

Abstract
Genetically obese fatty/fatty rats and obese/obese mice exhibit increased sensitivity to endotoxin hepatotoxicity, quickly developing steatohepatitis after exposure to low doses of lipopolysaccharide (LPS). Among obese animals, females are more sensitive to endotoxin liver injury than males. LPS induction of tumor necrosis factor α (TNFα), the proven affecter of endotoxin liver injury, is no greater in the livers, white adipose tissues, or sera of obese animals than in those of lean controls. Indeed, the lowest serum concentrations of TNF occur in female obese rodents, which exhibit the most endotoxin-induced liver injury. Several cytokines that modulate the biological activity of TNF are regulated abnormally in the livers of obese animals. After exposure to LPS, mRNA of interferon γ, which sensitizes hepatocytes to TNF toxicity, is overexpressed, and mRNA levels of interleukin 10, a TNF inhibitor, are decreased. The phagocytic activity of liver macrophages and the hepatic expression of a gene encoding a macrophage-specific receptor are also decreased in obesity. This new animal model of obesity-associated liver disease demonstrates that hepatic macrophage dysfunction occurs in obesity and suggests that this might promote steatohepatitis by sensitizing hepatocytes to endotoxin.
Keywords: cytokines, leptin, Kupffer cells, lipopolysaccharide
Obesity is a risk factor for a number of diverse diseases, including type II diabetes, hyperlipidemia, cardiovascular disease, osteoarthritis, and various infections (1). Although liver disease is not widely appreciated as a complication of obesity, epidemiologic evidence suggests that obesity increases the risk for cirrhosis. For example, in autopsy series, obesity was identified as the only risk factor for liver disease in 12% of cirrhotic subjects (2). Conversely, cirrhosis is approximately six times more prevalent in obese individuals than in the general population (2–4).
The pathogenesis of liver disease associated with obesity is unknown. Gradual progression from hepatic steatosis to steatohepatitis and, eventually, to cirrhosis is thought to occur (5–7). Hepatic steatosis is common in obese individuals and has been documented in individuals who are as little as 10% above ideal body weight (3). Although at least 40% of morbidly obese patients who undergo abdominal surgery to treat obesity exhibit hepatic steatosis (8, 9), only a fraction of obese individuals with steatosis develop cirrhosis. Disease progression appears to require one or more antecedent episodes of steatohepatitis. The histologic features of obesity-related steatohepatitis resemble those of alcohol-induced steatohepatitis (10, 11). Steatohepatitis is thought to trigger a fibrogenic response that is likely to result in significant deposition of collagen and hepatic nodularity, i.e., cirrhosis. This is supported by data that indicate that fewer than 10% of obese subjects with steatohepatitis exhibit normal liver morphology 5 years later and that almost 40% have become cirrhotic (5, 6). Steatohepatitis is also an important prerequisite for alcohol-related cirrhosis (12, 13), so it is tempting to speculate that, like alcohol, obesity predisposes individuals to steatohepatitis.
Experimental evidence suggests that gut-derived products, including bacterial lipopolysaccharide (LPS) and endotoxin, are involved in the pathogenesis of alcohol-related steatohepatitis (14). Of interest, jejunoileal bypass surgery, a procedure that increases portal endotoxemia, is no longer preferred as a treatment for morbid obesity because it is associated with an extremely high incidence of steatohepatitis and cirrhosis in these individuals (8, 9). Taken together, these observations suggest that obesity might predispose individuals to liver disease by increasing hepatic sensitivity to endotoxin. To test this hypothesis, two different strains of genetically obese rodents (and their lean littermates) were treated with low levels of LPS, after which appearance of liver-specific enzymes in the blood, liver histology, and survival rates were compared. Our results demonstrate increased hepatotoxicity and decreased survival after exposure to LPS in both of the obese strains and suggest two mechanisms—altered Kupffer cell function and increased hepatocyte sensitivity to tumor necrosis factor α (TNFα)—that might mediate obesity-related sensitivity to endotoxin.
EXPERIMENTAL PROCEDURES
Zucker fatty/fatty (fa/fa) rats, obese/obese (ob/ob) mice, and their lean littermates were from The Jackson Laboratory. Escherichia coli LPS was obtained from Sigma. Kits to measure TNFα protein and recombinant rat TNF standard were purchased from BioSource International (Camarillo, CA). Kupffer cell-specific gene (KCR) cDNA was provided by G. W. Hoyle (University of North Carolina, Chapel Hill, NC) (15). Oligonucleotide primers and probes for analysis of specific cytokine gene expression were synthesized to match GenBank sequences unique to TNFα, interleukin (IL) 10, IL-12, transforming growth factor (TGF) β-1, or interferon (IFN) γ.
Obese animals and their lean littermates were injected i.p. with LPS. Three different levels of LPS (500 μg of LPS/kg body weight, 200 μg of LPS/animal, and 100 μg of LPS/animal) were administered. Animals were killed before (n = 6/group) or at various times after (30 min, 1, 6, or 24 h) LPS treatment (n = 6–18/group/time point) to harvest serum, liver, and adipose tissue. All experiments were performed in accordance with National Institutes of Health and Johns Hopkins University guidelines to ensure humane treatment of animal subjects.
Serum concentrations of glucose, triglyceride, cholesterol, alanine aminotransferase (ALT), aspartate aminotransferase (AST), and alkaline phosphatase were measured by a multichannel automated analyzer in the clinical chemistry laboratory at the Johns Hopkins Hospital. Serum concentrations of TNF were assayed in triplicate aliquots of each serum sample by a commercial ELISA using rat recombinant TNFα protein as standard. Liver histology was evaluated after staining sections of paraffin-embedded, formalin-fixed tissues with hematoxylin and eosin.
Total RNA was isolated from liver and white adipose tissue according to the method of Chomczynski and Sacchi (16), and Northern blot analysis was performed as described (17). To ensure data reproducibility, at least four different Northern blot analyses, each containing RNA isolated from a different rat at each time point, were evaluated by densitometry. On each blot, KCR mRNA was normalized to Pu-1 mRNA, a constitutive monocyte gene product, at the same time point, and results from different blots were analyzed by ANOVA.
To assess treatment-related differences in cytokine gene expression, total RNA was reverse transcribed, and specific cytokine primers were used to amplify cytokine cDNAs as described (18). Conditions were established for each set of cytokine primers to ensure that each PCR reaction permitted semiquantitative amplification of a specific cytokine cDNA within a log-linear range (19). Reverse transcriptase PCR products were separated, blotted, and visualized by chemiluminescence after hybridization with cytokine-specific probes (18). Reverse transcriptase PCR assays were repeated with input RNA from at least three different rats from each group at each time point. The resultant Southern blots were evaluated by densitometry, and densitometric data were analyzed ANOVA.
Intravital microscopy was used to evaluate Kupffer cell function in obese and nonobese animals. A small, midline abdominal incision was performed under anesthesia, and the right lobe of the liver was exteriorized and placed on the microscope stage. fa/fa rats (n = 3) and their lean littermates (n = 3) were then injected i.v. with 1-μm fluorescent beads, and the time course and zonal pattern of fluorescent bead uptake by the liver were recorded continuously on videotape for the next hour. Computer-assisted analysis of videotapes was performed as previously described (20).
RESULTS
As expected, male fa/fa rats had significantly greater body weights, serum glucose concentrations, and serum lipid levels before LPS treatment than their lean male littermates (?/fa) (Table 1). Although total liver weights of the fa/fa rats were greater than those of lean controls, liver weight normalized to body weight was comparable in the two groups. Significant hepatic steatosis was not observed in the lean controls; however, liver steatosis (histologic grades 3–4) was evident in all fa/fa rats (compare Fig. 1 A and D). Serum markers of liver injury (AST and ALT) were slightly increased in the fa/fa rats compared with the lean controls before LPS was given (Table 1).
Table 1.
Characteristics of ?/fa and fa/fa rats before exposure to LPS
| ?/fa | fa/fa | P | |
|---|---|---|---|
| Body weight, g | 304 ± 55 | 496 ± 89 | <0.05 |
| Liver weight, g | 9.9 ± 1.6 | 15.2 ± 2.6 | <0.05 |
| Liver weight, % body weight | 3.26 ± 0.3 | 3.17 ± 0.4 | NS |
| Glucose, mg/dl | 185 ± 10 | 259 ± 26 | <0.03 |
| Triglycerides, mg/dl | 104 ±19 | 287 ±17 | <0.01 |
| Cholesterol, mg/dl | 70 ± 26 | 140 ±10 | <0.01 |
| AST, units/liter | 72 ± 2 | 74 ± 4 | NS |
| ALT, units/liter | 56 ± 2 | 86 ±6 | <0.05 |
NS, not significant.
Figure 1.
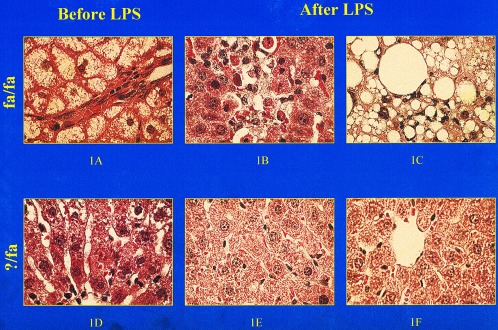
Liver histology before and after treatment with LPS. (A) Micro- and macrovesicular steatosis in a representative fa/fa rat before LPS treatment. Area of (B) hepatocyte apoptosis and (C) steatohepatitis in two distinct, but representative, fa/fa rats 24 h after treatment with LPS (0.5 μg/g). (D) Normal liver histology in a representative ?/fa rat before LPS. (E and F) Normal liver histology in two distinct, but representative, ?/fa rats 24 h after treatment with LPS (0.5 μg/g). (Final magnification ×400.)
Exposure to low levels of LPS led to dramatically different outcomes in male fa/fa and male ?/fa rats. Within 12 h of i.p. injection of LPS (0.5 mg/kg), 21 ± 2% of the fa/fa rats died (P < 0.001 vs. controls). By 24 h after treatment with this amount of LPS, all surviving fa/fa rats had developed steatohepatitis, i.e., histologic evidence of focal hepatocyte necrosis, hepatic inflammation (Fig. 1 B and C), and increased serum AST and ALT (Fig. 2A). In contrast, ?/fa rats treated with LPS exhibited little toxicity. None of these animals died or developed evidence of steatohepatitis (Figs. 1 E and F and 2A).
Figure 2.

Effect of LPS on serum activities of liver-associated enzymes. (A) AST and ALT in fa/fa rats and ?/fa controls 24 h after exposure to 0.5 μg/g LPS. (B) ALT in fa/fa and ?/fa rats 24 h after treatment with 100 or 200 μg of LPS. (C) AST and ALT in ob/ob and ?/ob mice 24 h after treatment with LPS (0.5 μg/g). (D) AST and ALT before and 24 h after treatment with LPS (0.5 μg/g) in female (F) and male (M) fa/fa rats. IU/L, units per liter for A–D.
Although the liver constituted ≈3% of total body weight in both fa/fa and ?/fa rats, fa/fa rats weighed considerably more than their lean littermates and thus received, on average, almost twice as much LPS per animal. Because this might have biased the results, subsequent experiments compared liver enzymes after treatment of obese and lean rats with identical amounts (100 or 200 μg) of LPS. No mortality occurred in either group. However, serum liver enzymes (Fig. 2B) were significantly greater in fa/fa rats than in ?/fa rats after treatment with either level of LPS. Thus, hepatotoxicity was significantly more pronounced in fa/fa rats than in ?/fa rats whether or not the amount of LPS administered was normalized to body weight.
To determine if similar heightened sensitivity to the hepatotoxic effects of endotoxin occurred in other models of genetic obesity, experiments were repeated with male ob/ob mice and their lean male (?/ob) littermates. As shown in Fig. 2C, serum AST and ALT increased substantially after LPS (0.5 mg/kg) in the ob/ob group to a level 10 times greater than in ?/ob mice. These increases in circulating liver enzymes correlated with histologic evidence of progression from steatosis to steatohepatitis in the ob/ob animals.
Alcohol-related steatohepatitis tends to be more severe in women than in men (13) and women might be more susceptible to obesity-associated steatohepatitis (2, 5, 6, 10, 11, 21, 22), so LPS sensitivity was compared in male and female fa/fa rats. Liver weight, body weight, and serum AST and ALT were similar in male and female fa/fa rats before LPS injection. However, 24 h after exposure to LPS, serum AST and ALT were significantly greater in female fa/fa rats than in male fa/fa rats (Fig. 2D).
Endotoxin is known to provoke the release of proinflammatory cytokines, including TNFα. TNF has been identified as a proximal mediator of endotoxin-induced injury to many tissues, including the liver (23, 24). Endotoxin delivered i.p. is normally cleared from portal blood by hepatic macrophages (Kupffer cells). This minimizes endotoxin stimulation of LPS-sensitive cells that reside in extrahepatic sites and attenuates systemic production of TNF but apparently does not preclude local TNF production by resident Kupffer cells because the latter are presumed to play an important role in the pathogenesis of endotoxin-mediated liver injury (25). To determine if fa/fa rats were more susceptible to LPS-induced liver injury because of increased TNF production in response to endotoxin, the expression of TNF was compared in fa/fa and ?/fa rats after LPS injection.
As shown in Fig. 3A, neither fa/fa nor ?/fa rats expressed appreciable TNF mRNA in the liver before treatment with LPS. However, within 1 h after LPS injection in both groups, the level of hepatic TNF transcripts increased significantly. Surprisingly, the induction of hepatic TNF mRNA by LPS was no greater in obese rats than in their lean littermates. Circulating levels of TNF protein also increased significantly after LPS injection in both fa/fa and lean rats, reaching a maximum 1 h after LPS administration in both groups. Consistent with the liver RNA results, TNF protein concentrations were not higher in male or female fa/fa rats than in the respective lean controls at any of the time points evaluated (i.e., at 30 min or 1, 6, or 24 h) after LPS treatment. Indeed, as shown in Fig. 3B, the lowest TNF concentrations occurred in female fa/fa rats, which exhibited the greatest hepatotoxicity after LPS. The ELISA method used might not distinguish biologically active TNF from that which is inactivated by circulating soluble TNF receptors, so the assay was repeated several times after diluting samples to dissociate potential inhibitors. However, dilution did not increase immunoreactive, “free” TNF in any serum sample, suggesting that circulating inhibitors did not account for the observed differences in TNF concentrations among the groups.
Figure 3.

TNFα expression in fa/fa and ?/fa rats pre- and 1 h posttreatment with LPS (0.5 μg/g). (A) Total liver RNA was isolated from three distinct rats/group at each time point, reverse transcribed, and amplified with TNF-specific primers. PCR products were separated on nondenaturing agarose gels, transferred to nylon membranes, and visualized by hybridization with TNF-specific probes. (B) Serum TNF concentrations were evaluated by ELISA using rat recombinant TNFα as standard. In both males and females, TNF expression was greater in serum and liver at 1 h after LPS than at any other time point (0, 0.5, 6, or 24 h) evaluated.
These results suggested that obese animals are more sensitive to the toxic effects of TNF. LPS is known to stimulate the release from macrophages of several cytokines that modulate the biological activity of TNF. These include IL-12, IFNγ, IL-10, and TGFβ (23, 26). Semiquantitative reverse transcriptase PCR analysis of liver RNA from obese and lean animals showed that obesity is associated with alterations in the profile of cytokines that regulate TNF, both before and after treatment with LPS. In the fa/fa rats, LPS induction of hepatic IFNγ mRNA is enhanced, TGFβ-1 mRNA expression is unaffected, induction of IL-10 mRNA is delayed, and expression of IL-12 mRNA is generally decreased (Fig. 4). Kupffer cells are important sources of TNF and TNF-related cytokines (23, 26, 27), so these results suggest that Kupffer cell function might be abnormal in the obese animals. Consistent with this theory, Northern blot analysis of liver RNA indicated that hepatic expression of KCR is decreased in the fa/fa rats compared with controls (Fig. 5A). Lean animals express high levels of KCR mRNA before and for the 1st h after treatment with LPS. Then, expression of this gene is down-regulated. In contrast, obese rats express few KCR mRNA at all time points evaluated.
Figure 4.

Hepatic expression of TNF regulatory cytokines pre- and posttreatment with LPS (0.5 μg/g). Liver RNA was isolated from three distinct rats/group either before (pre) or 1 and 24 h after LPS treatment and subjected to reverse transcriptase PCR analysis with sets of primers specific for IL-12, IFNγ, IL-10, or TGFβ-1 under semiquantitative conditions.
Figure 5.

Comparison of Kupffer cell function in fa/fa and ?/fa rats. (A) Total liver RNA was isolated from two distinct rats/group either before (pre) or 1 and 24 h after LPS (0.5 μg/g), separated by electrophoresis on denaturing polyacrylamide gels (20 μg RNA/lane), transferred to nylon membranes, and cohybridized with [32P]-labeled cDNAs for KCR and a constitutively expressed monocyte transcription factor (Pu-1). (B and C) Rats were injected intraportally with fluorescent beads, and intravital microscopy was used to evaluate (B) the distribution of beads in acinar zones 1 (periportal), 2 (midacinus), and 3 (pericentral) 30 minutes after injection and (C) the cumulative hepatic uptake of the beads over time. Results shown are representative of replicate experiments.
Obesity also is associated with abnormal Kupffer cell function. When rats were injected i.v. with fluorescent beads to evaluate Kupffer cell phagocytic function, striking differences in phagocytic activity were observed. In lean animals, most of the beads were taken up in zone 1 of the hepatic acinus, and the few remaining beads were phagocytosed in acinar zones 2 and 3. In contrast, phagocytosis in zones 1 and 2 was decreased in obese animals. This resulted in striking differences in the net hepatic uptake of fluorescent beads in the obese animals and their lean controls. As shown in Fig. 5 B and C, hepatic clearance of intraportally delivered fluorescent beads, a function of Kupffer cell phagocytic activity, is less than 50% of normal in fa/fa rats.
Kupffer cells are responsible for phagocytosing gut-derived endotoxin to prevent its escape into the systemic circulation, so these results suggest that obese animals might have baseline endotoxemia and, hence, evidence of TNF production in extrahepatic sites. To explore this possibility, the level of TNF mRNA in white adipose tissue was evaluated. Although TNF is not detected in white adipose tissue of lean animals until they are treated with LPS, obese mice and rats reproducibly express TNF in adipose tissue even before treatment with exogenous endotoxin. Consistent with these results, baseline serum concentrations of TNF protein were higher in male fa/fa rats (37.3 ± 6.6 pg/ml) than male ?/fa rats (16 ± 5.3 pg/ml) (P < 0.02).
DISCUSSION
Obesity has long been suspected of increasing the risk for cirrhosis (3). However, the mechanisms responsible for this are poorly understood. Observations that obesity is strongly associated with steatohepatitis, a pattern of liver injury that often progresses to cirrhosis in alcoholic individuals, provided a clue that the pathogenesis of liver disease related to obesity might be similar to that of alcoholic liver injury. Endotoxin or LPS is thought to play a role in liver injury induced by alcohol and several other hepatotoxins (14, 25, 28). The present study indicates that obesity increases susceptibility to endotoxin-mediated liver injury and, thus, identifies the common mechanism for both alcohol- and obesity-related liver diseases. In addition, this work identifies a new animal model that will be useful for elucidating the pathogenesis of steatohepatitis in general.
Considerable evidence suggests that the hepatotoxic effects of LPS are mediated through cytokines (especially TNF) (29, 30). Hepatocytes are normally resistant to the cytotoxic effects of TNF. However, two mechanisms, priming and sensitization, are known to increase hepatic susceptibility to TNF injury (23, 24). Priming occurs when TNF-producing cells, especially tissue macrophages, are conditioned, or primed, to release massive amounts of TNF in response to stimuli (e.g., small amounts of LPS) that normally cause only minimal TNF release. Priming is thought to mediate liver injury due to Corynebacterium parvum (31, 32), lead acetate (33), polyhalogenated hydrocarbons (34), ischemia reperfusion (35), cadmium, and choline deficiency (36) but is unlikely to explain obesity-related LPS hepatotoxicity because we found no evidence that LPS treatment induced greater expression of TNF in obese rodents than in lean controls. A second important mechanism of TNF-induced hepatic injury involves hepatocyte sensitization to normally innocuous amounts of TNF. Sensitization to the toxic actions of TNF might occur via several discrete mechanisms that culminate in defective adaptation to oxidant stress (37–39). The classic liver injury model of sensitization is that of galactosamine-induced hepatotoxicity, in which galactosamine is known to sensitize hepatocytes to TNF toxicity by depleting cellular uridine stores (40). Sensitization also appears to contribute to liver injury caused by other hepatoxins, including carbon tetrachloride, acetaminophen, and alcohol (41–43). Given that obese and lean male rodents experienced similar induction of TNF but dramatically different degrees of liver injury after treatment with LPS, it is reasonable to conclude that obesity promotes liver injury by a mechanism that involves sensitization of hepatocytes to TNF toxicity. Female obese rats developed significantly more liver injury after LPS than their male counterparts, despite less induction of TNF, so sensitization also appears to be an important component of gender-related susceptibility to LPS hepatotoxicity.
The molecular basis for obesity-related sensitization remains uncertain but might reflect, at least in part, relative overexpression of IFNγ after LPS exposure. IFNγ is known to augment TNF toxicity in isolated mouse hepatocytes (38) and is increased in many experimental models of TNF-related liver injury (32). IFNγ is produced by Kupffer cells, which are also sources of other cytokines (e.g., IL-12, IL-10, and TGFβ) that regulate tissue responses to TNF (44). The present study demonstrates that basal and/or LPS induction of IL-12 and IL-10 are also abnormal in obese rats, suggesting that there might be relatively global dysfunction of Kupffer cells in obesity. Consistent with this theory, this work has identified decreased Kupffer cell phagocytic function in obese rats. The latter is likely to have important implications for extrahepatic cytokine production because Kupffer cells provide the predominant barrier for egress of endogenous endotoxin from the portal to the systemic circulations (45). Indeed, our work has reproduced previously published evidence that basal adipose expression of TNF mRNA (46) and circulating concentrations of TNF protein (47) are relatively increased in obesity. Others have suggested that increased peripheral production of TNF might play a role in obesity-associated diabetes by interfering with insulin-dependent signaling in muscle and adipose tissue (48). Our results suggest that dysfunction of hepatic macrophages might be primarily responsible for abnormal, obesity-associated, cytokine production in peripheral tissues by permitting chronic, low grade endotoxemia.
The present work investigated animals in which obesity resulted from defective leptin-dependent signal transduction. Thus, it is uncertain if our results can be extrapolated to other situations that cause obesity and/or hepatic steatosis. Pertinent in this regard are data that suggest that choline deficiency, another cause of hepatic steatosis, promotes endotoxin liver injury by priming rather than by sensitization (36). Interesting to note, other investigators have reported differential sensitivity to endotoxin-induced glucose intolerance in leptin-defective rodents and in rats with streptozotocin-induced diabetes (48). Our observation that LPS caused relatively greater release of liver enzymes in ob/ob mice, which completely lack leptin (49–51), than in fa/fa rats, which overexpress leptin but have defective leptin receptors (52), is provocative because it suggests that competent leptin signaling might be requisite for normal tissue sensitivity to endotoxin-inducible cytokines. Given the importance of these cytokines in mediating insulin responsivity, lipid metabolism, bone turnover, vascular biology, inflammatory reactions to infections, and liver injury (23, 24), leptin modulation of cytokine actions could have important clinical consequences.
Acknowledgments
This work was supported by grants to A.M.D from the National Institute of Alcohol Abuse and Alcoholism, National Institutes of Health.
ABBREVIATIONS
- LPS
lipopolysaccharide
- TNFα
tumor necrosis factor α
- IL
interleukin
- TGF
transforming growth factor
- KCR
Kupffer cell-specific gene
- fa/fa
fatty/fatty
- ob/ob
obese/obese
- IFNγ
interferon γ
- AST
aspartate aminotransferase
- ALT
alanine aminotransferase
References
- 1.National Institutes of Health Consensus Development Panel on the Health Implications of Obesity. Ann Intern Med. 1985;103:147–152. [PubMed] [Google Scholar]
- 2.Wanless I R, Lentz J S. Hepatology. 1990;12:1106–1110. doi: 10.1002/hep.1840120505. [DOI] [PubMed] [Google Scholar]
- 3.Leevy C M. Medicine. 1962;41:249–258. doi: 10.1097/00005792-196209000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Silverman J F, O’Brien K F, Long S, Lettett N, Khazanie P G, Pories W J, Norris T, Caro J F. Am J Gastroenterol. 1990;85:1349–1355. [PubMed] [Google Scholar]
- 5.Lee R G. Hum Pathol. 1989;20:594–600. doi: 10.1016/0046-8177(89)90249-9. [DOI] [PubMed] [Google Scholar]
- 6.Powell E E, Cooksley W G E, Hanson R, Searle J, Halliday J W, Powell L W. Hepatology. 1990;11:74–80. doi: 10.1002/hep.1840110114. [DOI] [PubMed] [Google Scholar]
- 7.Bacon B, Fraavash M J, Janney C G, Neuschwander-Tetri B A. Gastroenterology. 1994;107:1103–1109. doi: 10.1016/0016-5085(94)90235-6. [DOI] [PubMed] [Google Scholar]
- 8.Haines N W, Baker A L, Boyer J L, Glagov S, Schneir H, Jaspan J, Ferguson D J. Hepatology. 1981;1:161–165. doi: 10.1002/hep.1840010212. [DOI] [PubMed] [Google Scholar]
- 9.Vyberg M, Ravn V, Andersen B. Liver. 1987;7:271–276. doi: 10.1111/j.1600-0676.1987.tb00355.x. [DOI] [PubMed] [Google Scholar]
- 10.Ludwig J, Viggiano T R, McGill D B, Ott B J. Mayo Clin Proc. 1980;55:434–438. [PubMed] [Google Scholar]
- 11.Diehl A M, Goodman Z, Ishak K G. Gastroenterology. 1988;95:1056–1061. [PubMed] [Google Scholar]
- 12.Galambos J. Gastroenterology. 1972;63:1026–1032. [PubMed] [Google Scholar]
- 13.Pares A, Caballeria J, Brugera M. J Hepatol. 1986;2:33–38. doi: 10.1016/s0168-8278(86)80006-x. [DOI] [PubMed] [Google Scholar]
- 14.McClain C J, Hill D B, Schmidt J, Diehl A M. Semin Liver Dis. 1993;13:170–182. doi: 10.1055/s-2007-1007347. [DOI] [PubMed] [Google Scholar]
- 15.Hoyle G W, Hill R L. J Biol Chem. 1991;266:1850–1857. [PubMed] [Google Scholar]
- 16.Chomczynski P, Sacchi N. Anal Biochem. 1987;162:156–161. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 17.Yin M, Yang S Q, Lin H Z, Lane M D, Chatterjee S, Diehl A M. J Biol Chem. 1996;271:17974–17978. doi: 10.1074/jbc.271.30.17974. [DOI] [PubMed] [Google Scholar]
- 18.Rai R, Yang S Q, McClain C, Karp C L, Klein A S, Diehl A M. Am J Physiol. 1996;270:G909–G918. doi: 10.1152/ajpgi.1996.270.6.G909. [DOI] [PubMed] [Google Scholar]
- 19.Karp C L, Wysocka M, Wahl L M, Ahearn J M, Cuomo P J, Trinchieri G, Griffen D E. Science. 1996;273:228–231. doi: 10.1126/science.273.5272.228. [DOI] [PubMed] [Google Scholar]
- 20.Rai R M, Zhang J X, Clemens M G, Diehl A M. Shock. 1996;6:243–247. doi: 10.1097/00024382-199610000-00003. [DOI] [PubMed] [Google Scholar]
- 21.Adler M, Schaffner F. Am J Med. 1979;67:811–817. doi: 10.1016/0002-9343(79)90740-x. [DOI] [PubMed] [Google Scholar]
- 22.Torosis J D, Barwick K W, Miller J D. Hepatology. 1985;9:237–242. [Google Scholar]
- 23.Tracey K J, Cerami A. Annu Rev Cell Biol. 1993;9:317–343. doi: 10.1146/annurev.cb.09.110193.001533. [DOI] [PubMed] [Google Scholar]
- 24.Vassalli P. Annu Rev Immunol. 1992;10:411–452. doi: 10.1146/annurev.iy.10.040192.002211. [DOI] [PubMed] [Google Scholar]
- 25.Nolan J P. Hepatology. 1989;8:232–236. [Google Scholar]
- 26.Pauli U. Crit Rev Eukaryotic Gene Expression. 1994;4:323–344. doi: 10.1615/critreveukargeneexpr.v4.i2-3.20. [DOI] [PubMed] [Google Scholar]
- 27.Grewe M, Gaussling R, Gyuflo K, Hoffmann R, Decker K. J Hepatol. 1994;20:811–818. doi: 10.1016/s0168-8278(05)80154-0. [DOI] [PubMed] [Google Scholar]
- 28.Tiegs G. J Hepatol. 1994;21:890–903. doi: 10.1016/s0168-8278(94)80256-4. [DOI] [PubMed] [Google Scholar]
- 29.Mohler K, Sleath P, Fitzner J. Nature (London) 1994;370:218–220. doi: 10.1038/370218a0. [DOI] [PubMed] [Google Scholar]
- 30.Ashkenazi A, Marsters A, Capon D J, Chamow S M, Figari I S, Pennica D, Goeddel D V, Palladino M A, Smith D H. Proc Natl Acad Sci USA. 1991;88:10535–10539. doi: 10.1073/pnas.88.23.10535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mizoguchi Y, Tsutsui H, Miyajima K. Hepatology. 1987;7:1184–1188. doi: 10.1002/hep.1840070603. [DOI] [PubMed] [Google Scholar]
- 32.Smith S, A, C, Bankowski J, Kenworthy-Bott L, Terminelli C. J Leukocyte Biol. 1993;54:23–29. doi: 10.1002/jlb.54.1.23. [DOI] [PubMed] [Google Scholar]
- 33.Honchel R, Marsano L, Cohen D, Shedlofsky S, McClain C. J Lab Clin Med. 1991;117:202–208. [PubMed] [Google Scholar]
- 34.Clark G C, Taylor M J, Tritscher A M, Lucier G W. Toxicol Appl Pharmacol. 1991;111:422–431. doi: 10.1016/0041-008x(91)90247-c. [DOI] [PubMed] [Google Scholar]
- 35.Colletti L M, Remick D G, Burtch G D, Kunkel S L, Strieter R M, Campbell D A J. J Clin Invest. 1990;85:1936–1943. doi: 10.1172/JCI114656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hill, D., Shedlofsky, S., McClain, C., Diehl, A. M. & Tsukamoto, H. Cytokines and Liver Disease (Dekker, New York).
- 37.Leist M, Gantner F, Bohlinger I, Germann P G, Tiegs G, Wendel A. J Immunol. 1994;153:1778–1788. [PubMed] [Google Scholar]
- 38.Adamson G, Billings R. Arch Biochem Biophys. 1992;294:223–229. doi: 10.1016/0003-9861(92)90161-o. [DOI] [PubMed] [Google Scholar]
- 39.Schulze-Osthoff K, Beyaert R, Vandevoorde V, Haegeman G, Fiers W. EMBO J. 1993;12:3095–3104. doi: 10.1002/j.1460-2075.1993.tb05978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tiegs G, Wolter M, Wendel A. Biochem Pharmacol. 1989;38:627–631. doi: 10.1016/0006-2952(89)90208-6. [DOI] [PubMed] [Google Scholar]
- 41.Hansen J, Cherwitz D L, Allen J I. Hepatology. 1994;20:461–474. [PubMed] [Google Scholar]
- 42.Czaja M J, Xu J, Alt E. Gastroenterology. 1995;108:1849–1854. doi: 10.1016/0016-5085(95)90149-3. [DOI] [PubMed] [Google Scholar]
- 43.Blazka M, Wilmer J L, Holladay S D, Wilson R E, Luster M I. Toxicol Appl Pharmacol. 1995;133:43–52. doi: 10.1006/taap.1995.1125. [DOI] [PubMed] [Google Scholar]
- 44.Decker K. Eur J Biochem. 1990;192:245–261. doi: 10.1111/j.1432-1033.1990.tb19222.x. [DOI] [PubMed] [Google Scholar]
- 45.Fox E S, Broitman S A, Thomas P. Lab Invest. 1990;63:733–736. [PubMed] [Google Scholar]
- 46.Kern P A, Saghizadeh M, Ong J M, Bosch R J, Deem R, Simsolo R B. J Clin Invest. 1995;95:2111–2119. doi: 10.1172/JCI117899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamakawa T, Tanaka S-I, Yamakawa T, Kiuchi Y, Isoda F, Kawamoto S, Okuda K, Sekihara H. Immunol Immunopathol. 1995;75:51–56. doi: 10.1006/clin.1995.1052. [DOI] [PubMed] [Google Scholar]
- 48.Hotamisligil G S, Peraldi P, Budavari A, Ellis R, White M F, Speigelman B M. Science. 1996;271:665–668. doi: 10.1126/science.271.5249.665. [DOI] [PubMed] [Google Scholar]
- 49.Campfield L A, Smith F J, Buisez Y, Devos R, Burn P. Science. 1995;269:546–549. doi: 10.1126/science.7624778. [DOI] [PubMed] [Google Scholar]
- 50.Halaas J L, Gajiwala K S, Maffei M, Cohen S L, Chait B T, Rabinowitz D, Lallone R L, Burley S K, Friedman J M. Science. 1995;269:544–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- 51.Pelleymounter M A, Cullen M F, Baker M B, Hecht R, Winters D, Boone T, Collins F. Science. 1995;269:540–543. doi: 10.1126/science.7624776. [DOI] [PubMed] [Google Scholar]
- 52.Ghilardi N, Ziegler S, Wiestner A, Wiestner A, Stoffel R, Heim M H, Skoda R C. Proc Natl Acad Sci USA. 1996;93:6231–6235. doi: 10.1073/pnas.93.13.6231. [DOI] [PMC free article] [PubMed] [Google Scholar]