkey anti-rabbit IgG coupled to horseradish peroxidase and chemiluminescence substrate (ECL reagent; Amersham) with a 15-min exposure. The monoclonal antibodies used were anti-LTβR, BDA8 [mouse IgG1 (10), a gift from J. Browning]; anti-Fas, CH11 (mouse IgM; MBL, Nagoya, Japan); anti-TNFR60, H398 (mouse IgG2a, Biosource, Camarillo, CA); and antibodies to intracellular adhesion molecule 1 (ICAM-1) (mouse IgG1, Chemicon, Temecula, CA).
TRAF3 Mutant and Transfection.
The TRAF3 deletion mutant encoding amino acids 368–568 was engineered by PCR amplification (Taq DNA polymerase) from TRAF3 cDNA using the following oligonucleotides: 5′ primer 5′-CCGGATCCATGGACTACAAGGACGACGATGACAAGAGCGCGGGGCAAGTG-3′, which introduces a BamHI site; and 3′ primer: 5′-CCCTCGAGCCTGAAAAACGCAGCC-3′, which introduces an XhoI site. The amplified product was purified, digested with BamHI and XhoI, and ligated into pcDNA3 (Invitrogen) and is referred to as TRAF3Δ1–367.
HT29.14S was cloned from HT29 cells (American Type Culture Collection) by limiting dilution in RPMI medium 1640 with 10% fetal bovine serum (27). HT29.14S cells were transfected with TRAF3Δ1–367 or empty pcDNA3 vector by electroporation and selected in medium with Geneticin (G418 sulfate) at 800 μg/ml (GIBCO). Transfected clones were used in assays between passages 4 and 10 and shifted into medium without G418 24 hr prior to assay. All cell lines tested free of mycoplasma by PCR analysis (Mycoplasma Primer Set, Stratagene). Cell viability in response to cytokines or antibodies was determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) dye reduction assay (52). The percentage cell viability was calculated as a ratio of the dye absorbance (570 nm) by cells cultured with cytokines or antibodies to medium alone. The A570 for individual lines in medium ranged from 1.1 to 1.6. Statistical analysis for calculations of IC50 and significance was carried out with prism and instat software (GraphPad, San Diego).
Binding and Immunoprecipitation Assays.
TRAF3Δ1–367 protein was detected by binding to a fusion protein between glutathione S-transferase (GST) and the cytoplasmic domain of the LTβR (LTβR-GST) which has been previously described in detail (34). Cells (70% confluent in a 75-cm2 flask) were metabolically labeled with [35S]methionine and [35S]cysteine as described (53), extracted in buffer containing nonionic detergent (1% Nonidet P-40/0.15 M NaCl/10 mM Tris, pH 7.4, containing 1 mM phenylmethanesulfonyl fluoride, 10 μg/ml leupeptin, 2 μg/ml aprotinin, and 0.1 mM dithiothreitol), and centrifuged for 5 min at 10,000 × g. The supernatant was incubated at 4°C overnight with 3 μl of GST bound to glutathione-Sepharose beads containing 20 μg of protein, the beads were removed, and the supernatant was precleared twice more. LTβR-GST beads (10 μg protein) were added to the precleared extracts and incubated for 2 hr. LTβR-GST or GST beads were washed in buffer, and the bound proteins were eluted, resolved by electrophoresis on a reducing SDS/12% polyacrylamide gel, and analyzed by PhosphorImager (Molecular Dynamics). For direct immunoprecipitation with anti-LTβR, detergent extracts were prepared from cells, precleared with 1 μl of preimmune goat serum, and immunoprecipitated with goat anti-LTβR (10 μg of IgG per ml of extract) and 20 μl of protein G-Sepharose.
NF-κB Gel-Shift Assay.
DNA-binding interactions were studied by gel-shift assays as described (54) with modification (39) using the κB sites in the HIV-1 enhancer. The composition of the activated NF-κB complex was examined by supershift analysis with antiserum to Rel family members (Santa Cruz Biotechnology).
RESULTS
TRAF3 Association with the LTβR Is Ligand Dependent.
LTα1β2 induction of TRAF3 association with the LTβR was investigated in the HT29.14S cell line model, in which LTα1β2 induces NF-κB activation and cell death (50). HT29.14S cells were treated with LTα1β2 for 15 min, the LTβR was immunoprecipitated from cell lysates with specific antibody, and TRAF3 was analyzed in the LTβR immunoprecipitate by Western blotting with an anti-TRAF3 serum. TRAF3 immunoprecipitation with the LTβR increased dramatically after treatment of the cells with recombinant soluble LTα1β2 or with agonistic LTβR monoclonal antibody (BDA8) and did not increase after treatment with TNF, LTα, or anti-Fas antibody (Fig. 1A). Thus, TRAF3 association with LTβR is ligand dependent. The less abundant antigen at ≈68–70 kDa is probably a modified form of TRAF3, since it specifically reacts with the anti-TRAF3 antiserum. The LTβR-TRAF3 complex assembled within a minute after ligand binding (Fig. 1B), indicating that the association is an early event in LTβR signaling. Multiple TRAF3 crossreactive proteins of very large size appeared during the 45 min after LTβR ligation. The LTβR·TRAF3 complex formation was optimal between 0.1 and 1 nM LTα1β2 (data not shown), concordant with the cell death response to this ligand in HT29 cells (27). These results are consistent with TRAF3 being a recruited component of a LTβR ligand–receptor signaling complex.
Figure 1.
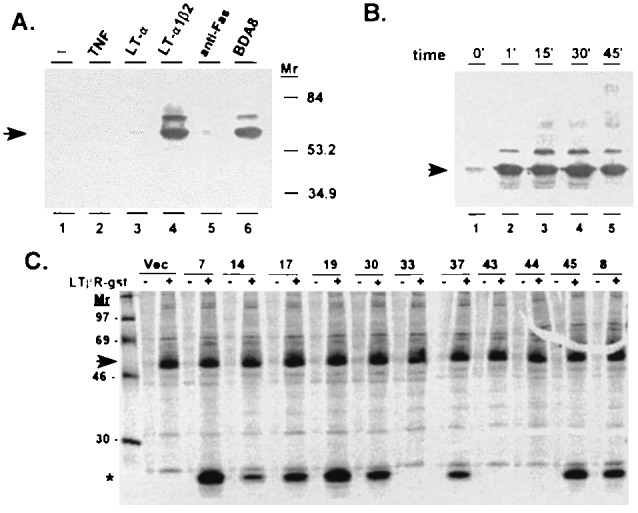
Association of LTβR with TRAF3. (A) HT29.14S cells (1 × 107) were incubated for 15 min at 37°C in one of the following: medium alone (lane 1); TNF (1 nM; lane 2); LTα (1 nM; lane 3); LTα1β2 (1 nM; lane 4); mouse anti-Fas IgM (100 ng/ml; lane 5); or anti-LTβR (BDA8; 5 μg/ml; lane 6). Cells were extracted immediately in cold buffer with nonionic detergent, LTβR was isolated by immunoprecipitation with goat anti-LTβR IgG, and TRAF3 was detected by Western blot analysis with rabbit anti-TRAF3. (B) Time course of TRAF3 recruitment. HT29.14S cells were incubated with LTα1β2 (1 nM) for the indicated times, and treated as in A. (C) Expression of TRAF3 and TRAF3Δ1–367 in HT29.14S cells. Drug (G418)-selected clones of HT29.14S transfected with TRAF3Δ1–367 or empty vector pool (Vec) were labeled with [35S]methionine and [35S]cysteine and proteins bound to GST (−) or LTβR-GST (+) were analyzed by SDS/PAGE and autoradiography using a PhosphorImager (range 30–600 pixels). The arrow notes the position of TRAF3 and the asterisk notes the position of TRAF3Δ1–367. Molecular mass standards (in kDa) are shown on the left in the first lane. The ratio of TRAF3Δ1–367 mutant to TRAF3 protein is as follows: 20 for clone 7; 7 for clone 8; 3 for clone 14; 6 for clone 17; 16 for clone 19; 7 for clone 30; 4 for clone 37; 11 for clone 45; and <0.1 for vector pool and clones 33, 43, and 44.
N-Terminally Truncated TRAF3 Inhibits LTβR-Ligand- but Not Fas-Antibody-Induced Cell Death.
TNF, Fas antibody, or LTα1β2 initiates death in interferon-γ-treated HT29.14S cells (27). To investigate the role of TRAF3 in cell death, an expression vector which confers G418 resistance or the vector with a putative dominant-negative TRAF3 mutant (TRAF3Δ1–367) under control of the cytomegalovirus immediate early promoter was transfected into HT29.14S cells and G418-resistant clones were selected. The resultant clones were analyzed for LTβR-ligand-induced death response and TRAF3Δ1–367 expression. The TRAF3Δ1–367 protein (molecular mass of 25 kDa) was expressed in 8 of 33 clones transfected with the TRAF3Δ1–367 expression vector, as detected on a phosphorimage of electrophoresed [35S]methionine/[35S]cysteine-labeled cell proteins which bound to a LTβR-GST fusion protein (Fig. 1C). The relative abundance of the mutant and wild-type TRAF3 proteins in the transfected cells was determined by PhosphorImager analysis and correction for the difference in methionine and cysteine content (2 Cys and 9 Met for TRAF3Δ1–367 and 28 Cys and 16 Met residues for wild-type TRAF3). The molar ratio of endogenous wild-type TRAF3 to TRAF3Δ1–367 in these clones varied from 1:3 to 1:20.
The eight G418-resistant, TRAF3Δ1–367 expressing, HT29.14S clones exhibited a significant attenuation of the cell death response to soluble LTα1β2 or to agonistic LTβR polyclonal antibody as measured by a shift in the dose–response curve when compared with control lines, including the parent HT29.14S cells (14S, Fig. 2), a pool of vector control-transfected cells (vec, Fig. 2), or to individual vector control-transfected clones (P < 0.002; Table 1). The initial pool of G418-resistant, TRAF3Δ1–367-transfected cells also had an attenuated response to LTα1β2 (IC50 = 2000 pM; data not shown), indicating that the eight clones are not rare in the initial population. However, the TRAF3Δ1–367-expressing clones were similar to the control lines in sensitivity to Fas antibody-induced apoptosis (Fig. 2 and Table 1). Interestingly, the TRAF3Δ1–367 expressing clones were somewhat attenuated in their sensitivity to TNF-induced cell death as compared with the control lines (P = 0.03; Table 1). Thus, TRAF3Δ1–367 inhibits LTβR-ligand-induced cell death, has no effect on Fas-induced cell death, and appears to have a small effect on TNF-induced cell death.
Figure 2.

A TRAF3 mutant inhibits cell death by LTβR. The HT29.14S clones expressing TRAF3Δ1–367 were incubated in medium containing either recombinant cytokines (soluble LTα1β2 or TNF) or receptor-specific antibodies (purified goat anti-LTβR IgG or anti-Fas IgM, CH11). Cells were plated at 104 cells per well in microtiter plates and cell viability was determined after 3 days by the MTT dye reduction assay. Each data point represent the mean ± SD of triplicate wells. The HT29.14S parental line (14S) and a pool of G418-resistant clones transfected with empty pCDNA3 plasmid (vec) were used as controls. The data shown was collected in one experiment. A summary of several determinations is shown in Table 1.
Table 1.
Effect of TRAF3Δ1–367 on ligand-induced cell death
| Transfected HT29.14S clones | IC50 for ligand
|
|||
|---|---|---|---|---|
| LTα1β2, pM | TNF, pM | Anti-Fas, ng/ml | Anti-LTβR, μg/ml | |
| Empty vector | 440 ± 350 (12) | 20 ± 10 (9) | 60 ± 58 (9) | 1.2 ± 1 (9) |
| TRAF3Δ1–367 | 5200 ± 3000** (8) | 200 ± 110* (8) | 30 ± 30 (8) | 9.5 ± 0.7** (8) |
Concentration of LTα1β2 and TNF, anti-Fas, or goat anti-LTβR IgG required to induce 50% loss of cell viability (IC50) as measured by MTT dye reduction assay. The IC50 values are given as the mean ± SD as determined for a number (n) of clones transfected with empty vector or with TRAF3Δ1–367 expression vector. TRAF3Δ1–367 expression vector clones are those which expressed TRAF3Δ1–367 as determined by binding to LTβR-GST. The statistical significance of the difference in IC50 between empty vector and TRAF3Δ1–367 clones was determined by the Student’s t test: ∗, P = 0.03; ∗∗, P < 0.002.
N-Terminally Truncated TRAF3 Does Not Inhibit LTβR-Ligand-Induced NF-κB Activation.
Two clones which express TRAF3Δ1–367 and are highly resistant to LTβR-ligand-induced cell death were compared with the pool of control vector-transfected cells for LTβR-ligand-induced NF-κB activation. The TRAF3Δ1–367-expressing clones did not differ from control vector-expressing cells in surface LTβR, Fas, or TNFR60 expression as measured by flow cytometry (data not shown). Stimulation of TRAF3Δ1–367-expressing or control HT29.14Svec cells for 15 min with LTα1β2 or antibodies to LTβR specifically induced similar levels of NF-κB activation as revealed by an electrophoretic mobility-shift assay (Fig. 3A). TNF was also similarly efficient at inducing activation of NF-κB in the TRAF3Δ1–367 expressing and control HT29.14Svec cells (Fig. 3A). Anti-Fas monoclonal antibody CH11 induced NF-κB poorly, although it is a very potent signal transducer for apoptosis in these cells, which is consistent with apoptosis and NF-κB activation being separate pathways in these cells. Antibodies to the p65 or p50 subunits of NF-κB, but not to c-Rel, Rel B, or p52, super-shifted the κB oligonucleotide, indicating that LTα1β2 activates a p65·p50 heterocomplex, similar to TNF (Fig. 3B). The expression of ICAM-1, an adhesion molecule regulated in part by NF-κB (55), is modestly enhanced on HT29.14S cells by LTα1β2 or TNF, with a shift in mean peak fluorescence of 50–80%, 14 hr after stimulation. TRAF3Δ1–367-expressing and control HT29.14Svec cells did not differ in LTα1β2-induced ICAM-1 expression (not shown). These results indicate that TRAF3Δ1–367 expression does not affect LTβR-ligand-induced NF-κB activation or ICAM-1 expression.
Figure 3.
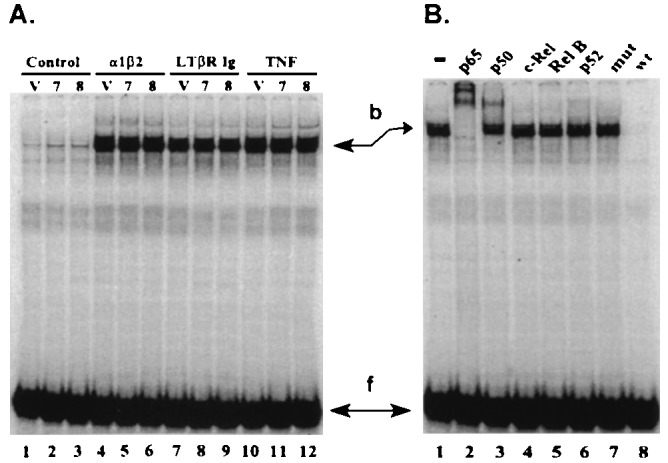
Activation of NF-κB by LTβR. (A) HT29.14S clones 7 and 8 transfected with TRAF3Δ1–367 mutant or empty pCDNA3 vector (V) were treated for 15 min with normal goat IgG (10 μg/ml) (lanes 1–3), LTα1β2 (1 nM) (lanes 4–6), goat anti-LTβR IgG (10 μg/ml) (lanes 7–9), or TNF (1 nM) (lanes 10–12). Nuclear extracts (4 μg) from these cells were incubated with 32P-labeled κB oligonucleotide and binding was analyzed by electrophoretic mobility-shift assay. Arrows indicate the positions of the bound (b) and free (f) 32P-labeled probe. (B) LTβR activates the NF-κB p65·p50 complex. HT29.14S cells were incubated with 1 nM LTα1β2 for 15 min. Antibodies to individual subunits of Rel family members (lanes 2–6) were added to the nuclear extracts for 15 min before electrophoresis. Extracts were preincubated with an excess of unlabeled mutant κB oligonucleotide (mut) (lane 7) or NF-κB binding site from the HIV-1 enhancer (wt) (lane 8). The results are representative of four similar experiments.
TRAF3Δ1–367 Expression Does Not Inhibit TRAF3 Recruitment to LTβR.
To investigate whether TRAF3Δ1–367 interferes with LTβR signaling by binding to the LTβR and preventing endogenous wild-type TRAF3 recruitment, ligand-dependent recruitment of TRAF3 and TRAF3Δ1–367 was compared in two clones which express TRAF3Δ1–367 and are highly resistant to LTβR-ligand-induced cell death and in the pool of control vector-transfected cells. When examined by LTβR immunoprecipitation and Western blotting with anti-TRAF3, LTα1β2 induced the same level of wild-type endogenous TRAF3 association with LTβR in TRAF3Δ1–367-expressing cells as in control cell lines (data not shown). To determine whether the mutant TRAF3 protein also associates with the LTβR, cell lines were labeled with [35S]methionine and [35S]cysteine, treated with LTα1β2, and lysed after 15 min, and the LTβR was immunoprecipitated. Both wild-type TRAF3 (60 kDa) and TRAF3Δ1–367 (25 kDa) immunoprecipitated with LTβR from the TRAF3Δ1–367-expressing cell lines after treatment with LTα1β2 (Fig. 4). As before, the amount of TRAF3 that coprecipitated with LTβR did not differ among TRAF3Δ1–367-expressing and nonexpressing cell lines. The ratio of TRAF3 to TRAF3Δ1–367 in the immunoprecipitates (after correction for the differences in Met and Cys residues) was 1.1 for both clones in two experiments. This result confirms the Western blotting analyses that TRAF3 association with LTβR is ligand dependent and unaffected by TRAF3Δ1–367 overexpression. Further, TRAF3Δ1–367 is present in the ligand–receptor complex in the TRAF3Δ1–367 overexpressing cells, in equimolar amounts to wild-type TRAF3, although overall TRAF3Δ1–367 is expressed in substantial molar excess over wild-type TRAF3 in these cells (Fig. 1C). Since TRAF3Δ1–367 can interact with TRAF3 at a high level in yeast two-hybrid assays (32, 33), one hypothesis which would be consistent with these data is that TRAF3Δ1–367 cannot compete with wild-type TRAF3 for ligand-clustered LTβR but can hetero-aggregate with wild-type TRAF3 bound to the receptor and thereby inhibit signal propagation.
Figure 4.
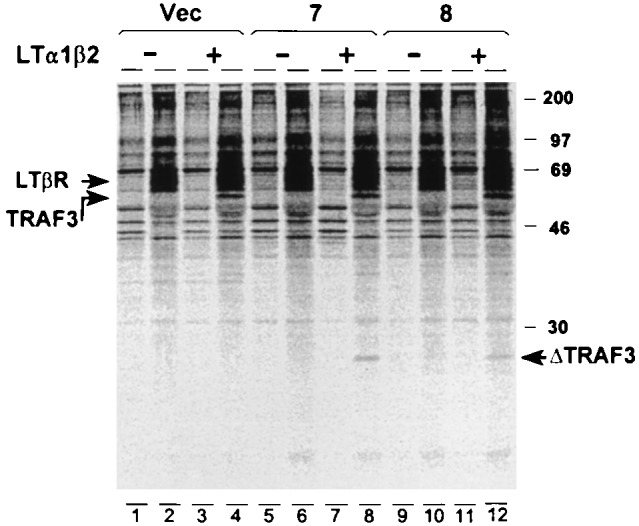
Recruitment of TRAFΔ1–367 to the LTβR signaling complex. Cell lines were labeled for 3 hr with [35S]methionine and [35S]cysteine and then treated with (+) or without (−) LTα1β2 at 1 nM for 15 min. The cell lysates were subjected to immunoprecipitation with goat anti-LTβR and separation of proteins by SDS/PAGE. Radioactivity was detected by a PhosphorImager (pixel range 30–600). Immunoprecipitates from the indicated cell lines formed with preimmune goat serum (lanes 1, 3, 5, 7, 9, and 11) or goat anti-LTβR (lanes 2, 4, 6, 8, 10, and 12). The density of LTβR bands measured at lower sensitivity were equivalent (<1% variation) between the cell lines.
DISCUSSION
The experiments presented here clearly establish the existence of two signaling pathways that emanate from the LTβR. One pathway leads to cell death and it depends on TRAF3-mediated signal propagation, while the second pathway leads to NF-κB activation and has previously been shown to depend on TRAF5-mediated signal propagation. Overexpression of an N-terminal deletion mutant of TRAF3, TRAF3Δ1–367, was used in these experiments to investigate the role of TRAF3 in signaling cell death. TRAF3Δ1–367 overexpression specifically blocked LTβR-dependent cell death, yet it did not affect NF-κB activation by the LTα1β2–LTβR complex. These results also highlight a functional difference between the role of TRAF3 in LTβR-dependent NF-κB activation and NF-κB activation mediated by other members of the TNFR family, since TRAF3 inhibits NF-κB activation by CD40 and TNFR80 (38). The TRAF3 mutant may not be able to inhibit LTβR-dependent NF-κB activation because it is unable to compete with NF-κB-activating TRAFs such as TRAF2 and TRAF5 for binding to LTβR. TRAF2 and TRAF5 may be more abundant than TRAF3 or they might have higher affinity than TRAF3 for LTβR. Alternatively, TRAF2 and TRAF5 may bind to a different site on LTβR than TRAF3.
The separation of NF-κB-inducing and cell death-inducing pathways in LTβR signaling is similar to the TNFR60 signal transduction mechanism. TNF-induced NF-κB activation can down-modulate the apoptotic effects of TNFR60 signaling (56–58). LTβR-induced NF-κB activation may provide a similar down-modulation of TRAF3-mediated cell death effects.
This work provides the first evidence we know of that TRAF3 association with a TNFR is ligand dependent. TRAF3 specifically associates with the LTβR within 1 min after treatment with LTα1β2 heterotrimer or agonistic antibodies. This result is consistent with the concept that ligand binding induces receptor aggregation, and receptor aggregation creates higher-affinity TRAF3-binding sites or higher-affinity binding sites for another protein that positively affects TRAF3 association with the receptor. So far, another protein is not evident in the analyses of ligand-induced LTβR-associated proteins. However, TRAF3 antibody detects crossreactive LTβR-associated proteins larger than TRAF3 which are indicative of ligand-induced modification of TRAF3 by another protein that could be receptor associated.
The finding that TRAF3 association with the LTβR is ligand dependent is consistent with the previous hypothesis that the Epstein–Barr virus (EBV) oncogene product, LMP1, mimics a constitutively activated TNFR (34, 48, 59). LMP1 has six hydrophobic transmembrane domains that enable it to constitutively homoaggregate in the plasma membrane. Genetic analyses indicate that the transmembrane domains are necessary for aggregation and for LMP1 activity in various transformation assays, including primary B lymphocyte transformation (60). In fact, the transmembrane domains and the part of LMP1 that engages TRAF3 and TRAF1 are sufficient for primary B lymphocyte growth transformation in the context of a specifically mutated EBV recombinant (61). In EBV-transformed primary B lymphocytes LMP1 is extensively associated with TRAF3 and TRAF1 (48). In fact, most of the TRAF3 and TRAF1 and a significant fraction of TRAF2 in EBV-transformed B lymphocytes is associated with LMP1 and most of the LMP1 is associated with TRAF3 or TRAF1. TRAF3 can negatively regulate NF-κB activation from the LMP1 TRAF-binding domain by displacement of TRAF1 and TRAF2.
An intriguing finding was that soluble anti-LTβR monoclonal antibody (BDA8) could induce formation of LTβR·TRAF3 complexes. This indicates that bivalent receptor aggregation is sufficient for efficient TRAF3 recruitment. However, BDA8 does not induce cell death in this system (27). TRAF3 recruitment therefore is not per se sufficient for induction of cell death. Additional events must occur after higher-order aggregation that are necessary to activate components of the death pathway.
Ligand-dependent recruitment of TRAF3 can be demonstrated in HT29 cells, which typically express a low density of receptors (≈103 to 104 per cell), but in systems that overexpress these components, the association between TRAFs and receptors is constitutive (non-ligand-dependent; ref. 39 and data not shown). Constitutive association is most likely a result of receptor or TRAF protein aggregation due to amplified protein levels. TRAF3 binds directly to LTβR-GST fusion protein (in the obvious absence of ligand), suggesting this fusion protein mimics the ligand–receptor complex, perhaps due to the multimeric structure of GST.
The results presented here indicate that the first 367 amino acids of TRAF3 are critical for signal propagation that leads to cell death from the LTβR. The LTβR–ligand complex associates with equimolar amounts of TRAF3 and TRAF3Δ1–367, and TRAF3Δ1–367 expression does not reduce the amount of TRAF3 that associates with the LTβR–ligand complex. The simplest model consistent with this finding is that TRAF3Δ1–367 is added onto the ligated LTβR–TRAF3 complex. The addition of TRAF3Δ1–367 may prevent the recruitment of an effector molecule by occupying its binding site on wild-type TRAF3. Alternatively, TRAF3Δ1–367 may alter the conformation of the binding site of an effector molecule on TRAF3. The fact that NF-κB activation is unaffected by TRAF3Δ1–367 overexpression indicates that the TRAF3Δ1–367 mutant does not globally affect the conformation and signaling properties of LTβR.
Collectively, these findings implicate a role for TRAF3 in cell death induced by LTβR and perhaps by other TNFR family members that bind TRAF3, such as TNFR80 (29), CD40 (28), and CD30 (30) and suggest that there may be alternate, non-death-domain, pathway for signaling cell death. This death-inducing pathway in HT29 cells has characteristics of a slow apoptotic process (27). In contrast to Fas ligand or TNF-induced cell death, which is usually evident within a few hours or 24 hr, respectively, LTβR-ligand-induced cell death requires 36–72 hr. Because of the time delay between TRAF3 association and cell death the effects of TRAF3 association with LTβR may be indirectly linked to the process of cell death. This is consistent with the fact that soluble anti-LTβR monoclonal antibody (BDA8) could induce LTβR–TRAF3 association but could not elicit cell death. The molecular mechanism by which TRAF3 signals cell death is unknown, but it may involve an interaction with a protein like TRADD that can interact with TRAFs (46) and death effector proteins like FADD (22, 23). Consistent with this possibility is our finding that TNF-induced cell death in HT29 cells is also partially inhibited by TRAF3Δ1–367.
The expression of LTα1β2 by cytotoxic T cells or natural killer (NK) cells is consistent with the possibility that cell death could be a biologically significant function of LTβR (8). LTβR appears to be important as a regulator of development and homeostasis of lymphoid organs, processes which involve both growth-promoting and apoptotic signaling. Mice lacking LTα or LTαβ do not form germinal centers during an immune response (4, 5). Alternatively, the TRAF3-mediated pathway may be an aberrant manifestation in tumor cells of signals emanating from the LTβR which would be growth-promoting in nontransformed cells (62).
Acknowledgments
We are grateful to Dr. Jeffrey Browning for insightful comments and to the LTβ project team at Biogen for their contributions of ligands, monoclonal antibodies, and the HT29.14S line. This work was supported in part by grants from the American Cancer Society (IM663 to C.F.W.), National Institutes of Health (AI33068 to C.F.W.; CA47006 to E.K.; and PO1CA69381 to J.C.R. and C.F.W.) and fellowships from the University of California University-wide AIDS Research Program (F95R-008, to W.R.F.) and the Leukemia Society of America (to G.M.).
ABBREVIATIONS
- GST
glutathione S-transferase
- LT
lymphotoxin
- LTβR
LTβ receptor
- NF-κB
nuclear factor κB
- TNF
tumor necrosis factor
- TNFR
TNF receptor
- TRAF
TNF receptor-associated factor
- ICAM-1
intracellular adhesion molecule 1
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
References
- 1.Ware C F, VanArsdale T L, Crowe P D, Browning J L. In: Pathways for Cytolysis, Current Topics in Microbiology and Immunology. Griffiths G M, Tschopp J, editors. Vol. 198. Basel: Springer; 1995. pp. 175–218. [DOI] [PubMed] [Google Scholar]
- 2.De Togni P, Goellner J, Ruddle N H, Streeter P R, Fick A, Mariathasan S, Smith S C, Carlson R, Shornick L P, Strauss-Schoenberger J, Russell J H, Karr R, Chaplin D D. Science. 1994;264:703–706. doi: 10.1126/science.8171322. [DOI] [PubMed] [Google Scholar]
- 3.Banks T A, Rouse B T, Kerley M K, Blair P J, Godfrey V L, Kuklin N A, Bouley D M, Thomas J, Kanangat S, Mucenski M L. J Immunol. 1995;155:1685–1693. [PubMed] [Google Scholar]
- 4.Matsumoto M, Lo S F, Carruthers C J L, Min J, Mariathasan S, Huang G, Plas D R, Martin S M, Geha R S, Nahm M H, Chaplin D D. Nature (London) 1996;382:462–466. doi: 10.1038/382462a0. [DOI] [PubMed] [Google Scholar]
- 5.Matsumoto M, Mariathasan S, Nahm M H, Baranyay F, Preschon J J, Chaplin D D. Science. 1996;271:1289–1291. doi: 10.1126/science.271.5253.1289. [DOI] [PubMed] [Google Scholar]
- 6.Ettinger R, Browning J L, Michie S A, van Ewijk W, McDevitt H O. Proc Natl Acad Sci USA. 1996;93:13102–13107. doi: 10.1073/pnas.93.23.13102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rennert P, Browning J L, Hochman P S. J Exp Med. 1996;184:1999–2006. doi: 10.1084/jem.184.5.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ware C F, Crowe P D, Grayson M H, Androlewicz M J, Browning J L. J Immunol. 1992;149:3881–3888. [PubMed] [Google Scholar]
- 9.Androlewicz M J, Browning J L, Ware C F. J Biol Chem. 1992;267:2542–2547. [PubMed] [Google Scholar]
- 10.Browning J L, Dougas I, Ngam-ek A, Bourdon P R, Ehrenfels B N, Miatkowski K, Zafari M, Yampaglia A M, Lawton P, Meier W. J Immunol. 1995;154:33–46. [PubMed] [Google Scholar]
- 11.Loetscher H, Pan Y, Lahm H, Gentz R, Brockhaus M, Tabuchi H, Lesslauer W. Cell. 1990;61:351–359. doi: 10.1016/0092-8674(90)90815-v. [DOI] [PubMed] [Google Scholar]
- 12.Schall T J, Lewis M, Koller K J, Lee A, Rice G C, Wong G H, Gatanaga T, Granger G A, Lentz R, Raab H, Kohr W J, Goeddel D V. Cell. 1990;61:361–370. doi: 10.1016/0092-8674(90)90816-w. [DOI] [PubMed] [Google Scholar]
- 13.Smith C A, Davis T, Anderson D, Solam L, Beckmann M P, Jerzy R, Dower S K, Cosman D, Goodwin R G. Science. 1990;248:1019–1023. doi: 10.1126/science.2160731. [DOI] [PubMed] [Google Scholar]
- 14.Crowe P D, VanArsdale T L, Walter B N, Ware C F, Hession C, Ehrenfels B, Browning J L, Din W S, Goodwin R G, Smith C A. Science. 1994;264:707–710. [PubMed] [Google Scholar]
- 15.Smith C A, Farrah T, Goodwin R G. Cell. 1994;76:959–962. doi: 10.1016/0092-8674(94)90372-7. [DOI] [PubMed] [Google Scholar]
- 16.Nagata S, Golstein P. Science. 1995;267:1449–1456. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- 17.Tartaglia L A, Ayres T M, Wong G H, Goeddel D V. Cell. 1993;74:845–853. doi: 10.1016/0092-8674(93)90464-2. [DOI] [PubMed] [Google Scholar]
- 18.Itoh N, Nagata S. J Biol Chem. 1993;268:10932–10937. [PubMed] [Google Scholar]
- 19.Cleveland J L, Ihle J N. Cell. 1995;81:479–482. doi: 10.1016/0092-8674(95)90068-3. [DOI] [PubMed] [Google Scholar]
- 20.Boldin M P, Mett I L, Varfolomeev E E, Chumakov I, Shemer-Avni Y, Camonis J H, Wallach D. J Biol Chem. 1995;270:387–391. doi: 10.1074/jbc.270.1.387. [DOI] [PubMed] [Google Scholar]
- 21.Hsu H, Xiong J, Goeddel D V. Cell. 1995;81:495–504. doi: 10.1016/0092-8674(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 22.Boldin M P, Varfolomeev E E, Pancer Z, Mett I L, Camonis J H, Wallach D. J Biol Chem. 1995;270:7795–7798. doi: 10.1074/jbc.270.14.7795. [DOI] [PubMed] [Google Scholar]
- 23.Chinnaiyan A M, Tepper C G, Seldin M F, O’Rourke K, Kischkel F C, Hellbardt S, Krammer P H, Peter M E, Dixit V M. J Biol Chem. 1996;271:4961–4965. doi: 10.1074/jbc.271.9.4961. [DOI] [PubMed] [Google Scholar]
- 24.Stanger B Z, Leder P, Lee T H, Kim E, Seed B. Cell. 1995;81:513–523. doi: 10.1016/0092-8674(95)90072-1. [DOI] [PubMed] [Google Scholar]
- 25.Boldin M P, Goncharov T M, Goltsev Y V, Wallach D. Cell. 1996;85:803–815. doi: 10.1016/s0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- 26.Chinnaiyan A M, O’Rourke K, Tewari M, Dixit V M. Cell. 1995;81:505–512. doi: 10.1016/0092-8674(95)90071-3. [DOI] [PubMed] [Google Scholar]
- 27.Browning J L, Miatkowski K, Sizing I, Griffiths D A, Zafari M, Benjamin C D, Meier W, Mackay F. J Exp Med. 1996;183:867–878. doi: 10.1084/jem.183.3.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Collins T, Read M A, Neish A S, Whitley M Z, Thanos D, Maniatis T. FASEB J. 1995;9:899–909. [PubMed] [Google Scholar]
- 29.Grell M, Douni E, Wajant H, Lohden M, Clauss M, Maxeiner B, Georgopoulos S, Lesslauer W, Kollias G, Pfizenmaier K. Cell. 1995;83:793–802. doi: 10.1016/0092-8674(95)90192-2. [DOI] [PubMed] [Google Scholar]
- 30.Lee S Y, Park C G, Choi Y. J Exp Med. 1996;183:669–674. doi: 10.1084/jem.183.2.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu H M, O’Rourke K, Boguski M S, Dixit V M. J Biol Chem. 1994;269:30069–30072. [PubMed] [Google Scholar]
- 32.Sato T, Irie S, Reed J C. FEBS Lett. 1995;358:113–118. doi: 10.1016/0014-5793(94)01406-q. [DOI] [PubMed] [Google Scholar]
- 33.Cheng G, Cleary A M, Ye Z S, Hong D I, Lederman S, Baltimore D. Science. 1995;267:1494–1498. doi: 10.1126/science.7533327. [DOI] [PubMed] [Google Scholar]
- 34.Mosialos G, Birkenbach M, Yalamanchili R, VanArsdale T, Ware C, Kieff E. Cell. 1995;80:389–399. doi: 10.1016/0092-8674(95)90489-1. [DOI] [PubMed] [Google Scholar]
- 35.Gedrich R W, Gilfillan M C, Duckett C S, Van Dongen J L, Thompson C B. J Biol Chem. 1996;271:12852–12858. doi: 10.1074/jbc.271.22.12852. [DOI] [PubMed] [Google Scholar]
- 36.Ansieau S, Scheffrahn I, Mosialos G, Brand H, Duyster J, Kaye K, Harada J, Dougall B, Hubinger G, Kieff E, Herrmann F, Leutz A, Gruss H-J. Proc Natl Acad Sci USA. 1996;93:14053–14058. doi: 10.1073/pnas.93.24.14053. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Rothe M, Wong S C, Henzel W J, Goeddel D V. Cell. 1994;78:681–692. doi: 10.1016/0092-8674(94)90532-0. [DOI] [PubMed] [Google Scholar]
- 38.Rothe M, Sarma V, Dixit V M, Goeddel D V. Science. 1995;269:1424–1427. doi: 10.1126/science.7544915. [DOI] [PubMed] [Google Scholar]
- 39.Nakano H, Oshima H, Chung W, Williams-Abbott L, Ware C, Yagita H, Okumura K. J Biol Chem. 1996;271:14661–14664. doi: 10.1074/jbc.271.25.14661. [DOI] [PubMed] [Google Scholar]
- 40.Ishida T, Tojo T, Aoki T, Kobayashi N, Ohishi T, Watanabe T, Yamamoto T, Inoue J-I. Proc Natl Acad Sci USA. 1996;93:9437–9442. doi: 10.1073/pnas.93.18.9437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lenardo M J, Baltimore D. Cell. 1989;58:227–229. doi: 10.1016/0092-8674(89)90833-7. [DOI] [PubMed] [Google Scholar]
- 42.Thanos D, Maniatis T. Cell. 1995;80:529–532. doi: 10.1016/0092-8674(95)90506-5. [DOI] [PubMed] [Google Scholar]
- 43.Rothe M, Pan M G, Henzel W J, Ayres T M, Goeddel D V. Cell. 1996;83:1243–1252. doi: 10.1016/0092-8674(95)90149-3. [DOI] [PubMed] [Google Scholar]
- 44.Rothe M, Xiong J, Shu H-B, Williamson K, Goddard A, Goeddel D V. Proc Natl Acad Sci USA. 1996;93:8241–8246. doi: 10.1073/pnas.93.16.8241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cheng G, Baltimore D. Genes Dev. 1996;10:963–973. doi: 10.1101/gad.10.8.963. [DOI] [PubMed] [Google Scholar]
- 46.Hsu H, Shu H B, Pan M G, Goeddel D V. Cell. 1996;84:299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- 47.Takeuchi M, Rothe M, Goeddel D V. J Biol Chem. 1996;271:19935–19942. doi: 10.1074/jbc.271.33.19935. [DOI] [PubMed] [Google Scholar]
- 48.Devergne O, Hatzivassiliou E, Izumi K M, Kaye K M, Kleijnen M F, Kieff E, Mosialos G. Mol Cell Biol. 1996;16:7098–7108. doi: 10.1128/mcb.16.12.7098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Browning J, Ribolini A. J Immunol. 1989;143:1859–1867. [PubMed] [Google Scholar]
- 50.Browning J L, Miatkowski K, Griffiths D A, Bourdon P R, Hession C, Ambrose C M, Meier W. J Biol Chem. 1996;271:8618–8626. doi: 10.1074/jbc.271.15.8618. [DOI] [PubMed] [Google Scholar]
- 51.VanArsdale T L, Ware C F. J Immunol. 1994;153:3043–3050. [PubMed] [Google Scholar]
- 52.Green L M, Reade J L, Ware C F. J Immunol Methods. 1984;70:257–268. doi: 10.1016/0022-1759(84)90190-x. [DOI] [PubMed] [Google Scholar]
- 53.Crowe P D, Walter B N, Mohler K M, Otten-Evans C, Black R A, Ware C F. J Exp Med. 1995;181:1205–1210. doi: 10.1084/jem.181.3.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Force W R, Tillman J B, Sprung C N, Spindler S R. J Biol Chem. 1994;269:8863–8871. [PubMed] [Google Scholar]
- 55.Hou J, Baichwal V, Cao Z. Proc Natl Acad Sci USA. 1994;91:11641–11645. doi: 10.1073/pnas.91.24.11641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Beg A A, Baltimore D. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 57.Wang C-Y, Mayo M W, Baldwin A S. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- 58.Van Antwerp D J, Martin S J, Kafri T, Green D R, Verma I M. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 59.Kaye K M, Devergne O, Harada J N, Izumi K M, Yalamanchili R, Kieff E, Mosialos G. Proc Natl Acad Sci USA. 1996;93:11085–11090. doi: 10.1073/pnas.93.20.11085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kaye K M, Izumi K M, Kieff E. Proc Natl Acad Sci USA. 1993;90:9150–9154. doi: 10.1073/pnas.90.19.9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kaye K M, Izumi K M, Mosialos G, Kieff E. J Virol. 1995;69:675–683. doi: 10.1128/jvi.69.2.675-683.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hochman P S, Majeau G R, Mackay F, Browning J L. J Inflammation. 1996;46:220–234. [PubMed] [Google Scholar]