

Abstract
Fabry disease is an X-linked inherited metabolic disorder that is caused by a deficiency of α-galactosidase A (α-Gal A). Progressive deposition of neutral glycosphingolipids that have terminal α-linked galactosyl moieties in vascular endothelial cells causes renal failure along with premature myocardial infarctions and strokes in patients with this condition. No specific treatment is available for patients with this disorder at this time. An animal model of this condition would be valuable for exploring therapeutic strategies for patients with Fabry disease. We report here the generation of α-Gal A deficient mice by gene targeting and an analysis of the resulting phenotype. The knockout mice display a complete lack of α-Gal A activity. The mice, however, appeared clinically normal at 10 weeks of age. Ultrastructural analysis revealed concentric lamellar inclusions in the kidneys, and confocal microscopy using a fluorescent-labeled lectin specific for α-d-galactosyl residues showed accumulation of substrate in the kidneys as well as in cultured fibroblasts. Lipid analysis revealed a marked accumulation of ceramidetrihexoside in the liver and the kidneys. These findings indicate the similarity of the pathophysiological process in the mutant mice and in patients with Fabry disease. The deficiency of α-Gal A activity and the accumulation of material containing terminal α-galactosyl residues in cultured embryonic fibroblasts derived from α-Gal A(−/0) mice were corrected by transducing these cells with bicistronic multidrug resistance retroviruses containing human α-Gal A cDNA.
Fabry disease is an X-linked inherited disorder of glycolipid metabolism resulting from deficient activity of the lysosomal enzyme, α-galactosidase A (α-Gal A; EC 3.2.1.22) (1). Neutral glycosphingolipids with terminal α-linked galactosyl moieties—globotriaosylceramide [ceramidetrihexoside (CTH): Galα1-4Galβ1-4Glcβ1-1Cer], and galabiosylceramide (Galα1-4Galβ1-1Cer)—accumulate in the liver, heart, spleen, kidney, vascular endothelial cells, and in plasma of the patients with this disorder. Major disease manifestations include paresthesias in the extremities, corneal dystrophy, angiokeratoma, and occlusive vascular disease of the heart, kidney, and brain, leading to premature mortality (for review, see ref. 2). Human α-Gal A cDNA (3, 4) and the genomic clone (5) have been isolated and mapped to Xq22 (6). Analysis of the α-Gal A gene in Fabry patients revealed heterogeneous molecular lesions such as point mutations and partial gene rearrangements (7).
There is no specific therapy for Fabry disease. Renal transplantation has been performed in Fabry patients with varying outcome (2). Enzyme replacement (8) and somatic gene therapy (9, 10) have potential as effective therapies for lysosomal storage diseases. We have reported efficient expression of the human α-Gal A in NIH 3T3 cells using bicistronic multidrug-resistant (MDR) gene retroviruses (11), and also in vitro correction of enzyme deficits in fibroblasts derived from Fabry patients using recombinant retrovirus (12). However, a suitable animal model for Fabry disease is required to evaluate the ex vivo and in vivo potential of these therapies. Although natural mutant animal models have been reported for other lysosomal disorders such as mucopolysaccharidosis I (13) and VII (14), no animal model has been identified for Fabry disease. An appropriate model of Fabry disease would be invaluable for the study of the molecular pathophysiology of this human genetic disease as well as for the development of effective therapeutic strategies.
To develop a mouse model for Fabry disease by disrupting α-Gal A gene by homologous recombination (15), we isolated and characterized the mouse α-Gal A gene (16). We found that the mouse and human α-Gal A genes are highly similar in size, gene organization, and nucleotide sequence of the coding regions (16). Here we report the generation and characterization of α-Gal A-deficient mice and demonstrate the correction of metabolic deficits in the α-Gal A-deficient fibroblasts by transducing them with bicistronic MDR retroviruses containing human α-Gal A cDNA.
MATERIALS AND METHODS
Gene Targeting and Generation of Mutant Mouse.
The targeting construct (TC) contains 7.5 kb of α-Gal A genomic sequence (16) in the pPNT vector (17) (Fig. 1A). The 1.5 kb of 5′ flanking fragment consisting of KpnI-BamHI fragment, and the 6 kb of EcoRI-EcoRI fragment (16) were subcloned into XhoI site and EcoRI site of pPNT, respectively. Tissue culture of J1 embryonic stem (ES) cells (18) and conditions for electroporation of the targeting construct were performed as described (19). Selection with 350 μg/ml G418 (Geneticin, GIBCO) and 2 μM ganciclovir (Syntex, Palo Alto, CA) was started 24 h after electroporation. After 8–10 days, resistant clones were picked, expanded, and analyzed as described (20). Briefly, genomic DNA from these clones was digested with EcoRI and hybridized with 5′ flanking probe isolated from the 0.5 kb of SacI-KpnI fragment (Fig. 1A). The sizes of the EcoRI fragments from wild-type (WT) and mutated (M) allele were 7.5 kb and 8.5 kb, respectively. The presence of a single integration event was confirmed by hybridization with neomycin phosphotransferase gene-specific probe (neo probe) using the same membrane after stripping the 5′ flanking probe (20, 21). Blastocyst injections were performed as described (22). Mice heterozygous for the α-Gal A gene deletion, α-Gal A(+/−), were generated by mating male chimeras with C57BL/6 females. α-Gal A(−/0) mutant male mice were generated from matings of α-Gal A(+/−) female and C57BL/6 male mice. To determine the genotypes of offspring, tail DNA was digested with EcoRI and hybridized with the 5′ flanking probe as described above. Embryonic fibroblasts from mice of each genotype were established using 14.5-16.5 E embryos as described (22).
Figure 1.

Targeted disruption of the α-Gal A gene in mouse ES cells and generation of α-Gal A(−/0) mice. (A) Schematic representation of the targeting construct (TC) and the wild-type (WT) and mutated (M) alleles of α-Gal A gene. The hatched bars indicate 5′ flanking probe (a) and neomycin resistance gene (neo) probe (b) used to identify targeted clones. Restriction enzyme sites are as follows: K, KpnI; B, BamHI; E, EcoRI; S, SacI. (B) A representative Southern blot analysis of tail DNA from α-Gal A(+/0), α-Gal A(+/−), and α-Gal A(−/0) mice hybridized with probe a. (C) α-Gal A activity in liver homogenates from α-Gal A(+/0) (n = 6) and α-Gal A(−/0) (n = 5) mice. Activities are expressed as nmol/h per mg protein. Bar = SD.
Histology.
For light microscopy, tissues were immersion-fixed for 4 days in 10% phosphate-buffered formalin, and embedded in paraffin, and 10 μm tissue sections were stained with hematoxylin-eosin. For electron microscopy, tissue blocks (<1 mm3) were fixed in 4% formaldehyde/1% glutaraldehyde in phosphate buffer at 4°C for 24 h. Samples were washed three times in 0.1 M cacodylate buffer with 0.2 M sucrose and postfixed in 1% osmium tetroxide buffered with symcollidine (pH 7.2) at 4°C for 1 h. After dehydration in serial alcohol and propylene oxide, samples were infiltrated with and embedded in Epon substitute. Thin sections were prepared and stained with uranyl acetate and lead citrate. For fluorescence staining with Griffonia (Bandeiraea) simplicifolia lectin (GSL1-B4 isolectin, Vector Laboratories), which is specific for α-d-galactosyl residues (23, 24), fibroblasts cultured on coverslips were fixed with 2% paraformaldehyde in PBS (pH 7.4) for 30 min, followed by permeablization with ice-cold methanol at −20°C for 6 min. Frozen kidney tissues were prepared by embedding in OCT compound, sectioned at 8 μm and allowed to air dry. After incubation with fluorescein isothiocyanate (FITC)-conjugated lectin in TBS (50 mM Tris/150 mM NaCl, pH 7.4) for 1 h at 4°C, slides were washed extensively with TBS. For the inhibition studies, sections were incubated with lectin in the presence of 200 mM d-galactose. Stained cells and tissues were examined with a confocal laser-scanning imaging system (Leica TCS 4-D, Leica, Deerfield, IL) using standard imaging analysis software in a fluorescence mode.
Lysosomal Enzyme Assays.
Tissues from each genotype were homogenized with 9 volumes of assay buffer (28 mM citric acid/44 mM dibasic sodium phosphate, pH 4.4) containing 0.5% sodium taurocholate and centrifuged at 14,000 rpm (15,000 × g) for 30 min. Cell pellets from cultured fibroblasts were resuspended in 8–10 volumes of the above buffer and sonicated on ice for 5 sec (twice) and then centrifuged as above. The supernatant solutions were assayed for α-Gal A activity by incubation with 5 mM 4-methylumbelliferyl-α-d-galactopyranoside in the presence of 100 mM N-acetylgalactosamine (25). This material was added as a specific inhibitor of α-galactosidase B activity. β-Hexosaminidase activity was determined (26) as a control for specificity. Protein concentrations were determined by the method of Lowry et al. (27).
Retrovirus Transduction.
Two bicistronic retroviruses, Ha-αGal-IRES-MDR and Ha-MDR-IRES-αGal, carrying human α-Gal A cDNA and MDR1 cDNA (11) were used to transduce fibroblasts from the embryos of the wild-type and α-Gal A mutant mice. After drug selection with the medium containing 25 ng/ml vincristine (Sigma) for 7–9 days, the resistant clones were isolated, expanded, and analyzed for α-Gal A activity. Clones were also subjected to fluorescence-labeled lectin staining as described above.
Analysis of Glycolipids.
Tissues were homogenized in 9 volumes of assay buffer without detergent and extracted with isopropanol and chloroform as described (28). Following partitioning, lipids in the lower phase were dried under a stream of nitrogen and redissolved in a minimum volume of chloroform:methanol (2:1). The mixtures were applied to high-performance thin-layer chromatography (HPTLC) and developed with chloroform:methanol:water (65:25:4). Glycolipids were identified by the characteristic purple color upon spraying with α-naphthol spray reagent (29).
RESULTS
Targeting of the α-Gal A Gene.
The targeting vector was constructed using the genomic clones of the mouse α-Gal A gene isolated from the 129/SvJ mouse genomic library (16). This vector carries a 1-kb deletion spanning part of exon III and intron III. At the site of the deletion, the neomycin resistance gene was inserted as a positive selection marker (Fig. 1A). Negative selection against random integration was conferred by a herpes simplex virus thymidine kinase (tk) gene (30). J1 ES cells were electroporated with the targeting vector DNA, and 109 double-resistant clones were isolated and screened by Southern blot analysis (20, 21). Three targeted clones were identified as positive for gene disruption based on the predicted size of the targeted allele. Additional integration of the targeting vector was excluded by hybridization with the neo probe (20, 21). Selected clones were injected into C57BL/6 blastocysts to generate several overt chimeras. Four male chimeras successfully transmitted the α-Gal A mutation through the germline (Fig. 1B).
To confirm the complete inactivation of the α-Gal A gene, we performed enzyme assays using various tissues and embryonic fibroblasts from α-Gal A mutant mice and wild-type littermate controls. α-Gal A activity was undetectable in liver homogenates from α-Gal A(−/0) mice, whereas it was readily demonstrated in those from wild-type littermates (Fig. 1C). Fibroblasts derived from α-Gal A(-/0) mice showed negligible α-Gal A activity (Table 1). A similar reduction in α-Gal A activity was observed in all other tissues analyzed (data not shown). β-Hexosaminidase activity remained unchanged in the liver and fibroblasts from the mutant mice (data not shown).
Table 1.
α-Gal A activity of fibroblast clones transduced with bicistronic MDR retroviruses containing human α-Gal A cDNA
| Clone | Genotype | Retrovirus | α-Gal A activity,* nmol/h per mg | Lectin (GSL-B4) staining† |
|---|---|---|---|---|
| N1 | N | — | 527.9 | − |
| N2 | N | — | 636.0 | − |
| K1 | KO | — | 1.4 | + |
| K2 | KO | — | 1.7 | + |
| KM-1 | KO | HaMDR | 38.8 | + |
| KM-2 | KO | HaMDR | 4.2 | + |
| KGM-1 | KO | Ha-αGal-IRES-MDR | 611.0 | − |
| KMG-1 | KO | Ha-MDR-IRES-αGal | 524.8 | − |
| KMG-2 | KO | Ha-MDR-IRES-αGal | 274.8 | − |
Primary fibroblasts from mice of either genotypes were established using 14.5–16.5 E embryos as described (22). Cultured fibroblasts were transduced with indicated retroviruses as described (11). N, normal; KO, knockout.
Data are expressed as means of three determinations.
Fluorescence-labeled lectin binding method as shown in Fig. 4.
Phenotypic Analysis of α-Gal A(−/0) Mice.
To obtain hemizygous mice, heterozygous female mice, α-Gal A(+/−), were mated with α-Gal A(+/0) male mice. Hemizygous male mice, α-Gal A(−/0), were born in the expected ratio and exhibited clinically normal phenotype at 10–14 weeks of age. Histopathological analysis of 10-week-old mice revealed no obvious histological lesions in hematoxylin-eosin stained sections of kidney, liver, heart, spleen, lungs, and brain. Electron microscopy revealed lipid inclusions with electron-dense concentric lamellar structures in the lysosomes of renal tubular cells typical of those seen in patients with Fabry disease (Fig. 2). Other cellular components appeared morphologically normal. Using fluorescent-labeled Griffonia (Bandeiraea) simplicifolia lectin, which selectively binds to α-d-galactosyl residues, we analyzed kidneys of 10-week-old mice and embryonic fibroblasts by confocal microscopy. This analysis revealed intense fluorescence in the kidneys of the mutant mice indicating significant accumulations of compounds containing α-d-galactosyl residues (Fig. 3C). The specificity of this lectin binding was confirmed by competitive inhibition with galactose (Fig. 3D). Kidney sections from the littermate α-Gal A(+/0) mice did not exhibit similar intensity of staining (Fig. 3 A and B). Cultured fibroblasts from α-Gal A(−/0) mouse embryos also displayed significant accumulation of α-Gal A substrates, as reflected by the granular staining with intense fluorescence in the cytoplasmic compartment (Fig. 4C). Fibroblasts from the littermate controls did not stain (Fig. 4A). The specificity of the lectin binding in cultured fibroblasts was confirmed by competitive inhibition with galactose (Fig. 4 B and D). Analysis of neutral sphingolipids in liver and kidneys of α-Gal A(−/0) mice showed a striking accumulation of CTH (Fig. 5). Due to heterogeneity in the fatty acid moiety of this class of lipids in mice, two spots are characteristically observed for CTH on HPTLC.
Figure 2.

Electron micrographs of kidneys from a 10-week-old α-Gal A(−/0) mouse. (A) Inclusions are seen in lysosomes of the renal tubular cells (arrows) (×6250). (B) Lysosomes contain inclusions of concentric lamellar structures (×20,000). (C) Higher magnification of inclusions in B (×60,000).
Figure 3.

FITC-labeled Bandeiraea simplicifolia lectin staining of kidneys from α-Gal A(+/0) (A and B) and α-Gal A(−/0) mice (C and D). (A) Kidney sections from a α-Gal A(+/0) mouse stained with FITC-labeled lectin. (B) Section as in A stained in the presence of 200 mM galactose. (C) Kidney section from a α-Gal A (−/0) mouse stained with FITC-labeled lectin. (D) Section as in C stained in the presence of 200 mM galactose. (Bar = 10 μm.)
Figure 4.

FITC-labeled lectin staining of embryonic fibroblasts from α-Gal A(+/0) (A and B) and α-Gal A(−/0) (C–F) mice. Slides were incubated with lectin only (A, C, and E) or lectin and galactose (B, D, and F). (D) After transducing with MDR-based retrovirus (Ha-MDR-IRES-αGal), the KMG-2 clone showed the correction of metabolic deficit as seen by clearance of accumulated material (E). (Bars = 10 μm.)
Figure 5.
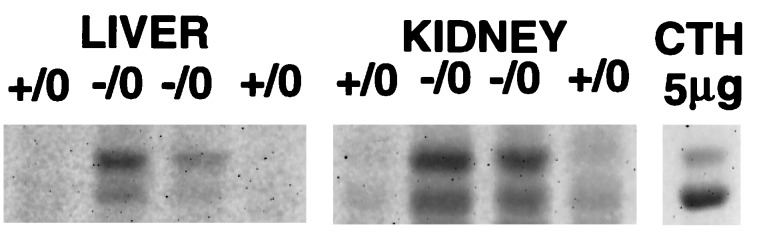
Accumulation of CTH in liver and kidney from α-Gal A(−/0) mice. Liver and kidneys from wild-type (+/0, n = 2) and α-Gal A-deficient (−/0, n = 2) 10-week-old mice were analyzed by HPTLC as described. Purified porcine CTH (5 μg; Matreya, Pleasant Gap, PA) was used as a marker.
Transduction of α-Gal A-Deficient Fibroblasts with Human α-Gal A.
To correct the enzyme deficits in fibroblasts, we used two bicistronic retroviruses carrying both human α-Gal A cDNA and MDR1 cDNA. The retroviruses, Ha-MDR-IRES-αGal and Ha-αGal-IRES-MDR, utilize an internal ribosome entry site (IRES; refs. 31 and 32) and generate a single transcribed mRNA of α-Gal A and MDR1 in different order (11). The α-Gal A-deficient fibroblasts transduced with either retroviruses showed significant corrections in the enzyme deficits compared with mutant fibroblasts transduced with the control virus, HaMDR (Table 1). Transduction of α-Gal A-deficient fibroblasts with Ha-MDR-IRES-αGal retrovirus corrected the deficiency of enzymatic activity and caused the clearance of accumulated material with α-galactosyl residues (Fig. 4E). Similar clearance was also observed in the mutant fibroblasts transduced with Ha-αGal-IRES-MDR retrovirus, but not in the fibroblasts transduced with HaMDR control (Table 1).
DISCUSSION
We have generated α-Gal A-deficient mouse lines by gene targeting. α-Gal A-deficient mice exhibit undetectable α-Gal A enzyme activity in the tissues and cultured fibroblasts, indicating a successful disruption of the α-Gal A gene. α-Gal A-deficient male and female mice appeared to be clinically normal at 10 weeks of age. However, ultrastructural analysis revealed typical lipid inclusions with lamellar structure in the lysosomes of renal tubular cells. Confocal microscopic analysis using Griffonia (Bandeiraea) simplicifolia lectin, which selectively binds to α-galactosyl residues, revealed intense staining in the kidneys and fibroblasts. Significant accumulation of CTH was observed in the liver and kidneys. Correction of the enzyme deficit and clearance of the accumulated residues occurred in the mutant fibroblasts that were transduced with MDR retroviruses containing human α-Gal A cDNA.
Electron microscopic analysis of 10-week-old mouse kidneys revealed inclusions typical of those seen in the human patients. Using specific lectin binding assay, we have demonstrated the accumulation of substrates with α-galactosyl residues in the kidneys as well as in the cultured fibroblasts. Most important, the HPTLC analysis revealed accumulation of CTH in the liver and kidneys. This reflects similarity in the pathophysiological events in α-Gal A-deficient mice and patients with Fabry disease. Although typical lamellar inclusions were seen in the lysosomes, other cellular components such as mitochondria and endoplasmic reticulum were well conserved in morphology. It is anticipated that as the α-Gal A-deficient mice age, CTH will continue to accumulate and that their phenotype will more closely resemble that of patients with Fabry disease in whom CTH has accumulated over decades. Early lethality of mouse models for other lysosomal disorders pose difficulties for their usage in developing human therapy (33). Slow progression of disease process in α-Gal A-deficient mice offers a broad window for trial and evaluation of therapeutic strategies.
Gene therapy holds strong potential for the treatment of metabolic disorders (9, 10). However, this approach is still hampered by difficulties in obtaining sufficiently higher gene expression in target organs (34). In clinical trials, long-term transgene expression in hematopoietic cells has been disappointingly low (35). An alternate strategy for overcoming this problem is to employ the selectable drug-resistance markers that allow enrichment of transduced cells in vivo. MDR1, which encodes P-glycoprotein, has been suggested as such a suitable gene for use in in vivo gene therapy (36). We have demonstrated the correction of enzyme deficits in the mutant cells transduced with the bicistronic MDR retroviruses containing human α-Gal A cDNA. Moreover, this enzyme correction leads to significant clearance of the accumulated substrates. Efficient expression of human α-Gal A using MDR retroviruses indicates the potential for the extension of these studies to ex vivo and in vivo gene therapy approaches in the α-Gal A-deficient mice.
In summary, α-Gal A-deficient mice generated by gene targeting are clinically normal at 10 weeks of age but exhibit accumulation of CTH and inclusions in the kidneys similar to those seen in Fabry patients. The correction of enzymatic and metabolic deficits in the α-Gal A-deficient fibroblasts transduced with MDR retroviruses containing human α-Gal A cDNA prompts detailed studies for in vivo correction of the α-Gal A deficiency. Experimental approaches to induce and treat the clinical phenotype in these mice should be valuable for the development of effective strategies for the treatment of patients with Fabry disease.
Acknowledgments
We thank S. Wahl and M. Young for critical reading of the manuscript, J. Ward for helpful comments on general pathology, C.-G. Huh for help with generating initial chimera, R. Mulligan for the pPNT vector, and Syntex for the gift of ganciclovir.
ABBREVIATIONS
- α-Gal A
α-galactosidase A
- MDR
multidrug resistance
- FITC
fluorescein isothiocyanate
- CTH
ceramidetrihexoside
- HPTLC
high-performance thin-layer chromatography
- ES cells
embryonic stem cells
- IRES
internal ribosome entry site
References
- 1.Brady R O, Gal A E, Bradley R M, Martensson E, Warshaw A L, Laster L. N Engl J Med. 1967;276:1163–1167. doi: 10.1056/NEJM196705252762101. [DOI] [PubMed] [Google Scholar]
- 2.Desnick R J, Ioannou Y A, Eng C M. In: The Metabolic and Molecular Beses of Inherited Disease. Scriver C R, Beaudet A L, Sly W S, Valle D, editors. New York: McGraw–Hill; 1995. pp. 2741–2784. [Google Scholar]
- 3.Bishop D F, Calhoun D H, Bernstein H S, Hantzopoulos P, Quinn M, Desnick R J. Proc Natl Acad Sci USA. 1986;83:4859–4863. doi: 10.1073/pnas.83.13.4859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsuji S, Martin B M, Kaslow D C, Migeon B R, Choudary P V, Stubblefield B K, Mayor J A, Murray G J, Barranger J A, Ginns E I. Eur J Biochem. 1987;165:275–280. doi: 10.1111/j.1432-1033.1987.tb11438.x. [DOI] [PubMed] [Google Scholar]
- 5.Bishop D F, Kornreich R, Desnick R J. Proc Natl Acad Sci USA. 1988;85:3903–3907. doi: 10.1073/pnas.85.11.3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Desnick R J, Astrin K H, Bernstein H S, Potluri R, Bishop D F. Clin Res. 1986;34:717A. (abstr.). [Google Scholar]
- 7.Eng C M, Resnick-Silverman L A, Niehaus D J, Astrin K H, Desnick R J. Am J Hum Genet. 1993;53:1186–1197. [PMC free article] [PubMed] [Google Scholar]
- 8.Brady R O, Tallman J F, Johnson W G, Gal A E, Leahy W R, Quirk J M, Dekaban A S. N Engl J Med. 1973;289:9–14. doi: 10.1056/NEJM197307052890103. [DOI] [PubMed] [Google Scholar]
- 9.Karlsson S. Blood. 1991;78:2481–2492. [PubMed] [Google Scholar]
- 10.Kay M A, Woo L C. Trends Genet. 1994;10:253–257. doi: 10.1016/0168-9525(94)90173-2. [DOI] [PubMed] [Google Scholar]
- 11.Sugimoto Y, Aksentijevich I, Murray G J, Brady R O, Pastan I, Gottesman M M. Hum Gene Ther. 1995;6:905–915. doi: 10.1089/hum.1995.6.7-905. [DOI] [PubMed] [Google Scholar]
- 12.Medin J A, Tudor M, Simovitch R, Quirk J M, Jacobson S, Murray G J, Brady R O. Proc Natl Acad Sci USA. 1996;93:7917–7922. doi: 10.1073/pnas.93.15.7917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wolfe J H, Sands M S, Barker J E, Gwynn B, Rowe L B, Vogler C A, Birkenmeier E H. Nature (London) 1992;360:749–753. doi: 10.1038/360749a0. [DOI] [PubMed] [Google Scholar]
- 14.Shull R M, Munger R J, Spellacy E, Hall C W, Constantopoulos G, Neufeld E F. Am J Pathol. 1982;109:244–248. [PMC free article] [PubMed] [Google Scholar]
- 15.Capecchi M. Science. 1984;244:1288–1292. doi: 10.1126/science.2660260. [DOI] [PubMed] [Google Scholar]
- 16.Ohshima T, Murray G J, Nagle J W, Quirk J M, Kraus M H, Barton N W, Brady R O, Kulkarni A B. Gene. 1995;166:277–280. doi: 10.1016/0378-1119(95)00592-7. [DOI] [PubMed] [Google Scholar]
- 17.Tybulewicz V L J, Crawford C E, Jackson P K, Bronson R T, Mulligan R C. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- 18.Li E, Bestor T H, Jaenisch R. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 19.Love P E, Tremblay M L, Westphal H. Proc Natl Acad Sci USA. 1992;89:9929–9933. doi: 10.1073/pnas.89.20.9929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kulkarni A B, Huh C-G, Becker D, Geiser A, Lyght M, Flanders K C, Roberts A B, Sporn M B, Ward J M, Karlsson S. Proc Natl Acad Sci USA. 1993;90:770–774. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohshima T, Ward J M, Huh C-G, Longenecker G, Veeranna, Pant H C, Brady R O, Martin L J, Kulkarni A B. Proc Natl Acad Sci USA. 1996;93:11173–11178. doi: 10.1073/pnas.93.20.11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robertson E J. In: Teratocarcinoma and Embryonic Stem Cells: A Practical Approach. Robertson E J, editor. Oxford: IRL; 1987. pp. 71–112. [Google Scholar]
- 23.Faraggiana T, Churg J, Grishman E, Strauss L, Prado A, Bishop D F, Schuchman E, Desnick R J. Am J Pathol. 1981;103:247–262. [PMC free article] [PubMed] [Google Scholar]
- 24.Robinson D, Khalfan H A. Biochem Soc Trans. 1984;12:1063. [Google Scholar]
- 25.Mayers J S, Scheere J B, Sifers R N, Donaldson M L. Clin Chim Acta. 1981;112:247–2. doi: 10.1016/0009-8981(81)90384-3. [DOI] [PubMed] [Google Scholar]
- 26.Tallman J F, Brady R O, Quirk J M, Villalba M, Gal A E. J Biol Chem. 1974;249:3489–3499. [PubMed] [Google Scholar]
- 27.Lowry O H, Rosenbrough N J, Farr A L, Randall R J. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 28.Rose H G, Oklander M. J Lipid Res. 1965;6:428–431. [PubMed] [Google Scholar]
- 29.Siakotos A N, Rouser G. J Am Oil Chem Soc. 1965;42:913–916. [Google Scholar]
- 30.McBurney M W, Sutherland L C, Adra C N, Lechair B, Rudnicki M A, Jardine K. Nucleic Acids Res. 1991;19:5755–5761. doi: 10.1093/nar/19.20.5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaufman R J, Davies M V, Wasley L C, Michnick D. Nucleic Acids Res. 1991;19:4485–4490. doi: 10.1093/nar/19.16.4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sugimoto Y, Aksentijevich I, Gottesman M M, Pastan I. Bio/Technology. 1994;12:694–698. doi: 10.1038/nbt0794-694. [DOI] [PubMed] [Google Scholar]
- 33.Tybulewicz V L J, Tremblay M L, LaMarca M E, Willemsen R, Stubblefield B K, Winfield S, Zablocka B, Sidransky E, Martin B M, Huang S P, Mintzer K A, Westphal H, Mulligan R C, Ginns E I. Nature (London) 1992;357:407–410. doi: 10.1038/357407a0. [DOI] [PubMed] [Google Scholar]
- 34.Miller A D. Blood. 1990;76:271–278. [PubMed] [Google Scholar]
- 35.Dunbar C E, Bodine D M, Sorrentino B, Dohahue R, McDonagh K, Cottler-Fox M, O’Shaughnessy J A. Ann NY Acad Sci. 1994;716:216–224. doi: 10.1111/j.1749-6632.1994.tb21714.x. [DOI] [PubMed] [Google Scholar]
- 36.Licht T, Pastan I, Gottesman M M, Herrmann F. Ann Hematol. 1996;72:184–193. doi: 10.1007/s002770050159. [DOI] [PubMed] [Google Scholar]