

Abstract
Development of effective drugs against the rhinovirus (HRV) responsible for the common cold remains a challenge because there are over 100 serotypes. This process could be significantly aided by an understanding of the atomistic mechanism by which such drugs work. We suggest that the most effective drugs against HRV-1A act by stiffening the pentamer channel of the viral coat through which the RNA is released, preventing the steps leading to uncoating. Using molecular dynamics methods we tested this Pentamer Channel Stiffening Model (PCSM) by examining the changes in strain energy associated with opening the pentamer channel through which the RNA is released. We find that the PCSM strain correlates well with the effectiveness of the WIN (Sterling–Winthrop) drugs for HRV-1A. To illustrate the use of the PCSM to predict new drugs and to prioritize experimental tests, we tested three modifications of the WIN drugs that are predicted to be nearly as effective (for HRV-1A) as the best current drug.
The major cause of common cold in humans is the rhinovirus (HRV), a member of the picornavirus family. HRV exhibits over 100 serotypes (1), making it difficult to obtain a general effective drugs. We are using atomistic force fields (FF) and molecular dynamics (MD) to help elucidate the atomic-level mechanism by which current drugs operate on HRV with the hope that this will lead to models useful in developing new drugs that operate on all serotypes.
The serotypes of HRV are classified into two groups. The major receptor group (including HRV-14 and HRV-16) binds to the intercellular adhesion molecule 1 receptors. The minor receptor group (including HRV-1A) binds to low density lipoprotein-type receptor molecules. Both HRV-14 and HRV-1A and their complexes with several drugs have been studied using x-ray crystallogaphy (2–7). The protein capsid of HRV consists of an icosahedral shell with 60 copies each of four proteins: VP1, VP2, VP3, and VP4 (see Fig. 1), totaling about 480,000 atoms (≈300 Å diameter). This protein capsid encloses a single-strand RNA (with about 7500 nucleotides) inside the cavity (≈100 Å diameter) of the icosahedral shell. The 5-fold sites consist of five VP1 molecules, and are referred to as the VP1 pentamer.
Figure 1.

HRV-1A icosahedral protein coat showing 466,080 atoms, each atom being shown as a point in the figure. VP1 is in red, VP2 in green, VP3 in blue, and VP4 in yellow. (This figure was made using mpsim/pts software developed by M. Iotov and K. T. Lim, Materials and Process Simulation Center). The diameter is ≈300 Å.
The pathway for HRV to infection is through a receptor-mediated endocytosis. After the binding of HRV to the cellular receptors, the VP4 clustered on the inner surface under the pentamer channel of the VP1 pentamer comes out through the pentamer channel. Subsequently, uncoating of the viral capsid leads to the release of the RNA in the cytoplasm. Although no precise sequence of events leading to virus uncoating, after penetration has yet been proposed, circumstantial evidence from experimental studies on poliovirus (same picornavirus family) justify the speculation that RNA is released through the center of the VP1 pentamer in the virus (1).
The x-ray structures for both HRV-1A and HRV-14 (6) show a canyon surrounding the pentamer site (formed from packing of five VP1 proteins) that is responsible for HRV receptor interaction. This receptor binding site is a highly conserved part on the virus shell. Several classes of compounds active against HRV have been identified (8) including isoxazole-derived drugs developed by Sterling–Winthrop (denoted WIN) and by the Janssen Research Foundation (denoted R) for HRV-14 and HRV-1A. For both HRV-14 and HRV-1A, the WIN drugs bind to the hydrophobic pocket on VP1 (known as the WIN pocket). Binding of the WIN drugs to HRV-14 disrupts the conformation of the residues lining the canyon floor (the WIN pocket is below the canyon floor) by about 4 Å (9). This prevents binding to the intercellular adhesion molecule 1 cellular receptors and thereby inhibits virus multiplication. Guha-Biswas et al. (10) analyzed the drug binding mechanism for HRV-14 and its WIN drugs using a conformational search procedure. They found that a large movement in the residues near the WIN pocket is theoretically possible, thus providing greater access for the entry of the drug into the cavity in HRV-14. For HRV-1A, the WIN pocket in the native virus is already occupied by a cofactor, possibly cellular in origin (3, 6). In this case the WIN drugs displace the cofactor but do not cause major conformational changes in the canyon floor. The crystal structure of HRV-1A has a conformation similar to that observed for drug-bound HRV-14. For drug-bound HRV-1A there is no blocking of receptor attachment. Because the drug is still effective, it has been speculated that the drug action must be due to the stabilization (increased stiffness) of the protein shell (11, 12).
We believe that interference with the uncoating process might be a good strategy for developing a drug to act on all serotypes of HRV, and hence we initiated FF and MD studies of this uncoating process. Based on the speculation that the drugs stiffen the viral capsid in HRV-1A (9) and that the RNA is released through the pentamer channel, we propose the Pentamer Channel Stiffness Model (PCSM): drug action on HRV-1A constricts or stiffens the pentamer channel sufficiently that the RNA and/or VP4 cannot exit, thus preventing uncoating. In this communication we report calculations testing for HRV-14 and HRV-1A and their drug complexes. We find that drug effectiveness correlates with increased stiffness of the pentamer channel, in support of the PCSM for HRV-1A. The WIN drugs do not stiffen the pentamer channel for HRV-14. We evaluated the effectiveness of modification of the WIN drugs using the PCSM and report three new drug candidates that are predicted to be as effective as WIN56291, one of the most effective drugs known for HRV1A.
The details of the calculations are described in Calculational Details, and the results are reported in the Results section. The Application of PCSM section contains the application of PCSM for the design of new drugs and a discussion of the results.
CALCULATIONAL DETAILS
Force Fields.
The forces between the atoms in the virus were described using the AMBER FF (13). In AMBER, hydrogen bonding is described in terms of Coulomb interactions (attractive) and a special two-body nonbond introduced to prevent unphysically short approach of the hydrogen to the acceptor atoms.
The forces involving drug molecules and cofactors were described using the DREIDING (14) FF, known to give good structures for such organic systems.
Structures.
The starting structures for native HRV-1A and HRV-14 and for the drug-bound systems were obtained from x-ray crystallography (based on exact icosahedral symmetry). The crystal structure of the asymmetric unit (by asymmetric unit we mean the protomer) of HRV-14 contains 6649 protein atoms (excluding hydrogens) and 272 crystallographic water molecules. Explicit hydrogens were added to nitrogen and oxygen atoms to allow for hydrogen bonds. The lone pair centers required by AMBER to describe the sulfurs of Met and Cys were added. The resulting structure has 8521 atoms in the asymmetric unit. The structure for several residues were missing from the x-ray structure due to disorder. We used the x-ray structure as available and added terminal hydrogens to the amino terminus with appropriate AMBER charges. Using the icosahedral symmetry elements the structures of the entire viral coats of HRV-14 and HRV-1A were derived.
Solvation HRV-1A and HRV-14.
The virus is in an aqueous environment and hence the simulations should consider the virus in a large box filled with water. Because of the large number of atoms, we chose to treat the water solvent implicitly. This is done by solvating each charged group in the protein using appropriate counterions. We consider that this, along with using a distance-dependent dielectric, represents the solvation effects induced by a real bath of water. Thus Asp and Glu are solvated by Na+ while Lys and Arg are solvated by Cl− using special parameters (15) for the solvated counterions Na+ and Cl−. This is done for every charged group unless there is a nearby residue of opposite charge (a salt bridge). The dielectric constant is taken as ɛ = ɛ0R, where ɛ0 = 1.
RNA.
The RNA does not have icosahedral symmetry and consequently it is not extracted from the crystallographic data. Consequently we ignored the RNA in all calculations. To determine whether this affects the structures, we carried out Hoover constant temperature dynamics on the full 512,760-atom protein capsid of HRV-14. We see no drift in the size of the capsid, suggesting that the structure of the capsid is stable in the absence of RNA.
The pentamer simulations.
To determine whether the drug affects the stiffness of the viral capsid, it is necessary to do MD simulations on at least the full pentamer. Therefore most calculations focused on the pentamer that included all atoms of the VP1, VP2, VP3, and VP4 proteins for all five asymmetric units. The resulting pentamer is solvated so that it is neutral, leading to 42,730 atoms for HRV-14 (including crystallographic waters and counterions) and 38,840 atoms for the HRV-1A pentamer.
Using the AMBER FF for HRV-1A and HRV-14, we calculated the optimum geometry [converged to rms force error of 0.1 (kcal/mol)/Å]. Table 1 shows that this leads to rms coordinate errors of 0.34 Å and 0.40 Å (excluding the waters and the counterions) compared with experiment for HRV-14 and HRV-1A, respectively.
Table 1.
RMS variation in coordinates (Å) between the starting x-ray structure and the minimized structure for pentamers with and without drugs
| Case | Protein | Drug | |
|---|---|---|---|
| HRV-1A | Empty | 0.398 | |
| With sucrose | 0.618 | 0.937 | |
| With WIN54954 | 0.628 | 0.824 | |
| With WIN56291 | 0.572 | 0.785 | |
| With R61837 | 0.649 | 0.684 | |
| HRV-14 | Empty | 0.344 | |
| With WIN52084 | 0.572 | 0.564 | |
| With r07 | 0.569 | 0.802 |
Crystallographic waters and counterions were excluded from the RMS.
The rms difference in coordinates between the starting structure (x-ray) and the minimized structure (theory) for various virus–drug complexes is given in Table 1, where we see agreement to about 0.6 Å for the protein and 0.6–0.8 Å for the drug. This is reasonable given the resolution of the experiments.
The difference in the crystal structures between HRV-1A and HRV-14 has been discussed by Kim et al. (6). The secondary structures of the four viral proteins are similar although the sequences are not (39% homology for VP1, 62% for VP2, and 52% for VP3). There is a great deal of sequence similarity in the canyon region of the various serotypes. The structure at the neutralizing immunogenic sites (antibody binding sites) and the GH loop of VP1 are the most variable loops in HRV-14 and HRV-1A.
The HRV-1A cofactor.
In HRV-1A the virus in its native state has a cofactor in the WIN pocket. The cofactor in HRV-1A was interpreted originally (3) as sucrose used in the purification of the virus. Because sphingosine or palmitate-like molecules have been found within the homologous pocket of polioviruses (16, 17), the cofactor is now interpreted as a 7-carbon fatty acid (6), (C6H13)C(O)OH. To predict the structure of the protein containing this fatty acid cofactor, we started with the crystal structure containing sucrose in HRV-1A. We converted the sucrose to the fatty acid, placing the carboxylate head near Asn-215 of VP1. The geometry of the cofactor was optimized by (i) conjugate gradient minimization of the cofactor while keeping the virus fixed until the RMS force changes were below 0.1 (kcal/mol)/Å, and (ii) minimization of all atoms of the full cofactor/protein complex.
The minimized structure for the fatty acid cofactor is shown in Fig. 2. The polar head of the carboxylic acid is near (see Fig. 2) Asn-215 (4.95 Å), Thr-102 (1.98 Å from the hydrogen of the threonyl oxygen), and Leu-103 (2.02 Å from its backbone nitrogen) (6) and hydrogen bonded to these residues. The hydrophobic tail of the cofactor is close to the Met-217 (4.55 Å) and Tyr-193 (4.21 Å from one of the carbons of the phenyl ring) of VP1 as predicted by Kim et al. (6). All these residues belong to VP1 of the viral capsid. (The coordinates of the fatty acid are in the supplementary material; see Fig. 2 legend.)
Figure 2.

Optimized structure of the (C6H13)C(O)OH fatty acid cofactor (red) in HRV-1A showing the distances of the COOH polar head of the fatty acid from the Asn-215 (purple), Leu-103 (green), and the threonyl oxygen of the Thr-102 (blue). (The coordinates of the fatty acid are in supplementary material that contains the coordinates of the virus HRV-1A with the fatty acid, and the coordinates of HRV-1A with the drugs WIN52035, WIN54221, and WIN56278. This material is in biograf format and is available at http://www.wag.caltech.edu.)
The drug molecules.
Fig. 3 shows the various WIN and Janssen drugs examined for HRV-1A and HRV-14 (the bottom two drugs in Fig. 3 are for HRV-14). There is crystallographic data on HRV-1A complex with WIN54954, WIN56291, or R61837. In these cases the pentamer was generated from the crystallographic data using symmetry conditions for icosahedral symmetry. The structure of the full drug/protein complex was optimized, leading to drug molecule/HRV structures with rms error shown in Table 1.
Figure 3.

WIN and Janssen drugs for HRV-1A. The minimum inhibitory concentration (MIC) values (in μM) are from refs. 6 and 18. The MIC values for MSC1, MSC2, and MSC3 shown in parenthesis are predicted values from Fig. 6a. The bottom two drugs are for HRV-14 (MIC values are from ref. 4).
The crystal structures were not available for WIN52035, WIN56278, and WIN54221. To predict the structure of WIN52035, we replaced the two chlorine atoms of WIN54954 with hydrogens and reoptimized the pentamer–drug complex. For WIN56278 we modified WIN54954 by adding a double bond in the alkyl chain (Fig. 3) and reoptimized the pentamer– drug complex. Similarly WIN54221 was built from WIN54954 by converting a single bond in the alkyl chain to a triple bond (Fig. 3) and reoptimizing the pentamer–drug complex.
To design new drug molecules, we used the known inhibitors as guides to build the molecules into the protein. We then minimized the structure keeping the protein structure fixed. This was followed by minimizing the full structure.
The Helium Balloon String Measurement of Pentamer Stiffness.
We assume that the protein decoating involves VP4 and/or RNA being released through the center of the VP1 pentamer site. To estimate how easy it is for this site to expand to the radius required by the VP4 and/or the RNA, we constructed a flexible string of 36 He atoms spaced by 2.0 Å. This 70-Å string (Fig. 4 a and b) is placed along the center of the pentamer site so that one tip is at the outer surface of the pentameric site (see Fig. 4a) while the other is near the VP4-VP3 complex (Fig. 4b). Starting with a vdW radius of RHe = 0.0, the He balloon string was then slowly expanded (allowing the He string to adjust to the shape of the pentamer channel) while the atoms of the protein were allowed to readjust. This was done by increasing the van der Waals radius RHe. The increase in energy (strain) is a measure of how difficult it is for the pentamer channel to open up as the VP4 and/or RNA is exported. As discussed below, we find significant differences in how various drugs affect the pentamer stiffness, with a good correlation with drug effectiveness.
Figure 4.

The pentamer of HRV-1A with WIN56291. [These figures were specially made by Terry Coley (Virtual Chemistry, San Diego) using vanimator software.] The He balloon string is shown in pink and the WIN56291 drug molecules are in cyan. VP1 is in red, VP2 is in green, VP3 is in blue, and VP4 is in yellow. (a) Top view of the pentamer. The diameter of the region shown is ≈90 Å. (b) Edge-on-view of the pentamer showing the He balloon string length (which is 70 Å long).
We used the following FF for the He. Each pair of adjacent He atoms has a harmonic bond [Ebond = ½ KR(R − Re)2] with Re= 2.0 Å and KR= 2000 (kcal/mol)/Å2. The bond angle terms is described as Eangle = Kθ(1 + cosθ) with a minimum at θ = 180° and kθ = 10 (kcal/mol)/rad2. The He van der Waals parameter was described as Evdw = Dv(ρ−12 − 2ρ−6), where ρ = R/Rv using Dv = 0.012 kcal/mol. Rv was varied from 0 to 20 Å. The interaction of the He with the protein was described with standard (geometric) combination rules: Rij = 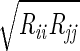 and Dij =
and Dij = 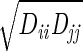 . However, all He · · ·He van der Waals interactions were ignored. The strain energies quoted exclude all terms involving He.
. However, all He · · ·He van der Waals interactions were ignored. The strain energies quoted exclude all terms involving He.
Programs.
MD calculations on the ≈40,000 atoms of the pentamer and particularly the ≈500,000 atoms of the full protein can become quite tedious and time-consuming, particularly because of the long-range nonbond interactions (electrostatic and vdW). In addition, the use of cutoffs in these nonbond interactions might bias the dependence of strain energies on RHe. Consequently, we used the cell multipole method (CMM) (19) for describing the nonbond interactions because it does not involve distance cut-offs. To rapidly carry out these calculations while retaining high accuracy, we used the mpsim program (18). mpsim was developed to efficiently carry out such accurate calculations on massively parallel computers. The calculations reported here used the Kendall Square Research 64-node machine in the Materials and Process Simulation Center. We used 60 processors for the full capsid (512,760 atoms) and 25 processors for the pentamers (≈40,000 atoms).
RESULTS
HRV-1A.
Using the He balloon string we tested the pentamer stiffness model (PCSM) for drug action on HRV-1A. The PCSM assumes that the drug stabilizes the viral shell, preventing uncoating. Experiments suggest that while the drug might stiffen the viral capsid for HRV-1A, these drugs inhibit cell binding in HRV-14 and it is not clear whether the drugs affect uncoating in HRV-14.
Fig. 5 shows how the strain in HRV-1A pentamer increases with the size of the He atoms. The energy required to open up the pentamer channel for native HRV-1A is much smaller than when drugs are present. For the native HRV-1A virus with either sucrose or the fatty acid, the energy does not increase rapidly until RHe = 20 Å, whereas with drugs WIN56291 (MIC value = 0.1μM), R61837 (MIC = 0.7 μM), WIN54954 (MIC = 2.5 μM), and WIN56278 (MIC = 2.25 μM) the energy increases rapidly above RHe = 10, 12, 14, and 14 Å, respectively. [MIC values were measured by experiments (6, 20).] This suggests that the effective drugs do indeed stiffen the pentamer site of the viral coat over that of the native state. For WIN52035 with MIC = 4.2 μM, the energy increases rapidly for RHe > 16 Å. This is stiffer than the native virus but is not as high as for WIN56291. The strain energy for WIN54221 (an inactive drug) behaves similar to the native HRV-1A with sucrose as the cofactor, again showing the correlation between the strain energy and the drug effectiveness.
Figure 5.

Pentamer stiffness for HRV-1A with various WIN and R drugs. The MIC values (in μM) in parenthesis are from refs. 6 and 20.
Fig. 6a shows the variation of strain energy at RHe = 18 Å with 1/MIC. The fit E(RHe = 18) = 1255 + 1200 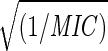 is shown in dotted lines and has been used to predict the MIC values for new drugs designed in the next section. Fig. 6b is the RHe at which Estrain = 1500 (kcal/mol) variation with 1/MIC. The fit Rcritical = 15.479 − 1.919 log(1/MIC) has also been used to predict the MIC values for new drugs along with Fig. 6a. The good overall correlation supports the PCSM hypothesis that stiffening of the VP1 pentamer site of HRV-1A correlates with drug efficacy. Based on these results we suggest that the PCSM be used to predict the likely drug effectiveness.
is shown in dotted lines and has been used to predict the MIC values for new drugs designed in the next section. Fig. 6b is the RHe at which Estrain = 1500 (kcal/mol) variation with 1/MIC. The fit Rcritical = 15.479 − 1.919 log(1/MIC) has also been used to predict the MIC values for new drugs along with Fig. 6a. The good overall correlation supports the PCSM hypothesis that stiffening of the VP1 pentamer site of HRV-1A correlates with drug efficacy. Based on these results we suggest that the PCSM be used to predict the likely drug effectiveness.
Figure 6.

Comparison of pentamer stiffness with MIC for various HRV-1A–drug complexes. (a) The strain energy at RHe = 18 Å. The values in parenthesis are the MIC values from reference 6 and 18. The dashed curve is the fit to Estrain(18 Å) = 1255 + 1200 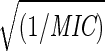 . For MSC1, MSC2, and MSC3 the predicted MIC values are shown on the fitted curve. (b) The radius at which Estrain = 1500 kcal/mol. The dashed curve is the fit to Rcrit = 15.479 − 1.919 log(1/MIC). The predicted MIC values for MSC1, MSC2, and MSC3 are shown on the fitted curve.
. For MSC1, MSC2, and MSC3 the predicted MIC values are shown on the fitted curve. (b) The radius at which Estrain = 1500 kcal/mol. The dashed curve is the fit to Rcrit = 15.479 − 1.919 log(1/MIC). The predicted MIC values for MSC1, MSC2, and MSC3 are shown on the fitted curve.
HRV-14.
The WIN drugs on HRV-14 have been suggested to have dual action of preventing cell attachment and preventing uncoating. However, it is not known if the drug prevents uncoating for HRV-14 because the WIN drugs impedes the cell attachment that triggers the uncoating process. In contrast, HRV-1A attaches to the cell in spite of bound WIN drugs, yet the virus does not uncoat, indicating that WIN impedes the uncoating mechanism in HRV-1A. We carried out the He balloon string studies on native HRV-14 and with the two known WIN drugs listed in Fig. 3 (along with their MIC values). This includes one good drug and one poor drug (high MIC value). (The WIN pocket in HRV-14 is longer than that of HRV-1A and hence the long alkyl chain drugs are more effective here than on HRV-1A; the WIN drugs in HRV-14 also bind in the hydrophobic WIN pocket as in HRV-1A.) The structure of the pentamer was minimized with the added counterions and the crystallographic waters. The rms difference in coordinates shows fairly good accuracy of these simulations for HRV-14 and its WIN drugs as shown in Table 1.
Fig. 7 shows the strain energy versus RHe for HRV-14 and its WIN drugs. According to PCSM, the HRV-14 virus shows no stiffening of the pentamer channel when the drug is present. Here we see no special correlation of the strain induced by r07 and WIN52084 with their MIC values. That is, although WIN52084 has a low MIC value of 0.03, the PCSM does not lead to stiffening greater than r07 (whose MIC is 2.4). These results suggest that inhibition of uncoating is not the major effect of the current drugs on HRV-14.
Figure 7.

Pentamer stiffness for HRV-14 with various WIN drugs. The MIC values (in μM) in parenthesis are from ref. 4.
Phelps and Post (21) used stochastic MD simulations to study the compressibility of HRV-14 with and without the WIN52084 drug. The compressibility was calculated using MD for about 3000 atoms surrounding the WIN pocket. They find a higher compressibility for the viral coat with WIN52084 than without the drug, suggesting that a more compressible coat leads to a better drug. The increase in compressibility in HRV-14 perhaps explains the lack of cell attachment.
APPLICATION OF PCSM
New Drugs.
To illustrate how one might use the atomic mechanism to design new drugs, we modified the best drug, WIN56291. It is known that WIN56291 is a better drug for HRV-1A than WIN54954 whereas the opposite is found for HRV-14. The difference between these drugs (Fig. 3) is that the length of the alkyl chain between the iso-oxazole ring and the phenyl oxazole rings is shorter for WIN56291. This difference in effectiveness probably arises because HRV-1A has a shorter (6) WIN pocket than HRV-14. Hence, a shorter drug might be more effective for HRV-1A than for HRV-14. Based on this we speculated that a new drug (denoted MSC1) where the alkyl chain is completely removed from WIN56291 (with only the ether like oxygen remaining in the alkyl chain) would be more effective for HRV-1A. To test this idea we optimized MSC1/HRV-1A and calculated the strain (Fig. 5). The energy increase starts at about the same radius as WIN56291 and Rcritical ≈ 14 Å for MSC1. The MIC value of MSC1 calculated from the fit to Fig. 6a is ≈0.13 μM. Thus according to PCSM, MSC1 should be comparable in effectiveness to WIN56291. This could be due to the removal of the flexible alkyl chain in WIN56291 by a rigid ether-like linkage (22) that makes MSC1 less volume filling.
A second new drug (MSC2) was built by removing the two chlorines on the phenyl ring of MSC1. The drug with the optimized viral coat of the MSC1 was further optimized for the structure of MSC2. This was done by first keeping the pentamer of the viral coat fixed, and MSC2 is relaxed during the minimization. Later a full minimization was done with both the viral coat and MSC2 movable. The strain energy reached the value of 1500 Kcal/mol at Rcritical = 14.5–15 Å (Fig. 5) unlike MSC1 which has a smaller Rcritical. The MIC value predicted from PCSM is ≈0.20 μM. This again is the expected trend as has been observed in WIN54954 and WIN52035. The phenyl ring of the drug is surrounded by residues such as like Cys-76, Tyr-240, and Lys-112 that have a high pKa. This cavity is favorable for the chlorine atoms on the phenyl ring (due to possible π bonding interactions with the Tyr-240) of MSC1 and hence MSC1 shows more stiffening than MSC2 which has no chlorines.
We thought that lengthening the chain in MSC1 by one CH2 group might improve drug effectiveness by making it more flexible. Hence we built and optimized MSC3 (Fig. 3). The PCSM correlation for MSC3 with RHe is also shown in Fig. 5. The predicted value of MIC for MSC3 is ≈0.16 μM, and MSC3 seems to be a better drug in stiffening the pentamer channel than MSC2.
Summary.
We carried out FF and MD calculations to test the pentamer stiffness model (PCSM) for uncoating of HRV. We find that the pentamer channel through which the RNA is released during uncoating is constricted when WIN drugs are bound to HRV-1A but not without the drug. The stiffness correlates with drug effectiveness. For HRV-14 we find that the effect of drugs on stiffness do not correlate with efficiency. These results support the PCSM and provide a correlation that could be used to aid drug design.
We designed three new molecules (MSC1, MSC2, and MSC3) and used the PCSM to assess their likely effectiveness on HRV-1A. We find that MSC1 leads to as good a constriction of pentamer channel of HRV-1A as the best known drugs. Of course an effective drug must have many properties not tested by the PCSM. It must be delivered to the appropriate site and it must displace whatever might already be in the site. However, we believe that the PCSM could be useful for rapid screening of new candidate drugs before subjecting them to more expensive experimental procedures. Because all serotypes of HRV must uncoat (even though they may bind to different cellular receptors), use of PCSM to develop drugs specifically directed toward pentamer stiffness might help develop drugs effective against all serotypes.
Acknowledgments
The research was funded by the U.S. Department of Energy–Biological and Chemical Technologies Research. The facilities of the Materials and Process Simulation Center are also supported by grants from National Science Foundation (CHE 95-22179 and ASC 92-100368), Chevron Petroleum Technology, Asahi Chemical, Aramco, Owens-Corning, Asahi Glass, Chevron Chemical Co., Chevron Research and Technology Co., Hercules, Avery Dennison, BP Chemical, and the Beckman Institute. Some of these calculations were carried out on the National Science Foundation Supercomputing Center at Illinois and on the Jet Propulsion Laboratory CRAY T3D.
ABBREVIATIONS
- HRV
human rhinovirus
- FF
force fields
- MD
molecular dynamics
- PCSM
Pentamer Channel Stiffness Model
- MIC
minimum inhibitory concentration
References
- 1.Rueckert R R. In: Fundamental Virology. Fields B N, Knipe D M, editors. New York: Raven; 1991. pp. 409–450. [Google Scholar]
- 2.Rossmann M G, Arnold E, Erickson J W, Frankenberger E A, Griffith J P, Hecht H J, Johnson J E, Kamer G, Luo M, Mosser A G, Rueckert R R, Sherry B, Vriend G. Nature (London) 1985;317:145–153. doi: 10.1038/317145a0. [DOI] [PubMed] [Google Scholar]
- 3.Kim S, Smith T J, Chapman M S, Rossmann M G. J Mol Biol. 1989;210:91–111. doi: 10.1016/0022-2836(89)90293-3. [DOI] [PubMed] [Google Scholar]
- 4.Badger J, Minor I, Kremer M J, Oliveira M A, Smith T J, Griffith J P, Guerin D M A, Krishnaswamy S, Luo M, Rossmann M G, McKinlay M A, Diana G D, Dutko F J, Fancher M, Rueckert R R, Heinz B A. Proc Natl Acad Sci USA. 1988;85:3304–3308. doi: 10.1073/pnas.85.10.3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bibler-Muckelbauer J K, Kremer M J, Rossmann M G, Diana G D, Dutko F J, Pevear D C, McKinlay M A. Virology. 1994;202:360–369. doi: 10.1006/viro.1994.1352. [DOI] [PubMed] [Google Scholar]
- 6.Kim K H, Willingmann P, Gong Z X, Kremer M J, Chapman M S, Minor I, Oliveira M A, Rossmann M G, Andries K, Diana G D, Dutko F J, McKinlay M A, Pevear D C. J Mol Biol. 1993;230:206–226. doi: 10.1006/jmbi.1993.1137. [DOI] [PubMed] [Google Scholar]
- 7.Rossmann M G, McKinlay M A. Infect Agents Dis. 1992;1:3–10. [PubMed] [Google Scholar]
- 8.Couch R B. In: Virology. 2nd Ed. Fields B N, Knipe D M, editors. New York: Raven; 1990. pp. 607–629. [Google Scholar]
- 9.Pevear D C, Fancher M J, Felock P J, Rossmann M G, Miller M S, Diana G D, Treasurywala A M, McKinlay M A, Dutko F J. J Virol. 1989;63:2002–2007. doi: 10.1128/jvi.63.5.2002-2007.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guha-Biswas M, Holder M, Pettitt B M. J Med Chem. 1993;36:3489–3495. doi: 10.1021/jm00075a001. [DOI] [PubMed] [Google Scholar]
- 11.Fox M P, Otto M J, McKinlay M A. Antimicrob Agents Chemother. 1986;30:110–116. doi: 10.1128/aac.30.1.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeichhardt H, Otto M J, McKinlay M A, Willingman P, Habermehl K-O. Virology. 1987;160:281–285. doi: 10.1016/0042-6822(87)90075-4. [DOI] [PubMed] [Google Scholar]
- 13.Weiner S J, Kollman P A, Case D A, Chandra Singh U, Ghio C, Alagona G, Profeta S, Jr, Weiner P. J Am Chem Soc. 1984;106:765–784. [Google Scholar]
- 14.Mayo S I, Olafson B D, Goddard W A., III J Phys Chem. 1990;94:8897–8909. [Google Scholar]
- 15.Singh U C, Weiner S, Kollman P. Proc Natl Acad Sci USA. 1985;82:755–759. doi: 10.1073/pnas.82.3.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Filman D J, Syed R, Chow M, Macadam A J, Minor P D, Hogle J M. EMBO J. 1989;8:1567–1579. doi: 10.1002/j.1460-2075.1989.tb03541.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yeates T O, Jacobson D H, Martin A, Wychowski C, Girard M, Filman D J, Hogle J M. EMBO J. 1991;10:2331–2341. doi: 10.1002/j.1460-2075.1991.tb07772.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lim, K. T., Brunett, S., Iotov, M., McClurg, R. B., Vaidehi, N., Dasgupta, S., Taylor, S. & Goddard, W. A., III (1997) J. Comp. Chem., in press.
- 19.Ding H Q, Karasawa N, Goddard W A., III J Chem Phys. 1992;97:4309–4315. [Google Scholar]
- 20.Diana G D, McKinlay M A, Brisson C J, Zalay E S, Miralles J V, Salvador V J. J Med Chem. 1985;28:748–752. doi: 10.1021/jm00383a010. [DOI] [PubMed] [Google Scholar]
- 21.Phelps D K, Post C B. J Mol Biol. 1995;254:544–551. doi: 10.1006/jmbi.1995.0637. [DOI] [PubMed] [Google Scholar]
- 22.Diana G D, Mallamo J P. J Mol Biol. 1993;230:226–227. [Google Scholar]