

Abstract
Phosphorylation of specific sites in the 2nd intracellular loop and in the C-terminal domain have previously been suggested to cause desensitization and internalization of the mu-opioid receptor (MOP-R). To assess sites of MOP-R phosphorylation in vivo, affinity-purified, phosphoselective antibodies were raised against either phosphothreonine-180 in the 2nd intracellular loop (MOR-P1) or the C-terminal domain of MOP-R containing phosphothreonine-370 and phosphoserine-375 (MOR-P2). We found that MOR-P2-immunoreactivity (IR) was significantly increased within the striatum of wild-type C57BL/6 mice after injection of the agonist fentanyl. Pretreatment with the antagonist naloxone blocked the fentanyl-induced increase. Furthermore, mutant mice lacking MOP-R showed only non-specific nuclear MOR-P2-IR before or after fentanyl treatment, confirming the specificity of the MOR-P2 antibodies. To assess whether MOP-R phosphorylation occurs following endogenous opioid release, we induced chronic neuropathic pain by partial sciatic nerve ligation (pSNL), which caused a significant increase in MOR-P2-IR in the striatum. pSNL also induced signs of mu opioid receptor tolerance demonstrated by a rightward shift in the morphine dose response in the tail withdrawal assay and by a reduction in morphine conditioned place preference (CPP). Mutant mice selectively lacking all forms of the β-endorphin peptides derived from the Pomc gene did not show increased MOR-P2-IR, decreased morphine antinociception, or reduced morphine CPP following pSNL. In contrast gene deletion of either proenkephalin or prodynorphin opioids did not block the effects of pSNL. These results suggest that neuropathic pain caused by pSNL in wild-type mice activates the release of the endogenous opioid β-endorphin, which subsequently induces MOP-R phosphorylation and opiate tolerance.
Keywords: Opiate Tolerance, Opioid peptides, neuropathic pain, enkephalins, dynorphins, antinociception
Introduction
The mu opioid peptide receptor (MOP-R) is the principal pharmacological target for clinically important opioid analgesics, including fentanyl and morphine; however, tolerance and dependence to these drugs are potentially serious consequences of their use. Tolerance to opioids can result from a variety of different mechanisms including receptor desensitization, compensatory changes in the neuronal circuit physiology, and NMDA-dependent learning mechanisms. Recent work has advanced our understanding of these underlying processes (Trujillo, 2000; Connor et al., 2004). Receptor phosphorylation by G protein receptor kinases (GRK) and subsequent β-arrestin activation and binding have been implicated as a general mechanism of G-protein coupled receptor desensitization (Gainetdinov et al., 2004), and MOP-R desensitization has been shown to be GRK3 and β-arrestin dependent in in vitro assays (Kovoor et al., 1998; Zhang et al., 1998; Bohn et al., 2000). Based on in vitro gene expression studies and using site-directed receptor mutagenesis, specific phosphorylation sites within the MOP-R sequence have been suggested (Arden et al., 1995; Yu et al., 1997; Celver et al., 2001; El Kouhen et al., 2001; Wang et al., 2002). In addition, agonist stimulation has been shown to increase phosphorylation of MOP-R in thalamic and striatal brain slices (Deng et al., 2001).
In transfected cell systems the heterologously expressed MOP-R becomes phosphorylated at specific serines and threonines, which then result in desensitization and internalization of the receptor in an arrestin-mediated manner (Gainetdinov et al., 2004). Although the pattern of receptor phosphorylation is complex, the threonine at position 370 and the serine at position 375 on the C-terminal tail of the rat receptor (corresponds to Ser377 in human) have been implicated in receptor internalization (El Kouhen et al., 2001), and Ser375 has been demonstrated to be required for morphine-mediated desensitization (Schulz et al., 2004). Mutation of the threonine at position 180 in the second intracellular loop of the MOP-R has also been shown to be sufficient for abolishing DAMGO-induced desensitization of mu opioid receptors expressed in Xenopus oocytes and AtT20 cells (Celver et al., 2001); however, other MOP-R domains mediate internalization in transfected HEK and AtT20 cell expression systems (Celver et al., 2004).
In an attempt to study the consequences of agonist-stimulated receptor phosphorylation in in vitro cellular models as well as in cells within brain, we have generated two antibodies against phospho-peptides corresponding to regions of the MOP-R predicted to be phosphorylated upon receptor activation. The first antibody was generated against a peptide corresponding to the second intracellular loop of the MOP-R and including a phosphorylated Thr180 (MOR-P1). The second antibody corresponds to a region of the MOP-R C-terminal tail that includes phosphorylated residues at Thr370 and Ser375 (MOR-P2). We have compared our results with a commercially available antibody directed against a single phosphorylated residue (p)-MOR(Ser375) (Narita et al., 2004). Ultimately, our aim with these antibodies will be to locate sites of mu opioid receptor activation in the brain under physiological conditions as we have done using phosphorylation-state selective kappa opioid receptor antibodies (Xu et al., 2004).
The striatum contains a dense expression of MOP-R (Mansour et al., 1994; Kitchen et al., 1997; Kaneko et al., 1995; Schulz et al, 1998) and is a vital area for the integration and regulation of motor and cognitive functions. MOP-R-expressing neurons seem to target the receptor preferentially to the somato-dendritic domain, and evidence suggests that MOP-R is located on neuronal dendrites in the striatum (Wang et al., 1996). The identity of the endogenous ligand for MOP-R is still uncertain as there is considerable cross-talk among the opioid peptide and receptor systems (Bodnar, 2004; Kieffer and Gaveriaux-Ruff, 2002); however, candidates include β-endorphin, endomorphin (Zadina et al., 1997) and enkephalin. Prodynorphin can also be processed to pentapeptide forms that bind MOP-R (Zamir et al., 1984).
The principal goal of the present study was to investigate the phosphorylation of MOP-R in response to activation of the endogenous opioid system within the mouse brain. We report that after injection of the mu opioid agonist, fentanyl, wild-type mice showed a significant increase in MOR-P2 immunoreactivity (IR) in the striatum. Chronic neuropathic pain induced by partial sciatic nerve ligation (pSNL) resulted in both tolerance to the pharmacological effects of morphine and increased striatal MOR-P2-IR. We report that deletion of endogenous β-endorphin selectively blocked the tolerance to acute morphine caused by pSNL and blocked the increase in MOR-P2-IR resulting from pSNL. The results suggest that β-endorphin released during chronic pain may act as an endogenous mu opioid agonist and that sustained β-endorphin release may induce mu receptor phosphorylation and opioid tolerance.
Experimental Procedures
Antibody Production and Purification
Peptides corresponding to unique regions of the muopioid receptor second loop CHPVKALDFRT(PO4)PRNA (amino acids 171-186) and C-terminal tail CARIRQNT(PO4)REHPS(PO4)TAN (amino acids 364-378) were synthesized and purified (PeptidoGenic Research, Livermore, CA). These peptides were conjugated to keyhole limpet cyanin (KLH) through the N-terminal cysteine and used to immunize rabbits (Cocalico Biologicals, Reamstown, PA) to develop polyclonal antiserum to the phosphorylated forms of the mu-opioid receptor. Affinity columns for purifying phospho-specific antibody were prepared by conjugating the MOR-P1 or MOR-P2 peptide to a sulfo-link resin (Pierce Biotechnology, Rockford, IL). The specificities of the raw sera and affinity-purified antibodies were quantified by ELISA as described previously (McLaughlin et al., 2003b).
Cell Culture and Receptor Mutagenesis
Stably transfected AtT20 Cells and HEK cells expressing the green fluorescent protein (GFP) conjugated mu-opioid receptor were used as described previously (Celver et al., 2004). Receptor mutants were created by designing PCR primers to introduce either a double amino acid (T370A/S375A) mutation in the C-terminal tail of the rat MOP-R cDNA or a C-terminal truncation after isoleucine-352. The constructs were ligated into pcDNA3-GFP37 vector provided by Dr. Kenneth Mackie (University of Washington, Seattle, WA) or pTARGET (Promega Biosciences, San Luis Obispo, CA), sequenced to confirm the mutagenesis and transfected into both HEK 293 and AtT20 cells using Lipofectamine (Invitrogen, Carlsbad, CA). Cells were maintained in DMEM supplemented with 2 mM L-glutamine, 100 unit/ml penicillin, 100 μg/ml streptomycin, and 10% horse serum (AtT20 Cells) or DMEM /F12 supplemented with 2 mM L-glutamine, Penicillin/Streptomycin, and 10% Fetal Bovine Serum (HEK 293 cells). Cells were grown at 37°C in 5% CO2 and were passaged when 90% confluent.
Confocal Imaging
Cells were plated at 75-90% confluency on poly-D-lysine coated cover slips 24 hours prior to treatment. Cells were treated as described in the text, fixed in 4% paraformaldehyde and incubated in 5 μg/ml primary antibody overnight at 4°C. Cover slips were incubated with a 1:10,000 dilution of Rhodamine conjugated anti-rabbit antibody (Jackson Immunoresearch Labs, West Grove, PA) for 2 hours at room temperature. Cover slips were allowed to air-dry, were then mounted to slides using Vectashield (Vector Labs, Burlingame, CA) and imaged with a Leica TCS SP/MP Laser Scanning Confocal Microscope.
Animal Care and Use
Protocols with mice were approved by the Institutional Animal Care and Use Committee in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (1996) and guidelines for the International Association for the Study of Pain. Mice were inspected regularly by veterinary staff to ensure compliance. Young adult male C57BL/6 mice (Charles River Laboratories, Wilmington, MA) or transgenic mice on a C57BL/6 genetic background were used in these experiments. Induced mutant mice having specific disruptions of the genes encoding MOP-R (-/-), prodynorphin (-/-), proenkephalin (-/-) and β-endorphin (-/-) (single nucleotide insertion into the Pomc gene that created a premature translational termination codon in place of the wild-type N-terminal tyrosine codon of β-endorphin) were generated as described (Schuller et al., 1999; Ragnauth et al., 2001; Rubinstein et al., 1996; Sharifi et al., 2001). Heterozygous breeding pairs, backcrossed >10 generations onto C57BL/6 or C57BL/6J backgrounds, were used to generate knockout (-/-) and wild-type littermate controls for this study. Pups were genotyped by PCR as described previously (McLaughlin et al., 2003a). Mice used were 16-28 weeks of age and weighed 25-35 gm at the time of the start of the procedures. All mice were housed in groups of two to four in plastic cages using Bed-o’Cob (Andersons, Maumee, OH) for home bedding within the University of Washington vivarium and maintained in pathogen-free housing units. The housing rooms were illuminated on a 12 hr light/dark cycle with lights on at 7 A.M.; lab chow and water were available ad libitum.
Immunoblotting
Adult C57Bl/6 mice (15-25 g) were decapitated 30 min after IP administration of 10 mg/kg fentanyl. Mouse brains were dissected on an ice-cold Petri dish, and the cerebella and corpora striata were removed. All samples were immediately frozen on dry ice and stored at -80°C until analyzed. On the day of the assay, tissue samples were homogenized in 3 ml of homogenizing buffer (25 mM Tris and 250 mM sucrose, pH 7.4, with Protease Inhibitor cocktail set 1 (500 mM 4-(2-Aminoethyl)benzenesulfonylfluoride HCl; 150 nM aprotinin; 1 mM E-64 protease inhibitor; 0.5 mM EDTA-Na2; 1mM leupeptin) (Calbiochem, San Diego, CA) and Phosphatase Inhibitor cocktail set 1 (bromotetramisole oxalate, cantharidin, and microcystin with orthovanadate) (Calbiochem, San Diego, CA) using a Dounce tissue homogenizer. Homogenized tissue was centrifuged at 3000x g for 30 min at 4°C. The supernatant was diluted with 100 mM NaCl, 0.2 mM MgSO4, followed by ultracentrifugation at 50,000x g for 25 min. The membrane pellet was resuspended in 100 μl solubilization buffer (50 mM Tris (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, and 1% Triton X-100, with protease and phosphatase inhibitor cocktails as described above) for 1 h on ice. Protein concentration for the mouse brain membrane fraction was determined using the BioRad assay kit (Hercules, CA). Approximately 40 μg/well mouse brain membrane were resolved by gel electrophoresis on a 10% NuPage pre-cast Bis-Tris gel (Invitrogen, Carlsbad, CA). Proteins were transferred to nitrocellulose and incubated for 2 hr in 10% nonfat dry milk (NFDM) in Tris-buffered saline (TBS, 50 mM Tris base and 150 mM NaCl, pH 7.4) to block nonspecific binding. The nitrocellulose membrane was then incubated at 4°C in 5% NFDM/TBS containing 10 μg/ml MOR-P2 antibody (raised in rabbits) and a 1:1000 dilution of MOR (Neuromics, Bloomington, MN) antibody (raised in guinea pig) for two nights at 4°C. Membranes were washed with TBST (Tris-buffered saline, 1% Tween 20) five times for 10 min each and incubated with 5% NFDM/TBS containing both Alexa Fluor680-labelled goat anti-rabbit IgG (1:5000) (Molecular Probes, Eugene, OR) and IR Dye-800-labeled anti-guinea pig IgG (1:5000) (Rockland Immunochemicals, Gilbertsville, PA) for 60 min at room temperature. The membranes were again washed five times in 5% TBST, then infrared fluorescence was detected using the Li-Cor Odyssey system (Li-Cor, Lincoln, NB).
Surgical procedures-neuropathic pain model
The partial sciatic nerve ligation (pSNL) model of chronic neuropathic pain was used as described previously (Xu et al., 2004; Seltzer et al., 1990). The right sciatic nerve was exposed at the gluteal region, and approximately one-third to one-half the diameter of the nerve was tightly ligated with 7-0 silk suture (Surgical Specialties Corporation, Reading, PA).
Immunohistochemical staining in the striatum
Adult mice were anesthetized with isoflurane (Sigma, St. Louis, MO) and intracardially perfused with a fresh solution of 4% paraformaldehyde in phosphate buffer (PB) (0.1 M sodium phosphate, pH 7.4). The brain was removed and postfixed in the same fixative for 1-2 hr. The tissue was then sunk in solution of 30% (w/v) sucrose in PB at 4°C over 24 hr. Transverse sections (40 μm) of the striatum at approximately bregma 0.74-1.32mm (according to Mouse Brain Atlas: C57BL/6J Coronal; Evan Williams and Rob Williams; http://www.mbl.org were cut on a sliding microtome and placed in PB until the tissue was processed for immunohistochemistry. Free-floating brain sections were rinsed in 0.1 M Tris buffer, pH 7.4, for 30 min, in 0.1 M Tris buffered saline (TBS), pH 7.4, for 30 min, blocked using 0.1% Triton X-100 and 4% normal goat serum in 0.1 M TBS for 60 min. The tissue sections were then incubated in 20 μg/ml affinity-purified rabbit anti-MOR-phosphoselective antibodies, MOR-P1 or MOR-P2. Antibodies were diluted in a solution containing 0.1% Triton X-100 and 4% normal goat serum in 0.1 M TBS. The tissue sections were rinsed in TBS for 60 min and incubated in Alexa Fluor-488 goat anti-rabbit IgG (secondary antibody) diluted 1:300 for 1 hr at 37°C. The sections were rinsed in TBS for 10 min, rinsed in TB for 20 min, and then mounted on gelatin-coated slides and viewed with a MRC 600 confocal microscope (Bio-Rad, Hercules, CA). For control sections, primary antibodies (preincubated with their cognate peptide) or no primary antibody was used to define non-specific staining. The average intensity of MOR-P2 labeling was analyzed using a computer-assisted imaging analysis system (Metamorph). Mean image intensity for each treatment group was performed with the investigator blind to condition, 3-5 sections per animal were assessed, and 5 independent animals were used to generate replicates. Quantitation was done for 4 groups of animals: sham-ligated wild-type; pSNL wild-type; sham-ligated β-endorphin KO; and pSNL β-endorphin KO. Data were normalized to the average pixel intensity above background in the wild-type, sham-ligated group.
Allodynia and hyperalgesia
Mice were habituated to handling and testing equipment for at least 1hr prior to behavioral testing. Allodynia was measured using a hand-held force transducer fitted with a 0.7 mm2 polypropylene tip (electronic von Frey anesthesiometer, IITC Inc., Life Science Instruments, Woodland Hills, CA, USA). The mice stood on a metal mesh covered with a plastic dome. The plantar surface of the hindpaw was perpendicularly touched by the polypropylene tip with a gradual increase in pressure until the threshold that induced paw withdrawal was found. Hyperalgesia was assessed using the Hargreaves radiant heat test (Hargreaves et al., 1988). Mice were placed on a glass surface (IITC Life Science Inc, Woodland Hills, CA, USA and a radiant heat source (50% light intensity) was positioned under the plantar surface of the hindpaw, and the latency to paw withdrawal was measured. A 20 sec maximum heat exposure was imposed to prevent tissue damage. To measure antinociception induced by morphine, latency of tail withdrawal from hot-water (55.0 ± 0.5 °C) immersion was assessed. “Cutoff time” in this test was set at 15 s to prevent tissue damage. Tail withdrawal latency was selected as an alternative measure of thermal sensitivity for morphine analgesia testing because the locomotor hyperactivity response of mice to morphine interfered with measurements of hind paw responses in the Hargreaves test.
Conditioned place preference (CPP)
We used a balanced and unbiased conditioned place preference paradigm to measure the rewarding properties of morphine (Li et al., 2006). Mice were conditioned in a three-compartment apparatus divided into two equal-sized outer sections (25 × 25 × 25 cm) joined by a small central compartment (8.5 × 25 × 25 cm) accessed through a single doorway (3 × 3 cm). The compartments differed in wall striping (vertical vs. horizontal alternating black and white lines, 1.5 cm in width), floor texture (Beta chips vs. Bed-o’Cob), and lighting intensity. On day 1 (4 days after pSNL surgery), mice were tested for initial bias by placing individual animals in the small central compartment and allowing them to freely explore the entire apparatus for 30 min. The testing apparatus was balanced; no significant difference in outer compartment preference was detected prior to conditioning. On day 2, mice were injected subcutaneously with saline (10 μl/gm body weight), immediately placed in one compartment (randomly selected) for 30 min, and then returned to their home cage. Four hours later, mice were injected (s.c.) with saline, morphine 2.5 mg/kg, or morphine 5 mg/kg and immediately confined for 30 min to the opposite outer compartment. On day 3 and day 4, mice again were conditioned first with saline, followed by saline or morphine 4 hours later, in the appropriate compartments. On day 5, mice were placed in the small central compartment and allowed to freely roam between the two outer compartments for 30 min. Time spent and locomotor activity in the compartments were evaluated by video tracking and motion analysis performed via a PC-based data acquisition system (Ethovision version 3.0, Noldus, Wageningen, The Netherlands) that received video input from a digital camera (Canon, ZR60). The camera was placed 3 m above the chamber, and the acquisition program collected 5 samples per second for each test. Conditioned place preference (CPP) was defined as the difference in time an animal spent on the morphine-paired side vs. the saline-paired side.
Chemicals
Fentanyl and naloxone were from Sigma (St. Louis, MO). DAMGO was purchased from Bachem (Torrance, CA). Morphine was obtained from NIDA (Bethesda, MD). Drugs were dissolved in 0.9% saline, unless otherwise specified.
Statistical analysis
Data are expressed as mean ± S.E.M. Differences in the mean intensities of MOR-P2-IR on the striatum of the mice after saline or fentanyl treatment were tested with Student’s t test for significant pair-wise comparisons. Group differences in the MOR-P2-IR on the striatum of mice after pSNL were assessed by analyses of variance (ANOVAs) followed by Bonferroni’s post-hoc test. Differences in locomotor activities between groups were assessed by the Student’s t-test. Group differences in paw withdrawal responses to thermal or mechanical stimuli and CPP times were assessed by analyses of variance (ANOVAs) followed by Fisher’s LSD post-hoc test. In all analyses, the null hypothesis was rejected at the <0.05 level of confidence.
Results
MOR-P Antisera Production and Characterization
Antisera against potentially phosphorylated regions of the rat mu-opioid receptor were produced by immunizing rabbits with KLH-conjugated phospho-peptides corresponding either to amino acids 171-186 (MOR-P1) or 364-378 (MOR-P2) (Fig. 1a). The raw antiserum developed against the MOR-P1 peptide demonstrated an increased titer for peptides corresponding to phosphorylated and unphosphorylated regions of the rat MOP-R (Fig. 1b). Raw sera from the MOR-P2 immunized rabbits showed a similar pattern (data not shown). MOR-P1 and MOR-P2 phospho-specific antibodies were affinity purified by absorption to phosphopeptide-linked sepharose as described in Experimental Procedures. The resulting affinity-purified MOR-P1 and MOR-P2 showed robust selectivities by ELISA for the phosphorylated peptide over the unphosphorylated peptide sequence and do not recognize a comparable phosphopeptide of different sequence (Fig. 1c, d). A commercially available mono-phospho-MOR antibody (Neuromics, Bloomington, MN) recognizes a similar region of the C terminal MOP-R sequence including a single phosphorylated serine at position 375 [(p)-MOR(Ser375)]. This antibody has a lower titer and less phospho-selectivity when compared to the MOR-P2 antibody (Fig. 1e).
Figure 1. Characterization of antisera selectivity.
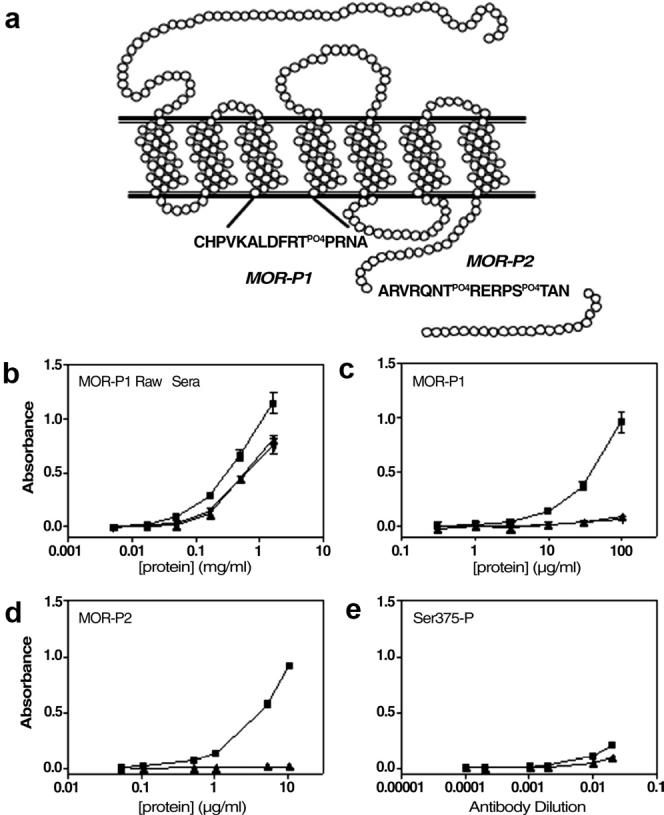
(A) Representation of the mu-opioid receptor (MOP-R) illustrating the regions of the receptor targeted by the phospho-selective antibodies MOR-P1 and MOR-P2. ELISA results demonstrating the specificity of MOR-P1 raw sera (B), affinity purified MOR-P1 (C), affinity purified MOR-P2 (D) and the commercial antibody recognizing (p)-MOP(Ser375) (E), against the specific phospho-peptide (■), specific non-phospho peptide (▲) and a non-specific phospho-peptide (RVRNTVQDPAS(PO4)MRD, matching amino acids 359-372 in the kappa opioid receptor sequence used to generate KOR-P (26) (▼).
HEK 293 cells and AtT20 cells transfected with MOP-R-GFP failed to show an increase in MOR-P1 immunoreactivity (IR) after 15, 30, 60 or 120 min treatment with agonist (0.1, 1 or 10 μM DAMGO or 0.1, 1 or 10 μM fentanyl) (data not shown). The results suggest that either the MOR-P1 antipeptide antibody does not recognize the full-length receptor or that this site does not change phosphorylation state following agonist treatment. In contrast, transfected HEK or AtT20 cells treated with 1 μM DAMGO showed a robust increase in MOR-P2-IR and translocation of the GFP-tagged MOP-R from plasma membrane into intracellular compartments, consistent with receptor internalization (Fig. 2). The increase in MOR-P2-IR was blocked by pretreatment with 1 μM naloxone, was not evident after treatment with 1 μM naloxone alone, and was not evident in cells treated with 1 μM DAMGO but not incubated with primary antibody (data not shown). Site directed mutagenesis of MOP-R at Thr370 and Ser375 MOP(T370A/S375A) abolished the DAMGO-induced increase in MOR-P2-IR (Fig. 2e). MOR-P2-IR was not increased for DAMGO treated cells expressing MOP-R truncated after isoleucine-352 MOP(Δ352)-GFP (Fig. 2f). Similarly, AtT20 cells expressing MOP(S375A)-GFP did not increase MOR-P2-IR following DAMGO treatment (data not shown). The single mutant construct MOP(T370A) did not efficiently express cell surface labeling in either AtT20 or HEK293 cells for reasons that we did not identify (data not shown). The results suggest that the MOR-P2 antibody was able to recognize phosphorylated MOP-R following agonist treatment.
Figure 2. DAMGO treatment increases MOR-P2 IR of transfected wild-type MOP-R-GFP but not MOP-(T370A/S375A)-GFP or MOP-(Δ352)-GFP.
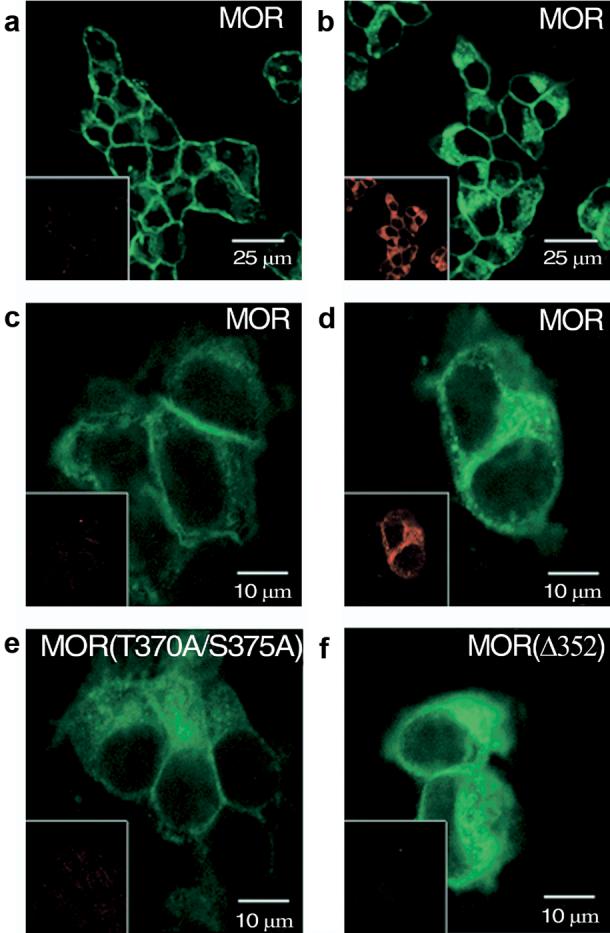
(A) Confocal images of GFP-conjugated MOP-R (green) with Rhodamine labeled MOR-P2-IR images (red, inset). MOR-P2 antibody specifically labels AtT20 cells transfected with the mu-opioid receptor in the presence (B) of 1 μM DAMGO for 30 min at 37°C. HEK293 cells transfected with wild-type MOP-R that were untreated (C) or treated with DAMGO as above (D) showed increased MOR-P2-IR (inset). A MOR-P construct with double amino acid substitution of MOP-R eliminating the two phosphorylation sites (T370A/S375A) was transfected into HEK 293 cells and showed no labeling of healthy cells in the presence of agonist (E). The C-terminal truncation mutant MOP-(Δ352) transfected into HEK 293 cells also showed no DAMGO-induced staining (F).
Using western analysis of brain membranes isolated from mice, we found that MOR-P2-IR labeling of a band at 70 kD was increased in striatum but not cerebellum after pretreatment with the efficacious opiate agonist fentanyl (10 mg/kg, i.p.) (Fig. 3). Under the same conditions, labeling with a non-phosphoselective antibody against MOP-R showed labeling of a band at 70 kD in the striatal but not cerebellar regions (Fig. 3). Fluorescence intensity analysis showed that the MOR-P2 labeling in fentanyl-treated mice significantly increased by 62±19% (n=4) compared to saline treated-striatal controls.
Figure 3. Mu opioid agonist induces an increase in MOR-P2 IR.
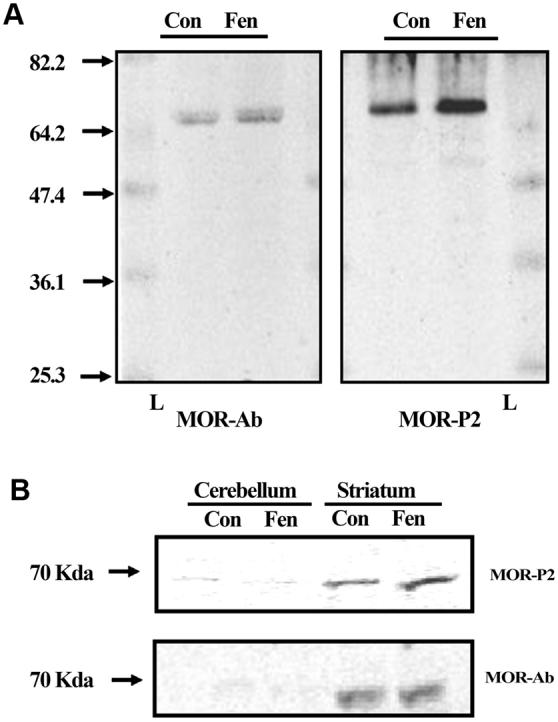
(A) Representative western blots of isolated mouse brain striatal membrane protein (40 mg/lane) taken from either fentanyl (10 mg/kg, i.p., Fen) or saline (Con) mice, and then incubated with either the non-phospho-MOR antibody (MOR-Ab, left) or the MOR-P2 (right) antibody. Fentanyl treatment significantly increased the MOR-P2 antibody labeling suggesting an increase in MOP-R phosphorylation. Both antibodies label a protein of approximately 70 kD as indicated by comparison to the molecular weight marker ladder (L). (B) Representative western blots of isolated mouse brain striatum and cerebellum for the non-phospho-MOR antibody or the MOR-P2 antibody. The samples from cerebellum demonstrate a lack of immunoreactivity for both the non-phospho-MOR antibody and MOR-P2 antibody further showing the selectivity of these antibodies. The images are each representative of 4 independent experiments. Two blots illustrate different points: A, showed the absence of nonspecific bands in the full length of the gel, and B, showed the direct comparison between MOR-P2-IR in cerebellum and striatum.
The utility of the MOR-P2 antiserum was next assessed using immunohistochemistry. Striata of mice pretreated with fentanyl (10 mg/kg, i.p.), 30 min prior to perfusion showed a significant increase in MOR-P2-IR compared with sections from saline pretreated controls (Fig. 4 a-b, p<0.05). The average intensity of MOR-P2-IR was 239 ± 16% greater after fentanyl than in saline-pretreated mice 100±4% (n=5 different animals in each group). The increased MOR-P2-IR was blocked by injection of naloxone (10 mg/kg, i.p.) given 30 min before fentanyl (Fig. 4d). Similarly, MOR-P2-IR did not increase after fentanyl treatment of MOP-R knockout mice (MOR-/-) (Fig. 4e). Additionally, controls showed a lack of any staining in the absence of primary antibody (Fig. 4f). Non-specific nuclear staining was evident in all samples. The attempt to reduce the nonspecific staining by preabsorbing the MOR-P2 antibody with liver powder extract was only partially successful (data not shown).
Figure 4. Mu opioid agonist induces an increase in MOR-P2 IR.

MOR-P2 IR within mouse striatum at approximately bregma 1.32-0.74 mm. The MOR-P2-IR (arrowheads) was significantly increased after injection of 10 mg/kg fentanyl 30 min before perfusion (B) compared to saline injection in wild-type mice (A). This increase in MOR-P2-IR was blocked by 10 mg/kg naloxone pretreatment (D) and was not evident in transgenic mice lacking mu opioid receptors (MOR-/-) (E). Additionally MOR-P2-IR in striatum was significantly increased in wild-type pSNL mice (C) but not in sham-ligated mice (data not shown). Negative controls showed the lack of specific staining in the absence of primary antibody (F). Scale bars: a-f, 50 μm. The results shown are representative images taken from more than three independent experiments.
We compared the distribution of MOR-P2-IR in striatum with the immunostaining evident using a non-phospho-MOP-R antibody (Neuromics, Bloomington, MN). The pattern of staining was not concordant (data not shown). The basis for the discrepancy was not identified, but possible reasons are discussed below. However, a second, commercial phospho-mu opioid receptor antibody that recognizes only a single phosphorylation site (p)-MOP(Ser375) also showed immunoreactivity that was completely concordant with MOR-P2 staining in the striatum (data not shown).
Having found that receptor phosphorylation could be increased by exogenous opioid treatment, we next assessed the effects of partial sciatic nerve ligation (pSNL) previously shown to induce the sustained release of endogenous opioid peptides (Xu et al, 2004). pSNL induces a robust neuropathic pain response that reaches its maximal intensity on day 7 (Seltzer et al., 1990; Xu et al., 2004). Interestingly, MOR-P2-IR was also increased by pSNL. On day 7 after pSNL, MOR-P2-IR within the striatum was significantly increased in wild-type pSNL mice (Fig. 4c) but not in sham-ligated mice (image not shown). The average intensity of staining was 224±14% greater than in sham operated mice 100±4% (n=5 different animals in each group).
Chronic neuropathic pain response after pSNL was confirmed in the present study by showing that mice displayed significant hyperalgesia; hindpaw withdrawal latencies were significantly reduced on both ipsilateral and contralateral sides compared to sham-ligated mice. Two-way measure ANOVA (factor ligation side + factor pSNL) revealed significant effects of both pSNL and ligation side, but no significant interaction effect (FpSNL/1,70=22.52, P<0.001; Fside/1,70=4.32, P<0.05, FpSNL×side/1,70=1.45, P>0.05). Four days after the pSNL, mice also displayed a significant decrease in mechanical threshold on the ipsilateral side. Two-way ANOVA (factor ligation side + factor pSNL) showed there were significant effects of pSNL and ligation side, as well as effect of interaction (FpSNL/1,70=50.44, p<0.001; Fside/1,70=6.62, p<0.05, FpSNL×side/1,70=13.01, p<0.001). The antinociceptive effects of morphine were tested using the tail-withdrawal assay because morphine-induced hyper-locomotion interfered with the von Frey allodynia and Hargreaves hyperalgesia testing. When tested on the fourth day after the surgical procedure no differences were observed in basal tail withdrawal latency. Morphine (2.5-10 mg/kg, s.c.) produced significant and dose-dependent antinociception in response to noxious thermal stimuli in both sham-ligated and pSNL mice. However, mice with pSNL displayed significantly less morphine analgesia than sham-ligated mice after 5 mg/kg of morphine. Two-way ANOVA (factor ligation side + factor pSNL) showed there were significant effects of pSNL and dose, but no significant interaction effect (FpSNL/1,65=4.69, p<0.05; Fdose/3, 65=32.85, p<0.001, FpSNL×dose/3,65=1.63, P>0.05, Fig. 5A). A cumulative dose-response curve in pSNL mice showed a clear rightward shift in the antinociceptive effects of morphine. The results suggest that pSNL induced tolerance to the acute effects of morphine.
Figure 5. pSNL reduced morphine analgesia.
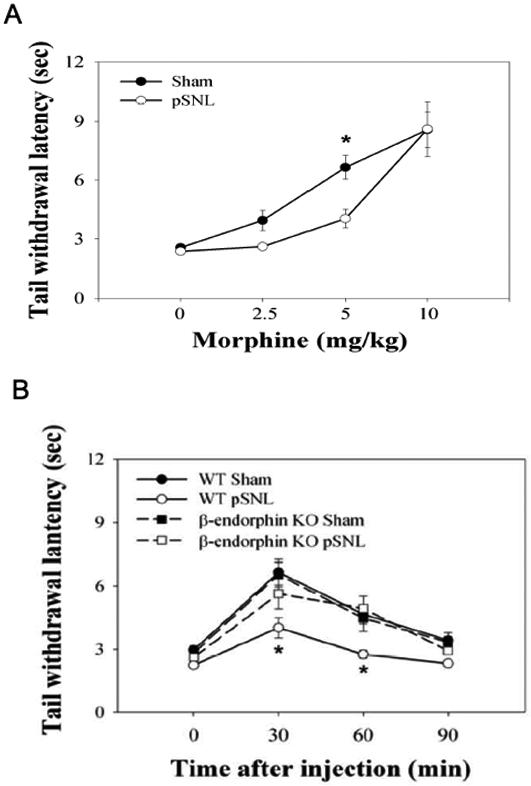
(A) The dose response of morphine-induced antinociception was obtained by measuring tail withdrawal latencies 30 min after morphine injection, 4 days after pSNL (saline group, sham n=10, pSNL n=10; morphine 2.5 mg/kg group, sham n=8, pSNL n=8; morphine 5 mg/kg group, sham n=10, pSNL n=9; morphine 10 mg/kg group, sham n=9, pSNL n=9). *, P< 0.05, compared to sham-ligated group by two-way ANOVA followed by Fisher’s LSD post-hoc test. (B) The time course of morphine-induced antinociception was obtained by measuring tail withdrawal latencies followed the injection of morphine at 5 mg/kg, 4 days after pSNL (WT sham group, n=10, WT pSNL n=9; KO sham n=5, KO pSNL n=5). Mice lacking expression of β-endorphin did not show decreases in morphine analgesic effects following pSNL. *, P< 0.05, difference between WT pSNL and β-endorphin KO pSNL groups by two-way ANOVA followed by Fisher’s LSD post-hoc test.
pSNL produced hyperalgesia and allodynia in β-endorphin KO mice that were equivalent to that produced in wild-type littermates. In the Hargreaves test, two-way ANOVA (factor pSNL + factor β-endorphin KO) showed that there were only significant effects of pSNL, but no significant effect of either β-endorphin KO or interaction. (FpSNL/1,25=10.55, P<0.001; F β-endorphin KO/1,25=0.04, P>0.05, FpSNL×side/1,25=0.05, P>0.05). Similarly in the von Frey allodynia test, pSNL produced a significant decrease in mechanical threshold on both WT and β-endorphin KO. Two-way measure ANOVA (factor dose + factor β-endorphin KO) showed there were only significant effects of pSNL, but no significant effects of either β-endorphin KO or interaction, (FpSNL/1,26=25.36, P<0.001; F β-endorphin KO/1,26=0.29, P>0.05, FpSNL×side/1,26=0.08, P>0.05). Moreover, the increases in tail-flick latencies caused by 5 mg/kg morphine administered to sham-ligated wild-type and β-endorphin KO mice were not significantly different. In contrast, β-endorphin KO mice did not show the reduced sensitivity to morphine evident in wild-type mice following pSNL (Fig. 5B). pSNL significantly reduced the analgesic effect of morphine in wild-type, however β-endorphin KO mice with pSNL were not less sensitive to morphine than sham-ligated mice (Fig. 5B). Two-way ANOVA (factor time + factor pSNL) demonstrated that there were significant effects of pSNL and time (FpSNL/1,68=38.17, p<0.001; Ftime/3,68=22.83, p<0.001). There was no significant difference between sham-ligated KO mice and pSNL KO mice.
In a second measure of morphine tolerance, we assessed the effects of pSNL on morphine conditioned place preference (CPP). Sham-ligated mice showed robust morphine conditioned place preference that was significant at the doses of 2.5 and 5 mg/kg (one-way ANOVA, F5,59 = 5.81, p<0.001, Fisher’s LSD test, p<0.05 vs. saline controls). Mice with pSNL showed significantly less time spent in drug-paired compartment compared to sham-ligated mice when mice were trained by 5 mg/kg of morphine (Fisher’s LSD Multiple-Comparison test, p<0.05, Fig. 6A), but interestingly pSNL had no significant effect on CPP induced by 2.5 mg/kg morphine. The decrease in morphine CPP was not due to an impairment of exploratory behavior. There was no significant difference in locomotor activity between sham-ligated mice and pSNL mice measured in the CPP chamber (p>0.05). pSNL did not cause a decrease in morphine CPP in mice lacking β-endorphin. There was no significant difference in morphine CPP between sham-ligated and pSNL β-endorphin KO mice (Fig. 6B).
Figure 6. pSNL decreased morphine CPP.
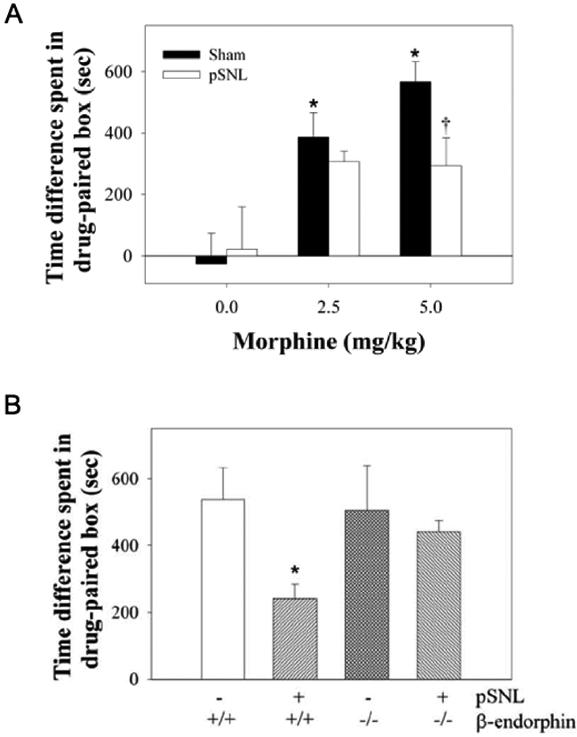
(A) Morphine-conditioned place preference in sham-ligated mice and pSNL mice. The results are plotted as the difference in the times spent on the morphine-paired side vs. the saline-paired side.*, P< 0.05 compared to saline-injected group and †, P < 0.05 compared to sham-ligated mice by ANOVA followed by Fisher’s LSD post-hoc test. (B) Morphine-conditioned place preference in WT mice and KO mice conditioned with 5 mg/kg morphine after pSNL or sham-ligation.*, P< 0.05 compared to WT sham (- pSNL) group and β-endorphin KO pSNL group by ANOVA followed by Fisher’s LSD post-hoc test.
The results suggest that pSNL induced mu opioid receptor tolerance by stimulating the sustained release of β-endorphin. To determine whether the increase in MOR-P2-IR was also caused by sustained β-endorphin release in wild-type mice, we compared the effects of pSNL on induced mutant mice selectively lacking either β-endorphin, proenkephalin, or prodynorphin opioid peptides. On day 7 after pSNL the average MOR-P2-IR intensity was significantly increased in wild-type mice, proenkephalin (-/-) mice and prodynorphin (-/-) mice (*p<0.05) compared to sham surgery (Fig. 7B,C). In contrast, β-endorphin KO mice did not show an increase in MOR-P2-IR after pSNL (Fig. 7A). To confirm that the mu opioid receptors in the β-endorphin knockout mice were still able to respond to agonist stimulation with an increase in MOR-P2-IR, we stimulated the mu opioid receptors by a single injection of fentanyl 10 mg/kg i.p., 30 min before perfusion. β-endorphin KO mice showed a significant 211±8.% increase in MOR-P2-IR. Similarly MOR-P2-IR increased in prodynorphin (-/-) mice by 200±2%, proenkephalin (-/-) mice by 222 ±14%, and wild-type 239±16% after injection of fentanyl (data not shown). These results confirm that the receptors were functional in the β-endorphin KO mice and suggest that chronic neuropathic pain activated the mu opioid receptors through the release of the endogenous opioid peptide β-endorphin in wild-type mice.
Figure 7. The average intensity of the MOR-P2-IR for wild-type mice, β-endorphin (-/-) mice, proenkephalin (-/-) mice and prodynorphin (-/-) mice on day 7 after pSNL or sham surgery.

Data for each bar represents the average pixel intensity for MOR-P2-IR quantified by Metamorph for an image field size of 450 μm × 300 μm. Data for ligated gene-deleted groups were normalized to the average pixel intensity in the wild-type, sham ligated group (100%). Data represent mean ± SEM, n=5 independent animals, *p<0.05, compared to the wild-type, sham-ligated group. For the β-endorphin (-/-) mice, two-way ANOVA (factor genotype + factor ligation) showed there were significant effects of genotype, ligation and interaction (Fgenotype/1,97=268.63, P<0.001; Fligation/1,97=91.34, P<0.001; Fgenotype×ligation/1,97=60.71, P <0.001). Bonferroni post-hoc test showed WT-sham was significantly different from WT-pSNL, whereas β-endorphin KO pSNL is significant different than WT-pSNL. No difference between KO SNL and KO sham group. For the prodynorphin(-/-) mice, two-way ANOVA (factor genotype + factor ligation) showed there was only significant effects of ligation, but no effects of genotype and interaction (Fgenotype/1,96=0.01, P>0.05; Fligation/1, 96=121.72, P<0.001; Fgenotype×ligation/1,96=0.31, P >0.05). Bonferroni post-hoc test showed WT-sham was significantly different than WT-pSNL, and DYN KO pSNL was not different from WT-pSNL. For proenkephalin (-/-) mice, two-way ANOVA (factor genotype + factor ligation) showed there was only significant effects of ligation, but no effects of genotype and interaction (Fgenotype/1,98=0.56, P>0.05; Fligation/1, 96=190.72, P<0.001; Fgenotype×ligation/1,96=3.52, P >0.05). Bonferroni post-hoc test showed WT sham was significantly different from WT-pSNL, and ENK KO pSNL was not different from WT-pSNL.
Discussion
The principal findings of this study were that a phosphoselective antibody directed against phosphothreonine 370 and phosphoserine 375 within the C-terminal domain of the mu-opioid receptor showed increased tissue labeling following agonist treatment and following sciatic nerve damage. The specificity of this reagent was established using transfected cell controls and mu opioid receptor knock out mice. Using this antibody and mutant mouse strains lacking expression of the different endogenous opioid peptides, we found that the increase in MOR-P2-IR after pSNL was selectively blocked by the absence of β-endorphin, but not of enkephalin or dynorphin peptides. These results suggest that MOP-R phosphorylation may be induced in vivo following release of β-endorphin under pathophysiological conditions.
Phosphorylation of the agonist-stimulated receptor has been shown to be one of the initial regulatory steps controlling receptor signaling and is vital to the desensitization and internalization of G-protein coupled receptors. Several studies have demonstrated that the phosphorylation events leading to desensitization and internalization can be distinguished (Lamey et al., 2002; Celver et al., 2004). For example, we previously found that mutation of the MOP-R at Thr180 abolished the DAMGO-induced desensitization of the receptor while leaving internalization intact (Celver et al., 2004). In contrast, agonist-stimulated phosphorylation of the MOP-R at Thr370 and Ser375 on the C-terminal tail has been shown to be important in receptor internalization (El Kouhen et al., 2001) as well as in the morphine- but not DAMGO-induced desensitization of the receptor (Schulz et al., 2004). Other potential phosphorylation sites within the receptor have also been reported (Connor et al., 2004); however the roles of specific MOP-R phosphorylation events in brain are not yet understood.
Phosphoselective antibodies provide a means to assess these possible phosphorylation events in vivo. The MOR-P1 antibody failed to support the hypothesis that T180 within MOP-R was a site for agonist-induced receptor phosphorylation; however, the MOR-P2 antibody results do support the suggestion that Thr370 and Ser375 on the C-terminal tail are phosphorylated. Parallel studies using a phosphoselective antibody (KOR-P) able to detect the Ser369-phosphorylated form of kappa opioid receptor in vivo also established the connection between opioid receptor phosphorylation within the C-terminal domain and kappa analgesic tolerance (McLaughlin et al., 2004).
The selectivity of the MOR-P2 antibody was assessed by ELISA data showing that affinity purified MOR-P2 was highly selective for the phosphorylated peptide. We also found that MOR-P2 recognized the agonist-stimulated receptor expressed in cells transfected with wild-type MOP-R-GFP but not MOP(T370A/S375A)-GFP mutated form of the MOP-R. Furthermore, receptor phosphorylation in the mouse brain was significantly enhanced by the selective agonist fentanyl and blocked by pretreatment with the antagonist naloxone. The absence of specific MOR-P2-IR in fentanyl-treated MOP-R(-/-) mice gives further confidence in the specificity of this reagent. Nevertheless, immunolocalization studies have inherent limitations, and direct demonstration of MOP-R phosphorylation is still required to establish that the MOR-P2-IR is actually caused by the phosphothreonine370, phosphoserine375 form of the MOP-R.
A discrepancy between MOP-R distribution determined by non-phospho-selective antibodies against MOP-R and MOR-P2-IR labeling may indicate that MOR-P2 detects only a subset of receptors. The phosphorylated receptors would need to be in a functional complex near the appropriate kinase and may not include newly synthesized or recently internalized receptors not positioned near a regulatory kinase. Immunolabeling conditions were optimized to distinguish specific from non-specific background signal, and sensitivity to low levels of receptor was not optimized. Thus, it seems likely that the pattern of MOP-R-IR may not have revealed all of the receptors present. Nevertheless, the expression of phosphorylated mu opioid receptors in striatum is consistent with previous localization studies.
We focused our analysis on the striatum because this region has been shown previously to be rich in MOP-R (Bruggemann et al., 2000; Kaneko et al., 1995; Kitchen et al., 1997; Mansour et al., 1994). With the MOR-P2 antibody, we also saw increased striatal labeling following pSNL. Results with MOR-P2 are consistent with those previously obtained in spinal cord by Narita and colleagues (Narita et al., 2004). Using the (p)MOP(Ser375), they found that pSNL increased (p)MOP(Ser375)-IR in the dorsal horn of spinal cord ipsilateral to the nerve lesion. This same antibody did not detect changes in immunoreactivity in striatum, suggesting that different phosphorylation events may be occurring in this region. The suggestion that phosphorylation sites of the mu opioid receptors in striatum may differ from that in spinal cord requires direct verification.
The lack of MOR-P1 staining of transfected cells or mouse brain sections following mu receptor activation suggests that either the MOR-P1 antipeptide antibody did not recognize the full-length receptor, that the 2nd intracellular loop of the MOP-R was occluded, or that the phosphorylation state of this site did not change following agonist treatment. While the absence of staining is not conclusive, it is consistent with other studies that fail to show receptor phosphorylation at this site (El Kouhen et al., 2001). This result suggests that while this site is clearly involved in the DAMGO-induced desensitization of the MOP-R (Celver et al., 2001), the block in receptor uncoupling caused by the T180A mutation may be due to an effect other than phosphorylation at this site.
Opioid peptides are endogenous neuromodulators that play a major role in the nociceptive pathway by interacting with opioid receptors. Several endogenous opioid peptides, including β-endorphin, enkephalin and dynorphin, are known to interact with mu opioid receptors. We report the novel finding that the increase in MOR-P2-IR in striatum was not evident in pSNL mice lacking the endogenous β-endorphin opioid system. MOR-P2-IR was significantly increased after pSNL in wild type mice, and gene deletion of either proenkephalin or prodynorphin did not block the increased MOR-P2-IR following pSNL. The cellular sources of β-endorphin that may be responsible for MOP-R activation were not defined in this study. Although β-endorphin has been detected in the striata of rats and mice (Gudehthlu et al., 1991; Bhargava et al., 1994, Rattan & Tejwani, 1996; Rogers et al., 1985), the diffusion distance from the sites of release to the sites of action have not been measured.
The results suggest that pSNL caused a sustained release of endogenous β-endorphin that resulted in mu opioid receptor tolerance. The reduction in morphine-CPP may have also been caused by opioid tolerance. Interestingly, the reduction in morphine CPP caused by pSNL indicates that the rewarding effects of opioids were reduced during chronic pain, suggesting that chronic pain may not increase opiate addiction risk. These results are consistent with prior studies showing that painful stimuli can induce endogenous opioid release. Naloxone-sensitive stress induced analgesia was first documented by Akil et al., (1976), and many subsequent studies have described the release of endogenous opioids by pain and stress. A specific role for β-endorphin in endogenous antinociceptive mechanisms was provided by Low and colleagues who showed that the introduction of a stop codon in place of the tyrosine 179 codon in the proopiomelanocortin gene resulted in mutant mice lacking β-endorphin (Rubinstein et al., 1996). These β-endorphin knockout mice showed normal morphine analgesia, but did not show naloxone-sensitive analgesia in response to mild swim stress (Rubinstein et al., 1996). Expression of other peptide products (e.g. ACTH and MSH) from the proopiomelanocortin gene was not affected. Direct demonstration of opioid peptide release in neuropathic pain models was provided by Yaksh and colleagues who showed that ligation of the L5/6 nerve roots evoked the biochemically measurable release of β-endorphin into cerebrospinal fluid in rats (Bach et al., 1995) supporting our conclusion that pSNL also releases β-endorphin.
Sustained activation of the endogenous opioid systems by neuropathic pain has previously been shown to produce opioid receptor tolerance. We have previously found, for example, that pSNL resulted in tolerance to the analgesic effects of a kappa opioid receptor agonist due to sustained release of the opioid peptide dynorphin (Xu et al., 2004; see also Nichols et al., 1997) and a subsequent desensitization of kappa opiate receptors (Xu et al., 2004). Our current results suggest that pSNL, similarly induces sustained β-endorphin release and thereby sustained mu opioid receptor activation, phosphorylation, and opiate receptor desensitization-induced morphine tolerance. Our finding of reduced opiate analgesia in animals following nerve injury is consistent with others demonstrating reduced analgesic potency of both systemic and intrathecal morphine in animal models of neuropathic pain (Ossipov et al., 1995; Idanpaan-Heikkila et al., 1997). Opioid compounds, including morphine, have also been reported to have reduced therapeutic efficacy in humans with neuropathic pain (Arnér and Meyerson 1988; Kupers et al., 1991; Bleeker et al., 2001).
In summary, the results presented in this study extend the in vitro characterization of phosphorylation sites in the mu opioid receptor that mediate desensitization in transfected cells to mechanisms underlying in vivo regulation of MOP-R. Understanding the mechanisms responsible for tolerance to opiate analgesics is relevant to understanding the regulation of sensitivity to endogenous opioids in pain.
Acknowledgements
Prodynorphin knockout mice were originally obtained as a gift from Dr. Hochgeschwender, and the proenkephalin knockout mice were originally obtained from Jackson Labs then bred locally. Joe Novak and Michael Lee performed the mouse genotyping. The work was supported by USPHS grants RO1-DA11676 and PO1-DA15916 (CC) and F32-DA16844 (TLG), and F32-DA20430 (MRB) from the National Institute on Drug Abuse. Dr. Michael Petraschka was supported by a fellowship from the German Research Foundation (DFG).
Abbreviations
- MOP-R
mu opioid peptide receptor
- WT
wild-type
- KO
disrupted-gene, or “knockout” mouse
- MOR-P1
a phosphoselective antibody directed against phosphorylated Thr180 within MOP-R
- MOR-P2
a phosphoselective antibody directed against phosphorylated Thr370 and Ser375 within MOP-R
- (p)-MOP(Ser375)
a phosphoselective antibody directed against a single phosphorylated residue within MOP-R
- GFP
green fluorescent protein
- DAMGO
D-Ala2, methyl-Phe4, glyol5] enkephalin
- GRK
G protein receptor kinase
- pSNL
partial sciatic nerve ligation
- IR
immunoreactivity
- Pomc
proopiomelanocortin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akil H, Mayer DJ, Liebeskind JC. Antagonism of stimulation-produced analgesia by naloxone, a narcotic antagonist. Science. 1976;191:961–962. doi: 10.1126/science.1251210. [DOI] [PubMed] [Google Scholar]
- Arden JR, Segredo V, Wang Z, Lameh J, Sadee W. Phosphorylation and agonist-specific intracellular trafficking of an epitope-tagged mu-opioid receptor expressed in HEK 293 cells. J Neurochem. 1995;65:1636–1645. doi: 10.1046/j.1471-4159.1995.65041636.x. [DOI] [PubMed] [Google Scholar]
- Arnér S, Meyerson BA. Lack of analgesic effect of opioids on neuropathic and idiopathic forms of pain. Pain. 1988;33:11–23. doi: 10.1016/0304-3959(88)90198-4. [DOI] [PubMed] [Google Scholar]
- Bach FW, Chaplan SR, Jang J, Yaksh TL. Cerebrospinal fluid beta-endorphin in models of hyperalgesia in the rat. Regul Pept. 1995;59:79–86. doi: 10.1016/0167-0115(95)00076-n. [DOI] [PubMed] [Google Scholar]
- Bhargava HN, Matwyshyn GA, Rattan AK, Koo KL, Tejwani GA. beta-Endorphin-like immunoreactivity in discrete brain regions, spinal cord, pituitary gland and peripheral tissues of U-50,488H-tolerant and -abstinent rats. J Pharmacol Exp Ther. 1994;268:856–861. [PubMed] [Google Scholar]
- Bleeker CP, Bremer RC, Dongelmans DA, van Dongen RT, Crul BJ. Inefficacy of high-dose transdermal fentanyl in a patient with neuropathic pain, a case report. Eur J Pain. 2001;5:325–329. doi: 10.1053/eujp.2000.0220. [DOI] [PubMed] [Google Scholar]
- Bodnar RJ. Endogenous opioids and feeding behavior: a 30-year historical perspective. Peptides. 2004;25:697–725. doi: 10.1016/j.peptides.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG. Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature. 2000;408:720–723. doi: 10.1038/35047086. [DOI] [PubMed] [Google Scholar]
- Bruggemann I, Schulz S, Wiborny D, Hollt V. Colocalization of the mu-opioid receptor and calcium/calmodulin-dependent kinase II in distinct pain-processing brain regions. Brain Res Mol Brain Res. 2000;85:239–250. doi: 10.1016/s0169-328x(00)00265-5. [DOI] [PubMed] [Google Scholar]
- Celver JP, Lowe J, Kovoor A, Gurevich VV, Chavkin C. Threonine 180 is required for G-protein-coupled receptor kinase 3- and beta-arrestin 2-mediated desensitization of the mu-opioid receptor in Xenopus oocytes. J Biol Chem. 2001;276:4894–4900. doi: 10.1074/jbc.M007437200. [DOI] [PubMed] [Google Scholar]
- Celver J, Xu M, Jin W, Lowe J, Chavkin C. Distinct domains of the mu-opioid receptor control uncoupling and internalization. Mol Pharmacol. 2004;65:528–537. doi: 10.1124/mol.65.3.528. [DOI] [PubMed] [Google Scholar]
- Connor M, Osborne PB, Christie MJ. mu-opioid receptor desensitization: Is morphine different? Br J Pharmacol. 2004;143:685–696. doi: 10.1038/sj.bjp.0705938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng HB, Yu Y, Wang H, Guang W, Wang JB. Agonist-induced mu opioid receptor phosphorylation and functional desensitization in rat thalamus. Brain Res. 2001;898:204–214. doi: 10.1016/s0006-8993(01)02179-5. [DOI] [PubMed] [Google Scholar]
- El Kouhen R, Burd AL, Erickson-Herbrandson LJ, Chang CY, Law PY, Loh HH. Phosphorylation of Ser363, Thr370, and Ser375 residues within the carboxyl tail differentially regulates mu-opioid receptor internalization. J Biol Chem. 2001;276:12774–12780. doi: 10.1074/jbc.M009571200. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Premont RT, Bohn LM, Lefkowitz RJ, Caron MG. Desensitization of G protein-coupled receptors and neuronal functions. Annu Rev Neurosci. 2004;27:107–144. doi: 10.1146/annurev.neuro.27.070203.144206. [DOI] [PubMed] [Google Scholar]
- Gudehithlu KP, Tejwani GA, Bhargava HN. Beta-endorphin and methionine-enkephalin levels in discrete brain regions, spinal cord, pituitary gland and plasma of morphine tolerant-dependent and abstinent rats. Brain Res. 1991;553:284–290. doi: 10.1016/0006-8993(91)90836-k. [DOI] [PubMed] [Google Scholar]
- Idanpaan-Heikkila JJ, Guilbaud G, Kayser V. Prevention of tolerance to the antinociceptive effects of systemic morphine by a selective cholecystokinin-B receptor antagonist in a rat model of peripheral neuropathy. J Pharmacol Exp Ther. 1997;282:1366–1372. [PubMed] [Google Scholar]
- Kaneko T, Minami M, Satoh M, Mizuno N. Immunocytochemical localization of mu-opioid receptor in the rat caudate-putamen. Neurosci Lett. 1995;184:149–152. doi: 10.1016/0304-3940(94)11192-l. [DOI] [PubMed] [Google Scholar]
- Kieffer BL, Gaveriaux-Ruff C. Exploring the opioid system by gene knockout. Prog Neurobiol. 2002;66:285–306. doi: 10.1016/s0301-0082(02)00008-4. [DOI] [PubMed] [Google Scholar]
- Kitchen I, Slowe SJ, Matthes HW, Kieffer B. Quantitative autoradiographic mapping of mu-, delta- and kappa-opioid receptors in knockout mice lacking the mu-opioid receptor gene. Brain Res. 1997;778:73–88. doi: 10.1016/s0006-8993(97)00988-8. [DOI] [PubMed] [Google Scholar]
- Kovoor A, Celver JP, Wu A, Chavkin C. Agonist induced homologous desensitization of mu-opioid receptors mediated by G protein-coupled receptor kinases is dependent on agonist efficacy. Mol Pharmacol. 1998;54:704–711. [PubMed] [Google Scholar]
- Kupers RC, Konings H, Adriaensen H, Gybels JM. Morphine differentially affects the sensory and affective pain ratings in neurogenic and idiopathic forms of pain. Pain. 1991;47:5–12. doi: 10.1016/0304-3959(91)90004-H. [DOI] [PubMed] [Google Scholar]
- Lamey M, Thompson M, Varghese G, Chi H, Sawzdargo M, George SR, O’Dowd BF. Distinct residues in the carboxyl tail mediate agonist-induced desensitization and internalization of the human dopamine D1 receptor. J Biol Chem. 2002;277:9415–9421. doi: 10.1074/jbc.M111811200. [DOI] [PubMed] [Google Scholar]
- Li S, Lee M, Bruchas M, Chan GC, Storm DR, Chavkin C. Reduction of Morphine Tolerance and Reward in Mice Lacking Calmodulin-Stimulated Adenylyl Cyclases. Mol Pharmacol. 2006;70:1742–1749. doi: 10.1124/mol.106.025783. [DOI] [PubMed] [Google Scholar]
- Mansour A, Fox CA, Burke S, Meng F, Thompson RC, Akil H, Watson SJ. Mu, delta, and kappa opioid receptor mRNA expression in the rat CNS: an in situ hybridization study. J Comp Neurol. 1994;350:412–438. doi: 10.1002/cne.903500307. [DOI] [PubMed] [Google Scholar]
- McLaughlin JP, Marton-Popovici M, Chavkin C. Kappa opioid receptor antagonism and prodynorphin gene disruption block stress-induced behavioral responses. J Neurosci. 2003a;23:5674–5683. doi: 10.1523/JNEUROSCI.23-13-05674.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin JP, Xu M, Mackie K, Chavkin C. Phosphorylation of a carboxyl-terminal serine within the kappa-opioid receptor produces desensitization and internalization. J Biol Chem. 2003b;278:34631–34640. doi: 10.1074/jbc.M304022200. [DOI] [PubMed] [Google Scholar]
- McLaughlin JP, Myers LC, Zarek PE, Caron MG, Lefkowitz RJ, Czyzyk TA, Pintar JE, Chavkin C. Prolonged kappa opioid receptor phosphorylation mediated by G-protein receptor kinase underlies sustained analgesic tolerance. J Biol Chem. 2004;279:1810–1818. doi: 10.1074/jbc.M305796200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita M, Kuzumaki N, Suzuki M, Narita M, Oe K, Yamazaki M, Yajima Y, Suzuki T. Increased phosphorylated-mu-opioid receptor immunoreactivity in the mouse spinal cord following sciatic nerve ligation. Neurosci Lett. 2004;354:148–152. doi: 10.1016/j.neulet.2003.09.077. [DOI] [PubMed] [Google Scholar]
- Nichols ML, Bian D, Ossipov MH, Malan TP, Jr, Porreca F. Antiallodynic effects of a CCKB antagonist in rats with nerve ligation injury: role of endogenous enkephalins. Neurosci Lett. 1996;215:161–164. doi: 10.1016/0304-3940(96)12964-5. [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Lopez Y, Nichols ML, Bian D, Porreca F. Inhibition by spinal morphine of the tail-flick response is attenuated in rats with nerve ligation injury. Neurosci Lett. 1995;199:83–86. doi: 10.1016/0304-3940(95)12026-z. [DOI] [PubMed] [Google Scholar]
- Ragnauth A, Schuller A, Morgan M, Chan J, Ogawa S, Pintar J, Bodnar RJ, Pfaff DW. Female preproenkephalin-knockout mice display altered emotional responses. Proc Natl Acad Sci U S A. 2001;98:1958–1963. doi: 10.1073/pnas.041598498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattan AK, Tejwani GA. Effect of chronic treatment with morphine, midazolam, and both together on beta-endorphin levels in the rat. Brain Res Bull. 1996;41:335–441. doi: 10.1016/s0361-9230(96)00022-6. [DOI] [PubMed] [Google Scholar]
- Rogers J, Shoemaker WJ, Morgan DG, Finch CE. Senescent change in tissue weight and immunoreactive beta-endorphin, enkephalin, and vasopressin in eight regions of C57BL/6J mouse brain and pituitary. Neurobiol Aging. 1985;6:1–9. doi: 10.1016/0197-4580(85)90064-8. [DOI] [PubMed] [Google Scholar]
- Rubinstein M, Mogil JS, Japón M, Chan EC, Allen RG, Low MJ. Absence of opioid stress-induced analgesia in mice lacking beta-endorphin by site-directed mutagenesis. Proc Natl Acad Sci U S A. 1996;93:3995–4000. doi: 10.1073/pnas.93.9.3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz S, Schreff M, Koch T, Zimprich A, Gramsch C, Elde R, Hollt V. Immunolocalization of two mu-opioid receptor isoforms (MOR1 and MOR1B) in the rat central nervous system. Neuroscience. 1998;82:613–622. doi: 10.1016/s0306-4522(97)00288-1. [DOI] [PubMed] [Google Scholar]
- Schulz S, Mayer D, Pfeiffer M, Stumm R, Koch T, Hollt V. Morphine induces terminal mu-opioid receptor desensitization by sustained phosphorylation of serine-375. EMBO J. 2004;23:3282–3289. doi: 10.1038/sj.emboj.7600334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seltzer Z, Dubner R, Shir Y. A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain. 1990;43:205–218. doi: 10.1016/0304-3959(90)91074-S. [DOI] [PubMed] [Google Scholar]
- Sharifi N, Diehl N, Yaswen L, Brennan MB, Hochgeschwender U. Generation of dynorphin knockout mice. Brain Res Mol Brain Res. 2001;86:70–75. doi: 10.1016/s0169-328x(00)00264-3. [DOI] [PubMed] [Google Scholar]
- Schuller AG, King MA, Zhang J, Bolan E, Pan YX, Morgan DJ, Chang A, Czick ME, Unterwald EM, Pasternak GW, Pintar JE. Retention of heroin and morphine-6 beta-glucuronide analgesia in a new line of mice lacking exon 1 of MOR-1. Nat Neurosci. 1999;2:151–156. doi: 10.1038/5706. [DOI] [PubMed] [Google Scholar]
- Trujillo KA. Are NMDA receptors involved in opiate-induced neural and behavioral plasticity? A review of preclinical studies. Psychopharmacology (Berl) 2000;151:121–141. doi: 10.1007/s002130000416. [DOI] [PubMed] [Google Scholar]
- Wang H, Moriwaki A, Wang JB, Uhl GR, Pickel VM. Ultrastructural immunocytochemical localization of mu opioid receptors and Leu5-enkephalin in the patch compartment of the rat caudate-putamen nucleus. J Comp Neurol. 1996;375:659–374. doi: 10.1002/(SICI)1096-9861(19961125)375:4<659::AID-CNE7>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Wang HL, Chang WT, Hsu CY, Huang PC, Chow YW, Li AH. Identification of two C-terminal amino acids, Ser(355) and Thr(357), required for short-term homologous desensitization of mu-opioid receptors. Biochem Pharmacol. 2002;64:257–266. doi: 10.1016/s0006-2952(02)01114-0. [DOI] [PubMed] [Google Scholar]
- Xu M, Petraschka M, McLaughlin JP, Westenbroek RE, Caron MG, Lefkowitz RJ, Czyzyk TA, Pintar JE, Terman GW, Chavkin C. Neuropathic pain activates the endogenous kappa opioid system in mouse spinal cord and induces opioid receptor tolerance. J Neurosci. 2004;24:4576–4584. doi: 10.1523/JNEUROSCI.5552-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Zhang L, Yin X, Sun H, Uhl GR, Wang JB. Mu opioid receptor phosphorylation, desensitization, and ligand efficacy. J Biol Chem. 1997;272:28869–28874. doi: 10.1074/jbc.272.46.28869. [DOI] [PubMed] [Google Scholar]
- Zadina JE, Hackler L, Ge LJ, Kastin AJ. A potent and selective endogenous agonist for the mu-opiate receptor. Nature. 1997;386:499–502. doi: 10.1038/386499a0. [DOI] [PubMed] [Google Scholar]
- Zamir N, Palkovits M, Weber E, Mezey E, Brownstein MJ. A dynorphinergic pathway of Leu-enkephalin production in rat substantia nigra. Nature. 1984;307:643–645. doi: 10.1038/307643a0. [DOI] [PubMed] [Google Scholar]
- Zhang J, Ferguson SS, Barak LS, Bodduluri SR, Laporte SA, Law PY, Caron MG. Role for G protein-coupled receptor kinase in agonist-specific regulation of mu-opioid receptor responsiveness. Proc Natl Acad Sci U S A. 1998;95:7157–162. doi: 10.1073/pnas.95.12.7157. [DOI] [PMC free article] [PubMed] [Google Scholar]