

Abstract
The class II transactivator (CIITA) is a master transcription regulator of gene products involved in the exogenous antigen presentation pathway, including major histocompatibility complex (MHC) class II, invariant chain, and DM. An extensive analysis of the putative functional domains of CIITA is undertaken here to explore the action of CIITA. Antibodies to CIITA protein were produced to verify that these mutant proteins are expressed. Both acidic and proline/serine/threonine-rich domains are essential for class II MHC promoter activation. In addition, three guanine nucleotide-binding motifs are essential for CIITA activity. Of these mutants, two exhibited strong transdominant-negative functions. These two mutants provide a plausible approach to manipulate MHC class II expression and immune responses.
Major histocompatibility complex (MHC) gene products DR, DP, and DQ play a critical role in the presentation of processed exogenous antigen to T cells (1–3). MHC class II molecules are constitutively expressed at high levels in B lymphocytes and dendritic cells and are induced in certain cell types such as macrophages, endothelial cells, astrocytes, and microglia upon treatment with interferon-γ (IFN-γ). The appropriate constitutive and inducible expression of MHC class II molecules is essential for normal immune response, whereas aberrantly high and low expression have been correlated with various autoimmune diseases (4, 5) and a type of severe combined immunodeficiency disease, the bare lymphocyte syndrome (BLS), respectively (6, 7). Patients with BLS lack MHC class II antigen expression on both constitutive and IFN-γ-inducible cells (8, 9). The lack of MHC class II antigen expression on cells from group A of type II BLS patients is primarily due to a defect in the transcription factor, class II transactivator (CIITA), initially identified as AIR-1 (10, 11).
The primary regulation of constitutive and IFN-γ-induced MHC class II genes is at the transcriptional level (9, 12, 13). The MHC class II, invariant chain, and DMA/DMB genes contain three highly conserved DNA cis-acting elements: the W, X, and Y boxes (14), also known collectively as the class II box. These three elements exhibit conservation in sequence, as well as spacing constraint and regimented stereospecific alignment (15, 16).
The transcription factors that directly bind to the MHC class II promoter elements are well studied (9, 12, 17), but expression of these proteins is generally ubiquitous and does not parallel MHC class II gene expression. In contrast, expression of CIITA closely parallels that of MHC class II gene expression (11). CIITA was cloned by its ability to complement RJ2.2.5, an in vitro-generated MHC class II negative cell derived from Raji (11, 18). Several groups, including our own, have shown that CIITA is induced by IFN-γ and that transfection of CIITA alone into cells is sufficient to activate MHC class II (19–21), invariant chain (19, 22), and HLA-DM genes (22).
A major issue in the field concerns the mode of action of CIITA. Although CIITA is a strong transactivator, it does not bind MHC class II promoter elements, nor does it appear to interact with transcription factors that bind these elements. In this report, we have undertaken an extensive domain analysis to delineate functional domains of CIITA. The results show that the proline-, serine-, and threonine-rich domains are critical for function. In addition, a GTP-binding domain consisting of phosphate-, Mg2+-, and guanine-binding motifs is crucial for CIITA function, strongly implicating a role for GTP binding. Two of the mutants are highly efficient transdominant-negative molecules that nearly abolished the normal function of CIITA. The biological implications of these observations are discussed.
MATERIALS AND METHODS
Cell Cultures and Transient Transfection.
2fTGH, G3A, Raji, Namalwa, and RJ2.2.5 cells were cultured as previously described (18, 19, 23). COS-7, a T antigen-transformed monkey kidney cell line, was maintained in DMEM-H medium containing 10% fetal calf serum (GIBCO/BRL). 2fTGH, G3A, and RJ2.2.5 cells were transfected as described previously (19, 24).
DNA Constructs.
The mammalian expression vector, pcDNA3.FLAG.CIITA8 (FLAG.CIITA8), contains the FLAG epitope (DYKDDDDK) upstream of the first methionine of CIITA8. All mutant constructs were generated by a published method (25) from the parental plasmid, FLAG.CIITA8, with a selection primer, 5′-AAATGCTTCAATgcTAGcgAAAAAGGAAG-3′, which mutated a SspI to NheI restriction site, and a mutagenic primer. All mutants were initially identified by the presence of an NheI site and later confirmed by sequencing.
Construction Of Proline/Serine/Threonine Deletion Mutants.
CIITA(Δ132–301) was constructed by creating ClaI restriction sites at nucleotides 511 (amino acids 132–133) and 1013 (amino acids 300–301) of CIITA8 using two mutagenic primers: 5′-533-GCCCAACTTCTGCTGGCATaTCgATACTCTCACCGATCAC-493-3′ and 5′-1036-GAGGTCAGGGCAGGTTCAtcgATGCTGGGCAGGTCAGTG-997-3′. The mutagenized construct was digested with ClaI to release the proline/serine/threonine-containing fragment, and the remaining sequence was in the plasmid religated. CIITA(Δ132–209) was constructed in the same way as mentioned above with mutagenic primers, 5′-533-GCCCAACTTCTGCTGGCATaTCgATACTCTCACCGATCAC-493-3′ and 5′-766-CAACGAGGAACTGGAGAAAtcgATGGGAATCTGGTCGGTTTTC-724-3′, and it contained a clean deletion of the proline/serine-rich domain. CIITA(Δ209–301) has deleted threonine-rich domain from amino acid residues of 209–301. This mutant construct was mutagenized with primers: 5′-766-CAACGAGGAACTGGAGAAAtcgATGGGAATCTGGTCGGTTTTC-724-3′ and 5′-1036-GAGGTCAGGGCAGGTTCAtcgATGCTGGGCAGGTCAGTG-997-3′.
Construction Of Nucleotide-Binding Motif Mutants.
CIITA-GTP1(ΔGK) and CIITA-GTP1(K→E) were mutated in the phosphate-binding motif. CIITA-GTP1(ΔGK) has a deletion of two amino acid residues, 420 and 421, by a “loop out” mutagenic primer: 5′-1395-CTCTTGCCCTGACCAGCCACAGCAATCACTCGTG-1353-3′. CIITA-GTP1(K → E) has a point mutation at residue 427 from lysine to glutamic acid. The mutagenic primer used to generate this mutant was 5′-1408-CCCAGCCCAATAGCTCTcGCCCTGACCAGCTTTGCC-1373-3′. CIITA-GTP2(ΔDAYG) and CIITA-GTP3(ΔSKAD) have deletions in the Mg2+- and guanine-binding motifs, respectively. CIITA-GTP2(ΔDAYG) was created by a loop out primer: 5′-1522-GAGCAGATCCTGCAGCCCCGGACGGTTCAAG-1480-3′. Mutagenic primer used to construct GTP3(ΔSKAD) was 5′-1813-CAGCTCAAATAGGGCCAGGCTCTGGACCAG-1777-3′.
Generation of Stable Transfectants Expressing Cell Surface MHC Class II Antigen with FLAG.CIITA8 Expression Vector.
Ten micrograms of FLAG.CIITA8 plasmid was transfected into G3A cell. The stable transfectants expressing cell surface MHC class II antigens were selected by the immunomagnetic selection, according to manufacturer is protocol (Dynal, Oslo). The cells underwent immunoselection three times for the next 4 weeks. Cells expressing MHC class II antigens were confirmed by fluorescence-activated cell sorting analysis (19).
Anti-CIITA (α-CIITA) Antibody Production.
Anti-human CIITA antibody was raised against a peptide, 726GEIKDKELPQYLALTPR742, crosslinked to KLH (keyhole limpet hemocyanin; Pierce). The antiserum was tested after numerous injections of the rabbit with the antigen.
Western Blot.
Raji, Namalwa, and RJ2.2.5 nuclear extracts were prepared according to Dignam et al. (26). Whole cell extract was prepared as described (27). The samples were analyzed by immunoblotting with α-FLAG (10 μg/ml; IBI–Kodak) or α-CIITA (1.5 μg/ml) antibodies using standard techniques (27). Immunoblots were detected by enhanced chemical luminescence (ECL; Amersham).
RESULTS
Generation and Characterization of CIITA Antibody.
Analysis of the primary amino acid sequence of CIITA did not show any homology to known conserved DNA-binding motif of transcription factors, and in vitro-translated CIITA apparently does not interact with DNA (11). In a separate study, our laboratory shows that CIITA is not detected in a complex that consists of proteins binding to the W, X, and Y elements (K. L. Wright, K.-C.C., and J.P.-Y.T., unpublished work). A structure–function analysis is undertaken here to identify the crucial domains of CIITA in an effort to understand its mode of action. To accurately assess the effects of mutagenesis on CIITA, it is essential to verify that mutant proteins are expressed to a similar level as wild-type controls. Several reagents were generated to assess this issue. First, a FLAG epitope-tagged CIITA was produced. FLAG was chosen because this epitope is small (8 aa) and is not likely to disrupt the native conformation of CIITA.
FLAG epitope-tagged CIITA, FLAG.CIITA8, functioned equally as well as wild-type CIITA to transactivate MHC class II promoter, as assessed by both the chloramphenicol acetyltransferase (CAT) reporter gene (Fig. 1A) and surface MHC class II antigen expression (Fig. 1B). The former was assessed by transfecting either FLAG-tagged or unmodified CIITA-expression vector together with DRA300CAT into a CIITA-defective cell line, G3A. pDRA300CAT (28) contains 267 bp of the DRA promoter linked to CAT reporter gene (28, 29). FLAG-tagged CIITA functioned properly in this transactivation assay (Fig. 1A). The latter was performed using similarly transfected G3A cells, and the results were analyzed by flow cytometry. The wild-type parent, 2fTGH, was responsive to IFN-γ (Fig. 1 Ba and Bb) as expected, while the G3A cell line (Fig. 1 Bc and Bd) was not. Transfection of FLAG-tagged CIITA into G3A cells resulted in high levels of surface MHC class II (Fig. 1 Be and Bf).
Figure 1.

The FLAG foreign epitope did not alter CIITA activity. (A) FLAG.CIITA8 activated the DRA promoter in G3A cells. G3A cells were cotransfected with the pDRA300CAT construct and an empty vector (lane 1), CIITA expression vector (lane 2), or FLAG.CIITA8 (lanes 3 and 4). The cells were harvested and analyzed for CAT activity 48 h after transfection. (B) FLAG.CIITA8 induces HLA-DRA expression on the MHC class II-negative G3A mutant cells. 2fTGH cells (Ba and Bb), G3A (Bc and Bd), and G3A (Be and Bf) stably integrated with FLAG.CIITA8 were not treated (Left) or treated (Right) with 500 units/ml of IFN-γ for 72 h. The cells were harvested and analyzed for surface HLA-DR antigen expression by a FACScan (Becton Dickinson).
The expression of recombinant CIITA protein in 2fTGH and COS-7 cells transfected with FLAG.CIITA8 was examined by Western blot analysis. Recombinant CIITA migrated at an apparent molecular weight of 145 kDa in SDS/PAGE and specifically was recognized by the α-FLAG antibody (Fig. 2A, lanes 1–4).
Figure 2.
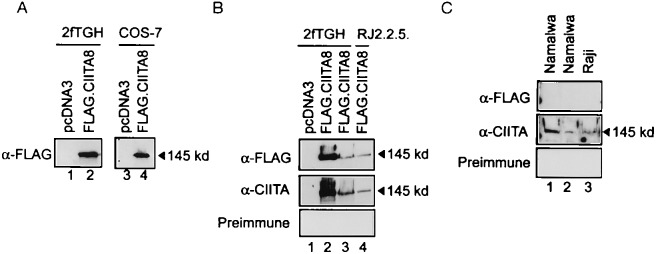
Detection of recombinant and endogenous CIITA. (A) Detection of transfected FLAG.CIITA8 by immunoblot. 2fTGH and COS-7 cells were transfected with 10 μg and 2 μg, respectively, of either FLAG.CIITA8 (lanes 2 and 4) or empty vector pcDNA3 (lanes 1 and 3). The cells were harvested 24 h after the transfection, and total lysates were prepared and separated on a 8% SDS/polyacrylamide gel. The gel was transferred onto nitrocellulose and the presence of recombinant CIITA was examined with 10 μg/ml α-FLAG antibody. (B) Transfected FLAG.CIITA8 was detected by both α-FLAG and α-CIITA antibodies. 2fTGH and RJ2.2.5 cells were transfected with either 10 μg (lanes 2 and 4) or 5 μg (lane 3) of FLAG.CIITA8 or control vector pcDNA3 (lane 1). Total lysates were prepared and examined with either α-FLAG antibody (Top), α-CIITA antibody (Middle), or preimmune serum (Bottom). (C) Anti-CIITA antibody specifically recognizes an endogenous CIITA, with an apparent molecular weight of 145 kDa. Two Namalwa and one Raji nuclear extracts were prepared and blotted with either the α-FLAG antibody (Top), α-CIITA antibody (Middle), or preimmune serum (Bottom).
A second serologic reagent, α-CIITA antibody, was generated against a CIITA peptide (see Materials and Methods). The specificity of this antibody was tested using extracts from 2fTGH and RJ2.2.5 cells transfected with pcDNA3 or FLAG.CIITA8. Whole cell extracts were made after transfection and analyzed by Western blot with α-FLAG and α-CIITA antibodies. Both antibodies recognized an ≈145-kDa molecule in FLAG.CIITA8 transfected 2fTGH and RJ2.2.5 cells (Fig. 2B Top and Middle, lanes 2–4). This band was not observed in 2fTGH cells transfected with the empty vector (Fig. 2B, lane 1) nor in a blot incubated with preimmune serum (Fig. 2B Bottom).
The recognition of endogenous CIITA by the α-CIITA antibody in two B cell extracts was a final test of this antibody. A 145-kDa band, which migrated at the same position as recombinant CIITA, was detected by the α-CIITA antibody in a Western blot (Fig. 2C, lanes 1–3). The same band was not observed with α-FLAG antibody or preimmune serum.
A Proline/Serine/Threonine-Rich Region Is Essential for the Transactivation Activity of CIITA.
The N terminus of CIITA contains an acidic domain (amino acids 30–160), followed by domains rich in proline (amino acids 163–195), serine (amino acids 209–237), and threonine (amino acids 260–322) (11). Acidic domain has been found in many transcription factors (30–34), and others (35, 36) have shown that the acidic domain of CIITA can function as an activation domain when fused to the GAL-4 DNA-binding domain. One of these reports (36) also replaced the acidic and proline-, serine-, and threonine-rich domains in the fusion construct with the acidic domain of HSV1 transcription activation factor (αTIF) and showed reduced function. This result suggests that either the acidic domain alone is not sufficient to activate the MHC class II promoter, or the heterologous HSV1 α-TIF acidic domain behaved differently from the CIITA acidic domain. Proline-, serine-, and threonine-rich domains have been found in many transcriptional factor and have a proposed role in protein–protein interaction (30, 37). Therefore, it was important to determine the contribution of each domain to the activity of native CIITA.
Mutant CIITA(Δ132–301) (Fig. 3A), which retains the acidic domain but deletes the proline-, serine-, and threonine-rich domains, was analyzed. This mutant did not exhibit any activation activity when transfected with pDRA300CAT (Fig. 3A, compare lanes 2 and 3). This indicates that the acidic domain alone is not sufficient to activate the MHC class II promoter, and intact proline-, serine-, and threonine-rich regions are also necessary. To better define the function of proline-, serine-, and threonine-rich domains of CIITA, two mutants, CIITA(Δ132–209) and CIITA(Δ209–301), were made. CIITA(Δ132–209) has a clean deletion of the proline-rich domains (amino acids 132–209), and this deletion did not affect the activation of DRA promoter by CIITA (Fig. 3A, lane 4). Deletion of the serine- threonine-rich domains in mutant CIITA(Δ209–301) also did not affect transactivation function (Fig. 3A, lane 5). One possible explanation is that the two subdomains may serve overlapping functions, although the analysis performed cannot rule out unexpected compensatory changes in protein fold in any of these mutants.
Figure 3.
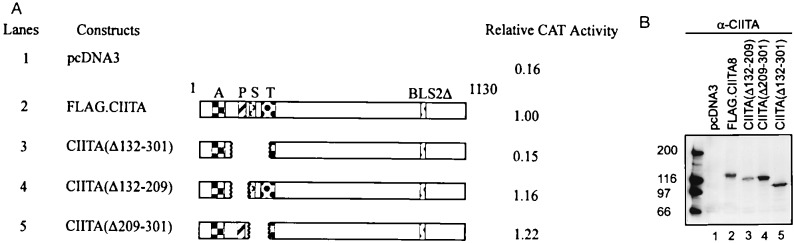
Requirement for proline-rich or serine/threonine-rich domain in addition to the acidic domain for CIITA activity. (A) G3A cells were cotransfected with the pDRA300CAT and pcDNA3 (lane 1), FLAG.CIITA8 (lane 2), or mutant constructs (lanes 3–5). The cells were harvested 48 h after transfection. CAT activity was analyzed and quantitated by PhosphorImager (Molecular Dynamics). The relative level of expression was calculated by using the wild-type control as a value of 1. These experiments were repeated three times. (B) Immunoblot analysis of wild-type FLAG.CIITA8 and mutant constructs. Cells transfected with FLAG.CIITA and mutant CIITA were harvested and lysed 24 h after transfection. Equivalent amount of total lysates from each sample was fractionated on 8% SDS-PAGE. The blot was examined with the α-CIITA antibody and visualized by ECL, according to the manufacturer’s specification. All of the mutant constructs were expressed at a comparable level to the FLAG.CIITA8. No band was observed in cells transfected with the empty vector pcDNA3 (lane 1).
To ascertain that the low activity of these mutants was not due to low expression of mutant proteins, cell extracts were made and blotted with the α-CIITA antibody. The mutants produced comparable levels of protein as the wild-type construct (Fig. 3B). These results can also be confirmed with the antibody against the FLAG epitope (data not shown).
CIITA Function Requires a GTP-Binding Motif.
Analysis of the primary amino acids of CIITA shows that a region of CIITA from residues 421 to 561 has a significant homology to a wide variety of GTP-binding proteins in the Ras superfamily, including the low-molecular weight Ras-like GTPase, the translation elongation factor (EF-TU), and the α subunits of heterotrimeric G protein (38). CIITA exhibits similarities to three consensus motifs found in other GTP-binding domains: the phosphate-binding motif GXXXXGKS, the Mg2+-binding motif DXXG, and the guanine-binding motif SKXD (Fig. 4). These sequence motifs are also referred to as G-1, G-3, and G-4, respectively. The G-1 region of CIITA (GX4GKS) has a perfect match to the conserved amino acid sequence of p21ras (10GX4GKS17). The ɛ-amino group of Lys16 (corresponding to Lys427 of CIITA) forms bonds with the α- and β-phosphate of GTP or GDP (39, 40). The G-3 region, 461DX2G464, of CIITA is also conserved and it corresponds to residues 57–60, 57DX2G60, of p21ras. In p21ras, the invariant aspartate (position 461 in CIITA) binds the catalytic Mg2+ through an intervening water molecule, while the amide proton of the invariant glycine (position 464 of CIITA) forms a hydrogen bond with γ-phosphate of GTP. The G-4 region of CIITA, 558SKXD561, is identical to the p21ras G-4 region (residues 116–119, 116NKXD119) except position 558 of CIITA has a serine residue instead of an asparagine or threonine residue and this change does not affect CIITA’s function (see below). Collectively, these homologies suggest that CIITA may exhibit guanine nucleotide-binding activity. To determine if these motifs are significant, each motif was individually mutated (Fig. 5A) and examined for transactivation activity. The mutants were cotransfected with the pDRA300CAT into G3A cells, and CAT activity was determined. CIITA-GTP1(ΔGK), which contained a deletion of residues 421–422 in the phosphate-binding motif, showed a reduced transactivation activity (22% above the basal activity and 38% of the wild-type controls; Fig. 5A, lanes 1–3). CIITA-GTP1(K → E) contains a substitution of Lys427 (K) to Glu (E) and also exhibits greatly reduced activity (lane 4). The transactivation capability of CIITA-GTP2(ΔDAYG) and CIITA-GTP3(ΔSKAD) constructs is also greatly diminished (Fig. 5A, lanes 5 and 6).
Figure 4.

Analysis of guanine nucleotide-binding motifs within CIITA. The amino acid residues of CIITA from 421 to 561 were aligned with the consensus sequence of GTPase and Ras. The bold letters are sequences within CIITA that are found in the GTPase consensus motifs. The number in the parentheses indicates the spacing between the motifs.
Figure 5.
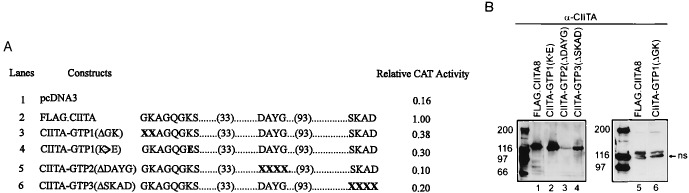
Guanine nucleotide-binding motifs are essential for CIITA activity. (A) G3A cells were cotransfected with the pDRA300CAT and pcDNA3 (lane 1), FLAG.CIITA8 (lane 2), or mutant CIITA (lanes 3 to 6). Cells were harvested 48 h later, and CAT activity was determined as described. The experiment was repeated three times. (B) Western blot to assess the level of protein expression by mutant CIITA genes (compare lane 1 and lanes 2–4; also compare lanes 5 and 6). An equal amount of total lysates was loaded, separated by SDS/PAGE, and transferred onto nitrocellulose. The blot was probed with the α-CIITA antibody. ns, Nonspecific band.
A Western blot was performed to ensure that all constructs expressed CIITA to a comparable level as the that of wild-type protein (Fig. 5B). The only exception is CIITA-GTP2(ΔDAYG) (lane 3), which showed a lower level of protein expression. This is consistent with reports which showed that this region is crucial for protein stability (41–43). These results, in total, strongly suggest that CIITA contains putative GTP-binding motifs.
Transdominant-Negative Mutants of CIITA.
CIITA represents a highly specific, yet potent, regulator of MHC class II genes. Any interference with its function is likely to significantly affect antigen presentation by MHC class II-mediated pathway and subsequent T cell activation, and such interference may be desirable in a number of diseases. The capacity of all CIITA mutants to serve as transdominant-negative mutants of CIITA function was tested in G3A. Two mutants exhibit transdominant-negative properties as shown in Table 1. The experiment consisted of the cotransfection of the pDRACAT300 reporter together with CIITA and an empty vector (pcDNA3), or a mutant form of CIITA. A titration of wild-type vs. mutant CIITA constructs established that a 1:1 ratio was sufficient to reveal transdominant-negative function (data not shown). Two mutants, CIITA(Δ132–301) and CIITA-GTP2(ΔDAYG) greatly reduced the function of wild-type CIITA, with maximal reduction of wild-type CIITA function by ≈96%. We also tested these two transdominant-negative mutants in another CIITA-deficient cell, RJ2.2.5 (13), and these two mutants also efficiently blocked the function of wild-type CIITA, as shown in Table 2. The effects of these transdominant mutants is gene-specific, since the TAP1 promoter (44) is not affected by them (data not shown).
Table 1.
Transdominant repression of DRACAT expression by CIITA mutants in G3A
| Parameters | 1 | 2 | 3 |
|---|---|---|---|
| pDRA300CAT | + | + | + |
| FLAG.CIITA | + | + | + |
| Control/mutant | pcDNA3 | CIITA (Δ132–301) | CIITA-GTP2 (ΔDAYG) |
| % inhibition* | — | 96.3 | 91.2 |
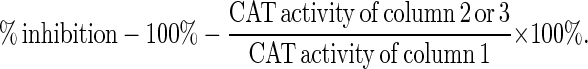 |
Table 2.
Transdominant repression of DRACAT expression by CIITA mutants in RJ2.2.5
| Parameters | 1 | 2 | 3 |
|---|---|---|---|
| pDRA300CAT | + | + | + |
| FLAG.CIITA | + | + | + |
| Control/mutant | pcDNA3 | CIITA (Δ132–301) | CIITA-GTP2 (ΔDAYG) |
| % inhibition* | — | 94.5 | 97.2 |
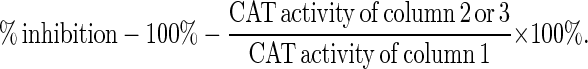 |
DISCUSSION
CIITA is a master regulator for all known genes involved in the class II antigen presenting pathway, including MHC class II (11, 19, 21), invariant chain (19, 22), and DM genes (22). CIITA transcript is expressed constitutively in B cells, and it is induced in cells such as fibroblasts, macrophages, and glioblastoma cells upon treatment with IFN-γ (19–21). The kinetic of CIITA induction by IFN-γ precedes the induction of MHC class II transcripts, and introduction of CIITA alone into a number of cell types is sufficient to activate class II genes. However, the mode of action of CIITA is not well understood. In this analysis, an extensive mutation analysis was undertaken, and the consequences on DRA promoter activation were assessed by introducing these mutants into a CIITA-negative, IFN-γ-defective mutant cell, G3A (19). The results show that the acidic domain alone is insufficient to activate the DRA promoter and requires an additional proline/serine-rich or threonine-rich domain to achieve activity. Most intriguing, guanine-nucleotide binding motifs within residues 420–561 are found to be essential for CIITA activity. Preliminary data have confirmed that CIITA can bind GTP (K.-C.C., J. Harton, and J.P.-Y.T., unpublished work) placing CIITA in a novel group of transcription factors. Two of these mutants are potent transdominant repressors of CIITA function. It may be possible to utilize these transdominant mutants to downregulate MHC class II expression and function in vivo.
Functional Role of Acidic and Proline/Serine/Threonine-Rich Domains of CIITA.
Acidic and proline/serine/threonine-rich domains are found in many activators or coactivators involved in gene expression (30, 37, 45, 46). Activation domains can be grouped into several categories by their amino acids content, including glutamine, acidic, proline, serine, and threonine domains. These domains have been shown to interact with the basal transcriptional machinery by in vitro and in vivo assays (30, 47). The interaction of the activation domain and basal transcription factors is believed to be responsible for gene activation by increasing the rate by which other basal transcription factors and RNA polymerase II bind to the TATA box and the initiator (48–52). The proline domain is found in CTF/NF1 and AP-2, among others, and it interacts with the TATA box-binding protein (TBP; ref. 30) and the TFIIB. For CIITA, two groups (35, 36) showed that the acidic domain can function as an activation domain when fused to a GAL-4 DNA-binding domain. The findings here, using native CIITA protein, reveal a role for both the acidic and the proline/serine/threonine-rich domain. This parallels findings with other transcription factors that contain an acidic domain (45, 53). In these factors, the acidic domain does not function by itself, but in conjunction with the proline- or serine/threonine-rich domains. One unique feature of CIITA that has to be considered in any working model is that CIITA does not appear to contact DNA. Thus, it is likely that CIITA uses the functional domains defined here to interact with basal transcription factor or other DNA-binding protein.
A Guanine Nucleotide-Binding Motif Is Unique to CIITA.
Guanine nucleotide-binding motifs play important roles in a large number of basic cellular functions including protein synthesis, signal transduction, and intracellular protein transport, but they have not been associated with transcription factors (54, 55). A comparison of the primary amino acids of CIITA to known guanine nucleotide-binding proteins reveals a region that is highly identical to the GTP-binding domain of RAS (Fig. 4), except a serine replaces the asparagine or threonine residue in CIITA. Point mutation of the serine to asparagine did not affect the activity of CIITA, indicating a flexibility of this residue. Mutation of any of the other three conserved motifs either reduces or abolishes the CIITA activity. In particular, CIITA-GTP1(K → E), which contains a single point mutation at the phosphate-binding motif, shows a drastically reduced transacting activity. In the Ras protein, this residue is shown by x-ray crystallography to interact with the α- and β-phosphate of GTP or GDP and is crucial for function. Our finding is in accordance with the findings in Ras (56).
GTP-binding proteins exist in two different states: an active GTP-bound form and an inactive GDP-bound form (57). The transition from the active GTP-bound to the inactive GTP-bound form is usually accompanied by a conformational change in the protein, and this change affects the recruitment or the interaction with downstream factors. GTP binding may similarly modify the function of CIITA. GTP-binding proteins are also involved in protein transport (58). Therefore, it is possible that a GTP bound CIITA may transport a positive factor(s) from the cytoplasm to nucleus or a negative nuclear regulator out of the nucleus. Our recent data showed that the wild-type CIITA can bind GTP, and mutation of any of the three GTP-binding motifs abrogated the CIITA’s GTP-binding activity (K.-C.C., J. Harton, and J.P.-Y.T., unpublished work).
Identification of CIITA Transdominant-Negative Mutants.
The identification of transdominant-negative mutants of CIITA has both practical and molecular implications. From a practical point of view, these mutants may be efficient in suppressing MHC class II promoter function and gene expression in an in vivo condition. For example, it may be possible to produce animals bearing such transdominant-negative CIITA molecules and use these for transplant purposes. From a scientific point of view, it is of interest that deletion/mutation of two specific domains resulted in potent and highly efficient transdominant-negative functions. Presumably, what remained nonmutated in these molecules is sufficient to block the function of wild-type CIITA, perhaps by interceding certain intracellular signals. The fact that the transdominant-negative function is observed at a 1:1 ratio of wild-type:mutant CIITA suggests that the mutant form is more efficient at interceding such a putative signal or function.
In summary, this report shows several functional domains important for the function of CIITA, including highly unusual domains that have not been previously associated with transcription factors. Mutations of CIITA result in potent transdominant molecules, which may be of practical and therapeutic use.
Acknowledgments
This work was supported by National Institutes of Health Grants AI29564 and CA48185 and a Multiple Sclerosis Grant RF1785. J.P.-Y.T. is an American Cancer Society Faculty Awardee.
ABBREVIATIONS
- MHC
major histocompatibility complex
- IFN-γ
interferon-γ
- CIITA
class II transactivator
- CAT
chloramphenicol acetyltransferase
References
- 1.Schwartz R H. Annu Rev Immunol. 1985;3:237–261. doi: 10.1146/annurev.iy.03.040185.001321. [DOI] [PubMed] [Google Scholar]
- 2.Kappler J W, Roehm N, Marrack P. Cell. 1987;49:273–280. doi: 10.1016/0092-8674(87)90568-x. [DOI] [PubMed] [Google Scholar]
- 3.Blackman M A, Marrack P, Kappler J. Immunol Res. 1989;244:214–217. doi: 10.1126/science.2784868. [DOI] [PubMed] [Google Scholar]
- 4.Bottazzo G F, Pujol-Borrell R, Hanafusa T, Feldmann M. Lancet. 1983;ii:1115–1119. doi: 10.1016/s0140-6736(83)90629-3. [DOI] [PubMed] [Google Scholar]
- 5.Massa P T, ter Meulen V, Fontana A. Proc Natl Acad Sci USA. 1987;84:4219–4223. doi: 10.1073/pnas.84.12.4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Preval C, Lisowska-Grospierre B, Loche M, Griscelli C, Mach B. Nature (London) 1985;318:291–293. doi: 10.1038/318291a0. [DOI] [PubMed] [Google Scholar]
- 7.Griscelli C, Lisowska-Grospierre B, Mach B. Immunodefic Rev. 1989;1:135–153. [PubMed] [Google Scholar]
- 8.Hume C R, Lee J S. Hum Immunol. 1989;26:288–309. doi: 10.1016/0198-8859(89)90007-4. [DOI] [PubMed] [Google Scholar]
- 9.Mach B, Steimle V, Martinez-Soria E, Reith W. Annu Rev Immunol. 1996;14:301–331. doi: 10.1146/annurev.immunol.14.1.301. [DOI] [PubMed] [Google Scholar]
- 10.Accolla R S, Jotterand-Bellomo M, Scarpellino L, Maffei A, Carra G, Guardiola J. J Exp Med. 1986;164:369–374. doi: 10.1084/jem.164.1.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steimle V, Otten L A, Zufferey M, Mach B. Cell. 1993;75:135–146. [PubMed] [Google Scholar]
- 12.Glimcher L H, Kara C J. Annu Rev Immunol. 1992;10:13–49. doi: 10.1146/annurev.iy.10.040192.000305. [DOI] [PubMed] [Google Scholar]
- 13.Ting J P. Immunol Res. 1993;12:65–77. doi: 10.1007/BF02918369. [DOI] [PubMed] [Google Scholar]
- 14.Benoist C, Mathis D. Annu Rev Immunol. 1990;8:681–715. doi: 10.1146/annurev.iy.08.040190.003341. [DOI] [PubMed] [Google Scholar]
- 15.Vilen B J, Cogswell J P, Ting J P. Mol Cell Biol. 1991;11:2406–2415. doi: 10.1128/mcb.11.5.2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vilen B J, Penta J F, Ting J P. J Biol Chem. 1992;267:23728–23734. [PubMed] [Google Scholar]
- 17.Ting J P, Painter A, Zeleznik-Le N J, MacDonald G, Moore T M, Brown A, Schwartz B D. J Exp Med. 1994;179:1605–1611. doi: 10.1084/jem.179.5.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Accolla R S. J Exp Med. 1983;157:1053–1058. doi: 10.1084/jem.157.3.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chin K C, Mao C, Skinner C, Riley J L, Wright K L, Moreno C S, Stark G R, Boss J M, Ting J P. Immunity. 1994;1:687–697. doi: 10.1016/1074-7613(94)90039-6. [DOI] [PubMed] [Google Scholar]
- 20.Steimle V, Siegrist C A, Mottet A, Lisowska-Grospierre B, Mach B. Immunol Res. 1994;265:106–109. doi: 10.1126/science.8016643. [DOI] [PubMed] [Google Scholar]
- 21.Chang C H, Fontes J D, Peterlin M, Flavell R A. J Exp Med. 1994;180:1367–1374. doi: 10.1084/jem.180.4.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang C H, Flavell R A. J Exp Med. 1995;181:765–767. doi: 10.1084/jem.181.2.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mao C, Davies D, Kerr I M, Stark G R. Proc Natl Acad Sci USA. 1993;90:2880–2884. doi: 10.1073/pnas.90.7.2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Basta P V, Sherman P A, Ting J P. Proc Natl Acad Sci USA. 1988;85:8618–8622. doi: 10.1073/pnas.85.22.8618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deng W P, Nickoloff J A. Anal Biochem. 1992;200:81–88. doi: 10.1016/0003-2697(92)90280-k. [DOI] [PubMed] [Google Scholar]
- 26.Dignam J D, Lebovitz R M, Roeder R G. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shuai K, Stark G R, Kerr I M, Darnell J E, Jr, Glimcher L H, Kara C J. Immunol Res. 1992;10:13–49. [Google Scholar]
- 28.Sherman P A, Basta P V, Moore T L, Brown A M, Ting J P. Mol Cell Biol. 1989;9:50–56. doi: 10.1128/mcb.9.1.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Basta P V, Sherman P A, Ting J P. J Immunol. 1987;138:1275–1280. [PubMed] [Google Scholar]
- 30.Xiao H, Pearson A, Coulombe B, Truant R, Zhang S, Regier J L, Triezenberg S J, Reinberg D, Flores O, Ingles C J. Mol Cell Biol. 1994;14:7013–7024. doi: 10.1128/mcb.14.10.7013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hori R, Carey M. Proc Natl Acad Sci USA. 1995;92:6047–6051. doi: 10.1073/pnas.92.13.6047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klemm R D, Goodrich J A, Zhou S, Tjian R. Proc Natl Acad Sci USA. 1995;92:5788–5792. doi: 10.1073/pnas.92.13.5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tong X, Drapkin R, Reinberg D, Kieff E. Proc Natl Acad Sci USA. 1995;92:3259–3263. doi: 10.1073/pnas.92.8.3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmitz M L, Stelzer G, Altmann H, Meisterernst M, Baeuerle P A. J Biol Chem. 1995;270:7219–7226. doi: 10.1074/jbc.270.13.7219. [DOI] [PubMed] [Google Scholar]
- 35.Riley J L, Westerheide S D, Price J A, Brown J A, Boss J M. Immunity. 1995;2:533–543. doi: 10.1016/1074-7613(95)90033-0. [DOI] [PubMed] [Google Scholar]
- 36.Zhou H, Glimcher L H. Immunity. 1995;2:545–553. doi: 10.1016/1074-7613(95)90034-9. [DOI] [PubMed] [Google Scholar]
- 37.Kim T K, Roeder R G. Proc Natl Acad Sci USA. 1994;91:4170–4174. doi: 10.1073/pnas.91.10.4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dever T E, Glynias M J, Merrick W C. Proc Natl Acad Sci USA. 1987;84:1814–1818. doi: 10.1073/pnas.84.7.1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pai E F, Kabsch W, Krengel U, Holmes K C, John J, Wittinghofer A. Nature (London) 1989;341:209–214. doi: 10.1038/341209a0. [DOI] [PubMed] [Google Scholar]
- 40.Milburn M V, Tong L, deVos A M, Brunger A, Yamaizumi Z, Nishimura S, Kim S H. Immunol Res. 1990;247:939–945. doi: 10.1126/science.2406906. [DOI] [PubMed] [Google Scholar]
- 41.Fasano O, Crechet J B, Parmeggiani A. Anal Biochem. 1982;124:53–58. doi: 10.1016/0003-2697(82)90218-4. [DOI] [PubMed] [Google Scholar]
- 42.Feuerstein J, Goody R S, Wittinghofer A. J Biol Chem. 1987;262:8455–8458. [PubMed] [Google Scholar]
- 43.Foster R, Hu K Q, Shaywitz D A, Settleman J. Mol Cell Biol. 1994;14:7173–7181. doi: 10.1128/mcb.14.11.7173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wright K L, White L C, Kelly A, Beck S, Trowsdale J, Ting J P. J Exp Med. 1995;181:1459–1471. doi: 10.1084/jem.181.4.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Radtke F, Georgiev O, Muller H P, Brugnera E, Schaffner W. Nucleic Acids Res. 1995;23:2277–2286. doi: 10.1093/nar/23.12.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bassel-Duby R, Hernandez M D, Yang Q, Rochelle J M, Seldin M F, Williams R S. Mol Cell Biol. 1994;14:4596–4605. doi: 10.1128/mcb.14.7.4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Truant R, Xiao H, Ingles C J, Greenblatt J. J Biol Chem. 1993;268:2284–2287. [PubMed] [Google Scholar]
- 48.Hori R, Carey M. Curr Opin Genet Dev. 1994;4:236–244. doi: 10.1016/s0959-437x(05)80050-4. [DOI] [PubMed] [Google Scholar]
- 49.Klein C, Struhl K. Immunol Res. 1994;266:280–282. doi: 10.1126/science.7939664. [DOI] [PubMed] [Google Scholar]
- 50.Kobayashi N, Boyer T G, Berk A J. Mol Cell Biol. 1995;15:6465–6473. doi: 10.1128/mcb.15.11.6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roberts S G, Choy B, Walker S S, Lin Y S, Green M R. Curr Biol. 1995;5:508–516. doi: 10.1016/s0960-9822(95)00103-5. [DOI] [PubMed] [Google Scholar]
- 52.Xiao H, Friesen J D, Lis J T. Mol Cell Biol. 1995;15:5757–5761. doi: 10.1128/mcb.15.10.5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Manet E, Allera C, Gruffat H, Mikaelian I, Rigolet A, Sergeant A. Gene Expression. 1993;3:49–59. [PMC free article] [PubMed] [Google Scholar]
- 54.Hall A. Immunol Res. 1990;249:635–640. [Google Scholar]
- 55.Quinn M T. J Leukocyte Biol. 1995;58:263–276. doi: 10.1002/jlb.58.3.263. [DOI] [PubMed] [Google Scholar]
- 56.Sigal I S, Gibbs J B, D’Alonzo J S, Temeles G L, Wolanski B S, Socher S H, Scolnick E M. Proc Natl Acad Sci USA. 1986;83:952–956. doi: 10.1073/pnas.83.4.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bourne H R, Sanders D A, McCormick F. Nature (London) 1991;349:117–127. doi: 10.1038/349117a0. [DOI] [PubMed] [Google Scholar]
- 58.Schlenstedt G, Wong D H, Koepp D M, Silver P A. EMBO J. 1995;14:5367–5378. doi: 10.1002/j.1460-2075.1995.tb00221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]