

Abstract
The Calvin Cycle is the primary conduit for the fixation of carbon dioxide into the biosphere; ribulose 1,5-bisphosphate carboxylase/oxygenase (RuBisCO) catalyzes the rate-limiting fixation step. Our goal is to direct the evolution of RuBisCO variants with improved kinetic and biophysical properties. The Calvin Cycle was partially reconstructed in Escherichia coli; the engineered strain requires the Synechococcus PCC6301 RuBisCO for growth in minimal media supplemented with a pentose. We randomly mutated the gene encoding the large subunit of RuBisCO (rbcL), co-expressed the resulting library with the small subunit (rbcS) and the Synechococcus PCC7492 phosphoribulokinase (prkA), and selected hypermorphic variants. The RuBisCO variants that evolved during three rounds of random mutagenesis and selection were over-expressed, and exhibited 5-fold improvement in specific activity relative to the wild-type enzyme. These results demonstrate a new strategy for the artificial selection of RuBisCO and other non-native metabolic enzymes.
Keywords: carbon dioxide fixation; horizontal transfer; in vitro evolution; metabolic engineering; ribulose 1,5-bisphosphate carboxylase oxygenase
Introduction
Ribulose 1,5-bisphosphate carboxylase/oxygenase (RuBisCO) catalyzes the conversion of carbon dioxide into biomass. It is the world’s most abundant enzyme, and virtually every carbon-containing molecule in the human body is a downstream product. Paradoxically, RuBisCO is perhaps the slowest metabolic enzyme in the contemporary biosphere (kcat ~3/s), and its competing oxygenase activity (photorespiration) ultimately leads to the release of 20–50% of the incorporated CO2 (Mann, 1999). It is not clear why such an essential enzyme is so slow and non-specific. The catalytic inefficiency of RuBisCO is thought to limit the efficiency of photosynthesis, so many have tried to improve this enzyme by rational design (John Andrews and Whitney, 2003).
Site-directed mutagenesis studies, which tend to focus upon residues in or near the active-site (Hartman and Harpel, 1994; Watson and Tabita, 1997; Spreitzer and Salvucci, 2002; Parry et al., 2003), have mostly produced catalytically compromised RuBisCO variants. Protein engineers have thus far failed to improve the efficiency (kcat/KM) or specificity (Ω = Vmax/KM of carboxylation divided by Vmax/KM of oxygenation) of cyanobacterial RuBisCO homologs to match those of naturally occurring plant homologs (Hartman and Harpel, 1994; Parry et al., 2003). The active-site loop six, for example, has been most intensively studied, but structure (and phylogeny) based rational design of this region has at best imparted modest (5–13%) improvements in specificity. Improvement of RuBisCO efficiency might therefore require the simultaneous mutation of multiple residues away from the active-site (Watson and Tabita, 1997; John Andrews and Whitney, 2003).
Random mutagenesis and genetic selection offer an alternative to site-directed mutagenesis. Previous workers have developed microbial selection systems for RuBisCO function based upon complementation of auxotrophic mutants of Chlamydomonas reinhardtii (Hong and Spreitzer, 1997; Du et al., 2000; Spreitzer et al., 2001), Synechococcus PCC6308 (Pierce et al., 1989; Amichay et al., 1993) or Rhodobacter capsulatus (Smith and Tabita, 2003). In the most recent published study, ~3300 random mutants of the Synechococcus PCC6301 RuBisCO were selected for their ability to accelerate the growth of R.capsulatus auxotrophs at high CO2 concentrations. The surviving clones were then screened for their ability to grow at lower CO2 concentrations. One clone (F342V) exhibited a ~2-fold improvement in the Michaelis constant of its complex with ribulose-1,5-bisphosphate (kRuBP) (Smith and Tabita, 2003). This result suggests that iterated cycles of random mutagenesis and selection (directed evolution) could impart further improvement.
The outcome of any directed evolution experiment is largely determined by the throughput, precision, sensitivity, and dynamic range of the screen or selection. Escherichia coli offer two advantages over normally photosynthetic microbes for directed evolution. First, the transformation efficiency of E.coli (>109 transformants per microgram of plasmid) is at least three orders of magnitude greater than those of photosynthetic microbes (Smith and Tabita, 2003). An E.coli-based genetic selection for RuBisCO function would enable the sampling of millions of different RuBisCO mutants in a single day. Second, the rate of carbon dioxide fixation in phototrophic cells is determined not by RuBisCO alone, but by its interactions with an array of biochemical accessories including activase, chaperonins (Gatenby and Ellis, 1990), modifying enzymes (Houtz and Portis, 2003), carboxysomes (Shively et al., 1998) and/or the C4 photosynthetic pathway. These accessories potentiate the activity of RuBisCO, but they might also obviate further evolution of its intrinsic catalytic parameters (Sage, 1999). E.coli does not ordinarily fix carbon dioxide, so RuBisCO variants evolved within this organism would be intrinsically more active in the absence of specialized accessories. E.coli naturally expresses 8 of the 11 Calvin Cycle enzymes and is amenable to metabolic engineering. It, therefore, seemed possible to engineer a strain that is dependent upon RuBisCO function for growth. Here, we describe the partial reconstruction of the Calvin Cycle in E.coli and its application in the directed evolution of RuBisCO hypermorphs.
Materials and methods
Materials
Synechococcus PCC6301 and PCC7942 cells were from the American Type Culture Collection (ATCC #27144 and #33912, respectively, Manassus, VA). E.coli strain K-12 was from the E.coli Genetic Stock Center (New Haven, CT). DH5Δlac(DE3) was previously described (Matsumura et al., 1999). The plant light bulb was from General Electric (cat #35753). pBR322, pACYC184 and pMAL-2px were from New England Biolabs (Beverly, MA); pET28a+ and pET30a+ were from Novagen (Madison, WI); pBAD myc his A was from Invitrogen (Carlsbad, CA); pBS KS+ was from Stratagene (La Jolla, CA). Primers were custom synthesized by IDT (Coralville, IA). B-PER was from Pierce (St Louis, MO). The chelating (iminodiacetic acid) sepharose fast flow resin and NaH14CO3 (0.1 mCi/mmole) were from Amersham Biosciences (GE Healthcare, Piscataway, NJ). The Bradford protein assay kit was from Bio-Rad (Hercules, CA); nitrocellulose membranes were from Schleicher and Schuell (Keene, NH). The rabbit anti-Synechococcus PCC6301 antibodies were the generous gift of Robert Tabita (Ohio State University). The IRD800-conjugated goat–anti-rabbit secondary antibody, and the IRD700-conjugated molecular weight standards were from LICOR (Lincoln, NE). BigDye 3.1 sequencing kits were from Applied Biosystems (Foster City, CA). DNA modifying enzymes, including restriction endonucleases, Taq and Vent polymerase and dNTPs were from New England Biolabs. 5-Bromo-4-chloro-3-indolyl-beta-galactopyranoside (X-gal) was from Gold Biotechnology (St Louis, MO). All other chemicals, including L-arabinose, antibiotics (chloramphenicol, ampicillin, kanamycin) and bacterial media [M9 (Sambrook and Russell, 2001) and BG-11 medium (Allen, 1968)], ribulose-1,5-bisphosphate (RuBP) were from Sigma-Aldrich (St Louis, MO).
Cloning and expression of Calvin Cycle genes
Synechococcus PCC6301 and PCC7942 cells were propagated by agitating them at room temperature in BG-11 medium (Allen, 1968) under ~2000 lux from a plant light bulb for 9 days. The cells (OD600 = 0.2) were harvested by centrifugation, washed in water and re-suspended in 1/1000 volumes of water. The prkA gene was amplified in a PCR from whole Synechococcus PCC7942 cells using Vent polymerase and primers 5′SprkA (5′-GGGCCCGAGCTCGTCACTGTCTC-GAGGGATCCTAT-3′) and 3′SprkA (5′-CCGGGAAGCTTA-GAGTCACACCGCTGAAAACAAGT-3′). The prkA gene was blunt-end cloned into EcoRV-cut pBS KS+, and sequenced to confirm its wild-type identity. It was subsequently subcloned into pET28a+ using restriction enzymes EcoRI and HindIII; this step fused the gene DNA sequence encoding an N-terminal hexa-histidine tag. The his6-prkA gene was subcloned into pBAD myc his A using restriction enzymes NcoI and HindIII. The araC-PBAD–his6-prkA expression cassette was subcloned into pACYC184 using SphI and HindIII. The final construct, PBAD-his6-prkA-pACYC184, contains the arabinose-inducible PBAD promoter, a fusion gene encoding a hexa-histidine-tagged phosphoribulokinase in the pACYC184 (low copy number, chloramphenicol-resistant) vector.
The rbcL/rbcS genes, which are adjacent in the Synechococcus PCC6301 chromosome, were PCR amplified together from whole cells using Vent polymerase and primers 5′SrbcL (5′-GGGCCCCATATGCCCAAGACGCAATCTGCCGCAG-G-3′) and 3′SrbcS (5′-CCCGGGGAGCTCAGGCTTTAG-TAGCGGCCGGGACG-3′). The rbcL/rbcS PCR product was cloned into two different vectors (pMAL-2px and pET30a+) in parallel using restriction enzymes NdeI and SacI (underlined), and sequenced to confirm its wild-type identity. (i) The rbcLS-pMAL-2px (trc promoter, ampicillin resistant, moderate copy number) was primarily employed for the high-throughput selections and secondary screens. (ii) The rbcLS-pet30a+ enabled more stable heterologous expression of the native (untagged) RuBisCO protein for in vivo characterization experiments; we later inserted DNA sequence encoding a C-terminal hexa-histidine tag by site-directed insertion mutagenesis (see ‘Purification of RuBisCO protein’ below).
DNA sequencing
The wild-type and variant rbcL, rbcS and prkA genes were sequenced using the ABI BigDye 3.1 protocol, and primers: rev10 (5′-AATTGTGAGCGGATAACAATTTCACACAG-3′), rbcL361 (5′-CGTGTTTGGCTTCAAAGCTATCCGT TC-3′), rbcL1196rev (5′-ACTGGAGAACGGAGTCATCACC GAGG-3′), rbcL1006 (5′-GATTCACTTCCGTGTCTTGGC-CAAGTG-3′), rbcL1113 (5′-TGCTGCCGGTTGCTTCCGGT GGTAT-3′), univ50 (5′-CCCAGTCACGACGTTGTAAAAC-GACG-3′), prkA614 (5′-CGACGAGCGCGTGCGTGAACT GCT-3′), prkA938 (5′-CCAGTGGACGCCCTGCGGTCG-TAA-3′). The sequencing reaction products were separated and analyzed with an ABI Prism 3100 capillary sequencer at the Emory/Georgia Tech FAME Center.
Random mutagenesis
The rbcL alleles were mutated in standard PCRs, which are slightly mutagenic (Cadwell and Joyce, 1992), before each round of evolution. In each reaction, 500 nM primers rev + 10 (5′-AATTGTGAGCGGATAACAATTTCACACAG-3′) and 3′SrbcS (5′-CCCGGGGAGCTCAGGCTTTAGTAGCGGCC-GGGACG-3′) were reacted with 50 ng rbcLS-pMAL-2px template in 60 mM Tris–HCl, pH 8.5, 15 mm (NH4)SO4, 2 mM MgCl2, 0.2 mM dNTP’s and 2.5 U Taq polymerase in a total volume of 50 μl; the temperatures were cycled 25 times at 94°C for 30 s, 65°C for 30 s and 72°C for 2 min. The PCR product was purified, digested with restriction enzymes NdeI and EcoRI, and subcloned into expression vector pMAL-2px. The resulting expression library was electroporated (Dower et al., 1988) into E.coli K-12 carrying PBAD-6his-prkA-pACYC184.
Genetic selection
The double transformants (rbcLS-pMAL-2px + PBAD-6his-prkA-pACYC184/K-12) were re-suspended in 90% M9/10% LB medium and propagated under the following selective conditions: M9 minimal agar plates including 0.2% glucose (Sambrook and Russell, 2001) supplemented with ampicillin, chloramphenicol, 0.2% L-arabinose, 0.5 mm isopropyl-beta-D-thiogalactopyranoside (IPTG) and exogenous CO2. We note that cells expressing phosphoribulokinase and RuBisCO can form colonies at ambient CO2 concentrations when streaked at very high cell densities (Figure 1b). We believe that cells cannibalize nutrients from their dead siblings under those conditions. Repetition of this experiment with serially diluted liquid cultures (rather than re-streaked colonies) showed that cells expressing prkA+ but not rbcL/rbcS produce micro-colonies under selective conditions only when plated at very high densities (>100 000 cells/plate). These micro-colonies were easy to distinguish from larger self-sustaining colonies, but we plated transformed cells at moderate densities (<10 000 colonies/agar plate) to reduce the risk of selecting false positives.
Fig. 1.
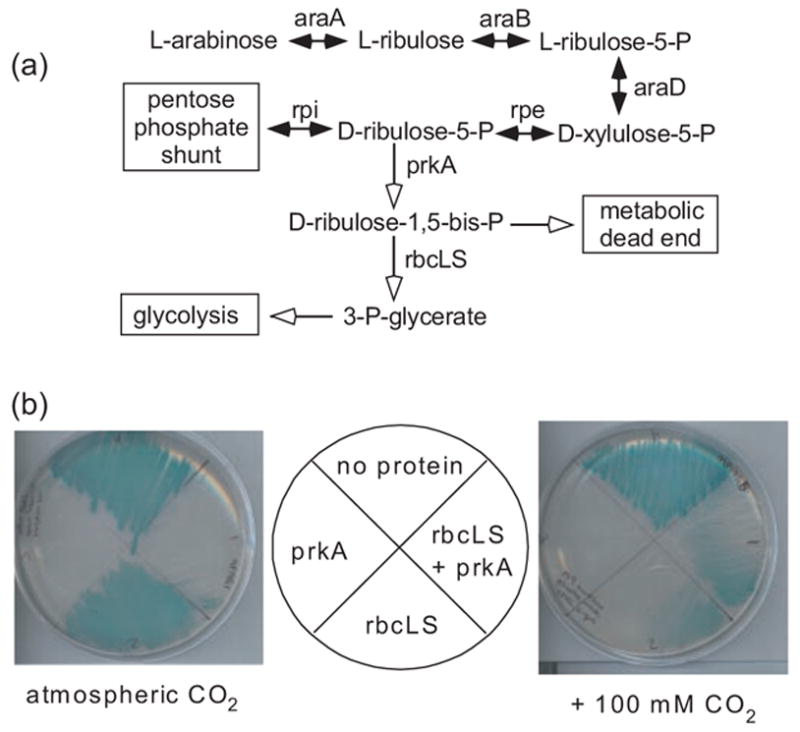
Metabolic engineering of E.coli. (a) The normal assimilation of L-arabinose into the pentose phosphate shunt is shown (black arrows). The heterologous expression of the Synechococcus PCC7942 phosphoribulokinase (prkA, white arrow) diverts the carbon flux and causes growth arrest. Co-expression of the Synechococcus PCC6301 RuBisCO genes (rbcL/rbcS, white arrow) rescues E.coli from prkA-mediated growth arrest. (b) Wild-type E.coli (strain K-12) were co-transformed with two plasmids: (i) A phosphoribulokinase (prkA) expression vector or pacyc184, PLUS (ii) A RuBisCO (rbcLS) expression vector or pBR322. Each of the four doubly transformed strains were streaked onto quadrants of an M9 minimal media agar plate supplemented with 100 μg/ml ampicillin, 34 μg/ml chloramphenicol, 0.2% L-arabinose, 0.5 mM IPTG and 0.008% X-gal. The plates were placed in quart-sized ziploc bags containing 0 or 5 g of dry ice, which quickly sublimated into gaseous CO2, and incubated for 24 h at 37°C.
Cell extract 14CO2 incorporation assay
The carboxylation activities of RuBisCO variants in E.coli cell extracts were assayed in two different instances. First, the clones isolated in the primary genetic screens (rbcLS-pMAL-2px + PBAD-6his-prkA-pACYC184/K-12) were compared in order to identify the best ones. Second, the rbcLS-pmal-2px vector was somewhat genetically unstable, so the wild-type and five best rbcL variants (1.15, 2.22, 2.24, 2.29, 3.54) were subcloned into rbcLS-pET30a+ E.coli; DH5Δlac(DE3) cells were transformed with the resulting constructs for a more precise and quantitative version of the assay. The assays are identical in design, with the modest technical differences noted below.
The transformed cells were propagated to mid-log phase at 37°C in 10 ml LB supplemented with ampicillin and chloramphenicol (rbcLS-pMAL-2px + PBAD-6his-prkA-pACYC184/K-12) or kanamycin [rbcLS-pET30a+/DH5Δlac(-lac(DE3)]. Expression of the rbcLS genes were induced by the addition of 0.5 mm IPTG, and the cultures were agitated for 37°C for 3 h (rbcLS-pMAL-2px) or 25°C overnight (rbcLS-pET30a+). The cells were harvested by centrifugation, re-suspended in TEM buffer (25 mm Tris, pH 8.0, 2 mM EDTA, 5 mM beta-mercaptoethanol), centrifuged again, re-suspended in 300 μl of RuBisCO buffer (50 mM Bicine, pH 8.0, 20 mM MgCl2, 5 mM beta-mercaptoethanol) and lysed by sonication. The RuBisCO protein was activated by incubation with excess NaH14CO3 (0.1 mCi/mmole stock: 5 mM final for rbcLS-pMAL-2px, 45 mM final for rbcLS-pET30a+) for 10 min at 25°C. Ribulose 1,5-bisphosphate (RuBP) was added to a final concentration of 0.6 mM (rbcLS-pMAL-2px) or 2.67 mM (rbcLS-pET30a+) in each extract, and the reaction was quenched after 1 min by the addition of HCl to 250 mm (rbcLS-pMAL-2px) or 1.375 N (rbcLS-pET30a+). The acidified reactions were incubated at 100°C in a fume hood to evaporate the unincorporated 14CO2; the acid-stable radioactivity was measured in a Beckman LS6500 scintillation counter.
Western blot
The wild-type and evolved rbcLS-pET30a+/DH5Δlac (DE3) cultures described above (cell extract assays) were harvested by centrifugation in a microfuge (3000 r.p.m. for 10 min). The supernatant was removed and the cell pellet was re-suspended in SDS/beta-mercaptoethanol buffer (Sambrook and Russell, 2001). The proteins in these extracts were electrophoretically separated in a 6% polyacrylamide gel and transferred to a nitrocellulose membrane. The membrane was blocked with 2% non-fat milk, reacted with polyclonal rabbit anti-RuBisCO antibodies and IRD800-labeled secondary antibodies. The immobilized antibody–antigen complexes were visualized and quantified using a LI-COR Odyssey Infrared imager.
Purification of RuBisCO protein
The evolved rbcL alleles were subcloned into expression vector rbcLS-pET30a+ (described above) using restriction enzymes NdeI and SacI. DNA sequence encoding a hexa-histidine tag was subsequently fused to the 3′ end of the rbcL by PCR amplification of the entire plasmid (Geddie and Matsumura, 2004) using 5′ phosphorylated primers 3his-3′rbcL (5′-CAT-CATCATTGACTATCGCTGGGGGAGTGAGCGT-3′) and 3his-3′rbcL rev (5′-ATGATGATGGAGCTTGTCCATCGT TTCGAATTCGAAC-3′) where the underlined letters encode histidines. E.coli DH5Δlac (DE3) were transformed with the rbcL-6his-rbcS-pET30a+ expression vectors and spread on LB–kanamycin agar plates. The transformants were propagated in 350 ml LB-kanamycin at 37°C until mid-log phase. Protein expression was induced by the addition of 0.5 mM IPTG, and the cultures were agitated overnight at 25°C. The cells were harvested by centrifugation, washed and lysed by re-suspension in B-PER and 1 mg chicken lysozyme. The RuBisCO-6his proteins were purified by immobilized metal affinity chromatography as recommended by the manufacturer (Novagen). The protein was dialyzed overnight in 25 mM Tris–HCl pH 8.0, 5 mM beta-mercaptoethanol, 2 mM EDTA. The protein was stored at 4°C; enzyme activity generally diminished over the course of ~2 weeks under these conditions, so all of the RuBisCO variants were purified in parallel and assayed the next day.
14CO2 incorporation assay of purified RuBisCO
The purified wild-type and mutant RuBisCO proteins (5 μg) were separately activated by incubation with 24 mM NaH14CO3 (0.1 mCi/mmole) for 10 min at 25°C. We added RuBP to a final concentration of 2 mM and quenched the reaction after 1 min by the addition of HCl to 420 mM. The acidified reactions were incubated at 100°C to evaporate the unincorporated 14CO2, and the acid-stable radioactivity was measured in a Beckman LS6500 scintillation counter.
Results
E.coli-based genetic selection for RuBisCO function
We developed a simple but sensitive genetic selection for RuBisCO function in E.coli as follows. The Synechococcus PCC7942 phosphoribulokinase and the Synechococcus PCC6301 RuBisCO were employed because they are active when expressed in E.coli, and have been purified and characterized (Newman and Gutteridge, 1993; Kobayashi et al., 2003). This cyanobacterial RuBisCO is less CO2-specific (Ω = 40) than plant homologs (Ω = 80–100) (Spreitzer and Salvucci, 2002), but has become a model for mutagenesis studies (Watson and Tabita, 1997) because the plant RuBisCOs do not properly assemble when expressed in bacteria (Spreitzer and Salvucci, 2002). The Synechococcus PCC7942 phosphoribulokinase gene (prkA) was cloned into the L-arabinose-inducible expression vector PBAD-6his-prkA-pACYC184. The genes encoding the Synechococcus PCC6301 RuBisCO large and small subunits (rbcL and rbcS, respectively) were cloned into the IPTG-inducible moderate copy number rbcLS-pMAL-2px; this construct is very similar in design to the most efficient expression system reported to date (Kleman et al., 1996).
Wild-type E.coli cells normally convert L-arabinose into D-ribulose-5-phosphate (Lin, 1987) (Figure 1a). Heterologously expressed phosphoribulokinase should irreversibly convert this intermediate into D-ribulose-1,5-bisphosphate (RuBP). E.coli does not normally make or use this compound, so the phosphoribulokinase would effectively divert the carbon flux from the pentose phosphate shunt into a metabolic dead end. As predicted, E.coli K-12 cells expressing prkA alone do not grow in M9 minimal medium supplemented with L-arabinose (Figure 1b). We believe that these cells are unable to produce aromatic amino acids and pyridoxal phosphate (Zhao and Winkler, 1994). The addition of casamino acids to the minimal medium rescues prkA-expressing cells from growth arrest (data not shown), suggesting that the accumulation of RuBP is not intrinsically or irreversibly toxic. The expression of rbcL/rbcS alone appears somewhat toxic to E.coli, particularly in the presence of CO2 (Figure 1b). We tentatively postulate that RuBisCO reacts with some essential native metabolite at a low but physiologically detectable rate.
E.coli cells expressing prkA, rbcL and rbcS form colonies when streaked on M9 minimal medium agar plates supplemented with L-arabinose, but only when incubated with elevated concentrations of gaseous carbon dioxide (Figure 1b). In other words, phosphoribulokinase, RuBisCO and carbon dioxide are each deleterious (to different extents) on their own, but growth occurs when all three are present. The Synechococcus PCC6301 RuBisCO is relatively slow at low CO2 concentrations [kC ~ 180 μm (Smith and Tabita, 2003)]; the CO2-dependence of colony formation suggests that RuBisCO activity, rather than some other indirect mechanism, rescues the cells from induced growth arrest. The modest growth rates of cells rescued by the wild-type Synechococcus RuBisCO (Figure 1b) leaves room for improvement by directed evolution or other means.
Directed evolution of RuBisCO hypermorphs
Random mutations were introduced into the wild-type rbcL gene by PCR amplification; the cumulative error rate of Taq polymerase in a PCR is ~10−3/nt (Cadwell and Joyce, 1992), which for the ~1.4 kb rbcL gene corresponds to 1.4 mutations/allele. The sequence of the RuBisCO large subunit is unusually well-conserved in nature (Dean et al., 2002), so a conservative mutagenesis strategy seemed appropriate for this initial evolution experiment. The resulting library was subcloned into the rbcLS-pMAL-2px expression vector, downstream of the trc promoter. Wild-type K-12 E.coli were co-transformed with the library and the PBAD-6his-prkA-pACYC184 expression vector, and 200 000 transformants were propagated under selective conditions (Table I). The fastest-growing colonies were picked, re-streaked and propagated under selective conditions (secondary screen). Many of the selected clones were not reproducibly faster-growing; we now believe that the parental rbcLS-pMAL-2px expression vector was genetically unstable (Keasling, 1999) and somewhat imprecise within these screening conditions.
Table I.
Directed evolution of Synechococcus PCC6301 rbcL
| Round | [CO2]a | 1° Selectionb | 2° Screenb | 3° Screenb | Hypermorphs |
|---|---|---|---|---|---|
| 1 | 9–54 mM | 2 × 105 | 70 | 26 | 1 |
| 2 | 9–54 mM | 7 × 104 | 44 | 12 | 6 |
| 3 | 9–27 mM | 3 × 105 | 70 | 12 | 5 |
Each round of selection was carried out under different concentrations of carbon dioxide. The actual intracellular CO2 concentration is not known, but assumed to reflect the weight of dry ice sublimated in 4 l ziploc bags. All of these values are >100× atmospheric partial pressures.
Estimated number of clones evaluated at each step.
The most promising clones were evaluated in the following tertiary screen. The transformed E.coli were grown to mid-log phase in liquid LB-ampicillin/chloramphenicol, induced for 3 h with 0.5 mM IPTG, harvested by centrifugation, washed, re-suspended in assay buffer and lysed by sonication. The extract was reacted with ribulose 1,5-bisphosphate (RuBP) and NaH14CO3 for 90 s and quenched with HCl. The unincorporated 14CO2 was evaporated, and the amount of acid-stable product (presumably radiolabeled 3-phosphoglycerate) was measured in a scintillation counter. Those clones that exhibited increased 14CO2 incorporation (hypermorphs, data not shown) were the source of template for the next round of random mutagenesis and selection.
Characterization
We isolated 12 rbcL hypermorphs in three rounds of random mutagenesis and selection (Table I). DNA sequence analysis revealed five unique sequences (Table II). The wild-type and evolved (1.15, 2.22, 2.24, 2.29, 3.54) rbcLS alleles were subcloned into pET30a+. The resulting rbcLS-pET30a+ plasmids exhibited more reproducible behavior than the corresponding rbcLS-pMAL-2px plasmids in our cell extract assays (Figure 2). E.coli DH5Δlac(DE3) were separately transformed with each of the rbcLS-pET30a+ expression vectors, and the transformed cells were propagated in parallel to mid-log phase, induced overnight with 0.5 mM IPTG at room temperature, harvested by centrifugation and lysed. The cell extracts were analyzed by western blot analysis using RuBisCO specific antibodies (Figure 2a). All of the evolved rbcLS genes produced 2- to 5-fold more RuBisCO protein than the wild-type, even when all were driven by the ‘new’ T7 promoter (Figure 2b). Side-by-side 14CO2 incorporation assays of the same rbcLS-pet30a+-derived cell extracts showed a reproducible 2- to 7-fold increase in RuBisCO activity among the artificially evolved clones (Figure 2c).
Table II.
Sequences of selected rbcL alleles
| g22t (Ala8Ser) | c489t | t702c | t776c (Met259Thr) | c783t | t799c | t1025c (Phe342Ser) | |
|---|---|---|---|---|---|---|---|
| 1.15 | X | X | X | ||||
| 2.19 | X | X | X | ||||
| 2.22 | X | X | X | X | |||
| 2.24 | X | X | X | X | |||
| 2.29 | X | X | X | X | |||
| 2.30 | X | X | X | ||||
| 2.44 | X | X | X | ||||
| 3.18 | X | X | X | X | |||
| 3.22 | X | X | X | X | |||
| 3.40 | X | X | X | X | |||
| 3.54 | X | X | X | X | X | ||
| 3.57 | X | X | X |
Fig. 2.
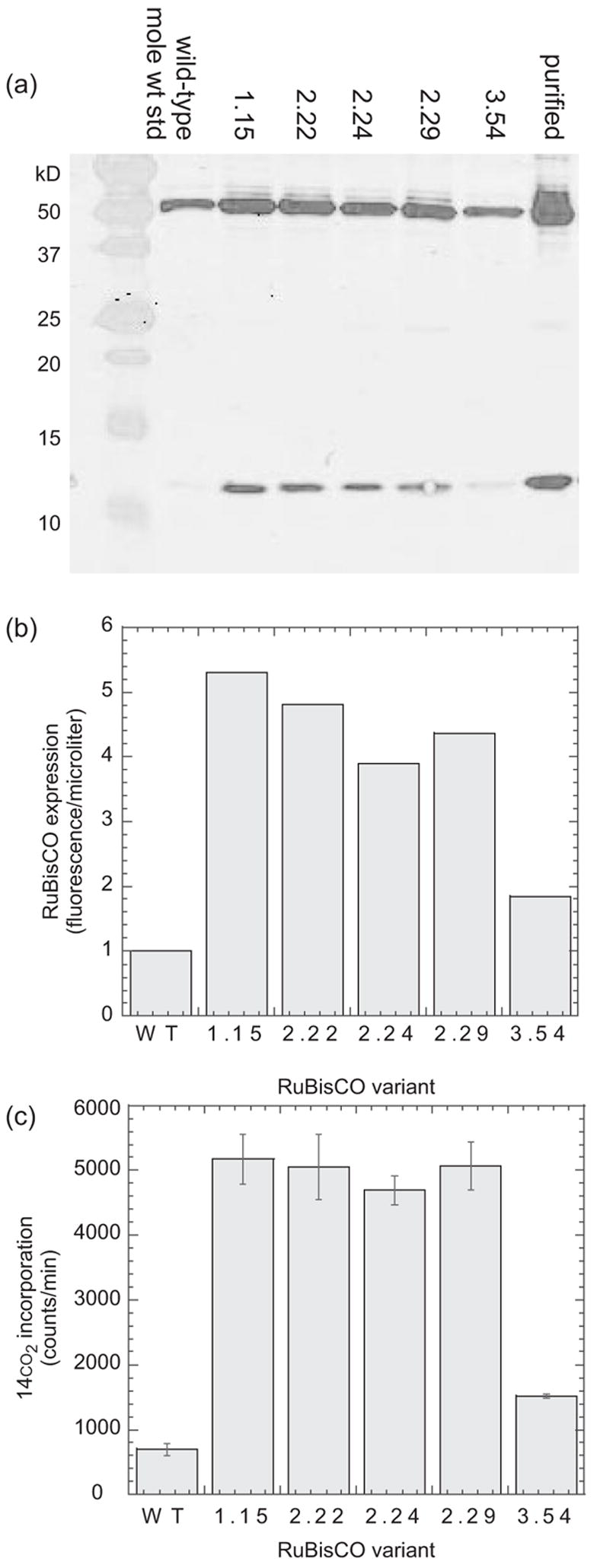
rbcL mutations increase protein expression and enzyme activity in vivo. (a) The wild-type and evolved Synechococcus rbcL variants were co-expressed in E.coli with the wild-type rbcS. The cells were lysed and the proteins in equal volumes of the soluble fraction (0.125 μl in lanes 2–7, 36 ng pure RuBisCO in lane 8) were separated in a 6% polyacrylamide gel, transferred to a nitrocellulose membrane and probed with rabbit anti-RuBisCO polyclonal antibodies and secondary antibodies conjugated to the fluorescent IRD800 dye. (b) The fluorescence of the bands in the western blot were quantified using the Odyssey software. (c) The cell extracts were reacted with ribulose 1,5-bisphosphate and NaH14CO3. The reactions were quenched with hydrochloric acid, and the unincorporated 14CO2 was evaporated in a fume hood. The acid-stable radioactivity (incorporated 3-phosphoglycerate and downstream products) was measured in a scintillation counter.
We sought to characterize the purified RuBisCO variants. Each rbcL allele was fused to a DNA sequence encoding a C-terminal hexa-histidine tag. We expressed the wild-type and artificially evolved RuBisCO proteins, and purified them by immobilized metal affinity chromatography. SDS-polyacrylamide gel analysis (Figure 3a) showed highly purified bands corresponding to the large and small subunits (52 kDa for large subunit, 13 kDa for small subunit), even though the latter was not his-tagged. Both bands cross-reacted with the polyclonal anti-RuBisCO antibody (Figure 2a, lane 8). Bradford protein assays showed that the purification yields of the evolved proteins were ~4- to 15-fold better than that of the wild-type (Figure 3b). These differences were reproducible from batch to batch, and basically consistent with the pattern of over-expression observed in the western blot (Figure 2a and b). Equal quantities (5 μg) of purified wild-type and evolved RuBisCO protein were characterized in side-by-side 14CO2 incorporation assays. All three of the variants (2.24 = A8S/M259T, 2.29 = M259T, 3.54 = M259T/F342S) exhibited ~5-fold greater specific activity than the wild-type (Figure 3c). Several interpretations of this result are discussed below.
Fig. 3.
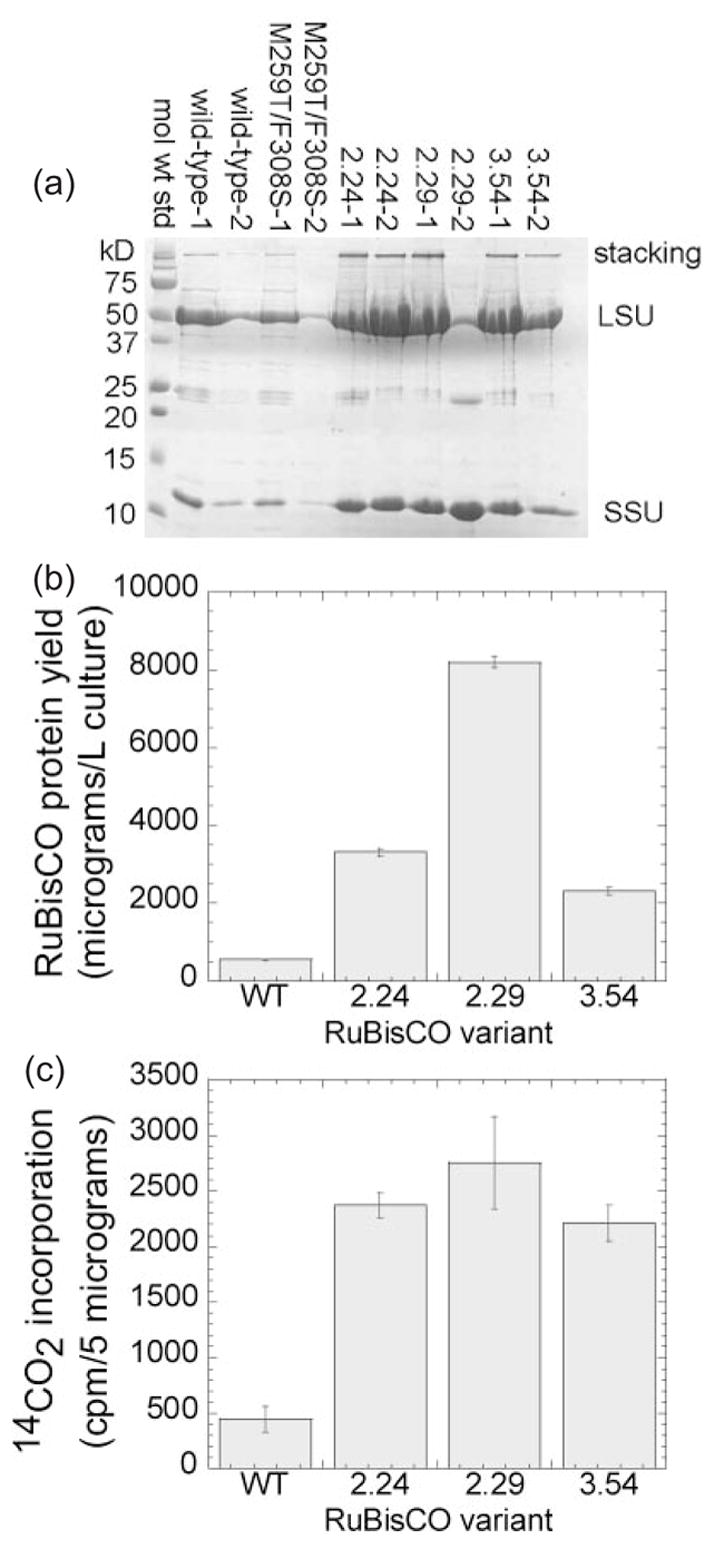
Mutations in rbcL improve purification yields of RuBisCO protein and specific activity in vitro (a) Hexa-histidine-tagged versions of the wild-type and three evolved Synechococcus rbcL variants were co-expressed in E.coli with the wild-type rbcS. The cells were lysed, and the RuBisCO was purified by IMAC and analyzed by SDS–PAGE as described in the text. (b) The purification yields of the C-terminal his-tagged RuBisCO clones (a) were determined by the Bradford protein assay, using BSA as a standard. (c) Equal quantities (5 μg) of each purified RuBisCO were reacted with ribulose 1,5-bisphosphate and NaH14CO3; the reactions were quenched with HCl; the unincorporated 14CO2 was evaporated and the rate of 3-phosphoglycerate product formation was measured in a scintillation counter.
Discussion
The evolved RuBisCO variants exhibited improvement over the wild-type, in terms of over-expression of untagged protein (Figure 2a), increased activity of the untagged enzymes within cell extracts (Figure 2c), increased purification yield of his6-tagged protein (Figure 3a and b) and increased specific activity of purified his6-tagged protein (Figure 3c). For each of these properties, the wild-type RuBisCO and each of the evolved variants were assayed with respect in parallel to ensure fair comparisons. The activity of RuBisCO in crude extract was not equal to the product of expression levels and specific activity, but this can be attributed at least in part to differences in the technical details of each assay. Escherichia coli cell extracts may contain compounds that differentially inhibit or destabilize the RuBisCO variants. Moreover, the his6-tag fused to the C-terminus of the large subunit reduces the activity of the assembled holoenzyme (Spencer Whitney, Personal Communication). We were therefore not surprised about the non-additive nature of the observed improvements in expression and activity.
The evolved RuBisCO variants exhibited ~5-fold improvement in specific activity (Figure 3c). Protein engineers have effected vast improvements in the kinetic properties of a wide variety of enzymes, but a 5-fold increase in kcat would be unprecedented for RuBisCO (Spreitzer and Salvucci, 2002; Parry et al., 2003). We, therefore, tentatively propose a more cautious interpretation. The M259T mutation may enhance the conformational stability of the RuBisCO protein, both within the cell (Figure 3b) and in vitro during dialysis and storage (Figure 3c). Partial unfolding of the purified wild-type RuBisCO before the enzyme assay would decrease its apparent specific activity relative to those of the evolved mutants. This mechanism would still enhance the overall RuBisCO activity within a cell, and would likely have the same effect upon fitness as an improvement in specificity or catalytic efficiency. We are currently dissecting the effects of individual mutation upon the kinetic and biophysical properties of RuBisCO in an effort to determine the precise mechanism(s) of adaptation. Regardless of mechanism, these hypermorphs demonstrate that our E.coli selection system, in conjunction with the more precise 14CO2 incorporation assay, can be used to identify RuBisCO clones with increased activity. It may also be possible to generalize our underlying metabolic engineering strategy to evolve other non-native enzyme activities. The challenge is to identify growth conditions that select against transgenic cells expressing partially constructed metabolic pathways.
The E.coli-based RuBisCO selection system demonstrated here is very sensitive and high in throughput. It should enable genetic dissections of RuBisCO structure and function, including the identification of amino acid differences that underlie differences in specificity, catalytic efficiency and thermostability between the natural homologs. The current version of the selection (using rbcLS-pMAL-2px) is, however, somewhat imprecise and limited by the frequency of false positives (clones that survived selection with decreased RuBisCO activity). We believe that these shortcomings are manifestations of plasmid instability (Keasling, 1999), and can be reduced or eliminated by integration of the prkA expression cassette into the E.coli chromosome. In addition, the selection in its current form does not favor RuBisCO variants with greater CO2-specificity (Ω). Escherichia coli uses 2-phosphoglycolate phosphatase (encoded by gph) to metabolize the oxygenation product of RuBP (Pellicer et al., 2003). It should therefore be possible to evolve more specific variants through selections in engineered gph auxotrophs.
The selected amino acid changes (A8S, M259T, F342S) were similar or identical to those isolated from the published R.capsulatus selection experiment (M259T, F342V, F342I) (Smith and Tabita, 2003). This apparent convergence further supports the utility of the E.coli-based selection system and encourages us to speculate further about the evolutionary potential of RuBisCO. First, it is consistent with our presumption that RuBisCO hypermorphs identified in our E.coli-based selection system would retain their function in other types of cells. Second, it suggests that a limited number of beneficial mutations are accessible by transition-biased whole gene mutagenesis (effected by either PCR or the XL-1 Red mutator strain). Others have predicted that further improvement will require multiple, synergistic mutations (Watson and Tabita, 1997; John Andrews and Whitney, 2003). We are currently constructing RuBisCO libraries by more sophisticated random recombination techniques (Crameri et al., 1998; Ness et al., 2002) in an effort to capture these epistatic mutation combinations.
Acknowledgments
DG showed that rbcLS-pET30 is more reliable (genetically stable) than rbcLS-pMAL-2px. K.K.W. helped develop the genetic selection (Figure 1b) and CO2 incorporation assay. IM cloned the wild-type rbcL, rbcS and prkA genes and made the wild-type and mutant rbcL-6his-rbcS-pET30 constructs. We thank Ms Lori Rowe and Mr Charles O’Brien for sequencing them. We are grateful to Dr Tabita for providing the polyclonal anti-RuBisCO antibodies (Figure 2a), Drs Robert Tabita and Robert Houtz for reading the manuscript and to Dr Spencer Whitney for his helpful comments. M.R.P. was supported by NSF BIO/MCB (#0109668); D.N.G. by NIH training grant (T32 GM008367); I.M. by NIH/NIAID (1 R21AI054602-01).
References
- Allen MM. J Phycol. 1968;4:1–4. doi: 10.1111/j.1529-8817.1968.tb04667.x. [DOI] [PubMed] [Google Scholar]
- Amichay D, Levitz R, Gurevitz M. Plant Mol Biol. 1993;23:465–476. doi: 10.1007/BF00019295. [DOI] [PubMed] [Google Scholar]
- Cadwell RC, Joyce GF. PCR Methods Appl. 1992;2:28–33. doi: 10.1101/gr.2.1.28. [DOI] [PubMed] [Google Scholar]
- Crameri A, Raillard SA, Bermudez E, Stemmer WP. Nature. 1998;391:288–291. doi: 10.1038/34663. [DOI] [PubMed] [Google Scholar]
- Dean AM, Neuhauser C, Grenier E, Golding GB. Mol Biol Evol. 2002;19:1846–1864. doi: 10.1093/oxfordjournals.molbev.a004009. [DOI] [PubMed] [Google Scholar]
- Dower WJ, Miller JF, Ragsdale CW. Nucleic Acids Res. 1988;16:6127–6145. doi: 10.1093/nar/16.13.6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du YC, Hong S, Spreitzer RJ. Proc Natl Acad Sci USA. 2000;97:14206–14211. doi: 10.1073/pnas.260503997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatenby AA, Ellis RJ. Annu Rev Cell Biol. 1990;6:125–149. doi: 10.1146/annurev.cb.06.110190.001013. [DOI] [PubMed] [Google Scholar]
- Geddie ML, Matsumura I. J Biol Chem. 2004;279:26462–26468. doi: 10.1074/jbc.M401447200. [DOI] [PubMed] [Google Scholar]
- Hartman FC, Harpel MR. Annu Rev Biochem. 1994;63:197–234. doi: 10.1146/annurev.bi.63.070194.001213. [DOI] [PubMed] [Google Scholar]
- Hong S, Spreitzer RJ. J Biol Chem. 1997;272:11114–11117. doi: 10.1074/jbc.272.17.11114. [DOI] [PubMed] [Google Scholar]
- Houtz RL, Portis AR., Jr Arch Biochem Biophys. 2003;414:150–158. doi: 10.1016/s0003-9861(03)00122-x. [DOI] [PubMed] [Google Scholar]
- John Andrews T, Whitney SM. Arch Biochem Biophys. 2003;414:159–169. doi: 10.1016/s0003-9861(03)00100-0. [DOI] [PubMed] [Google Scholar]
- Keasling JD. Trends Biotechnol. 1999;17:452–460. doi: 10.1016/s0167-7799(99)01376-1. [DOI] [PubMed] [Google Scholar]
- Kleman GL, Horken KM, Tabita FR, Strohl WR. Appl Environ Microbiol. 1996;62:3502–3507. doi: 10.1128/aem.62.9.3502-3507.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi D, Tamoi M, Iwaki T, Shigeoka S, Wadano A. Plant Cell Physiol. 2003;44:269–276. doi: 10.1093/pcp/pcg048. [DOI] [PubMed] [Google Scholar]
- Lin EC. In: Escherichia coli and Salmonella typhimurium: Cellular and Molecular Biology. Neidhardt FC, editor. American Society for Microbiology; Washington DC: 1987. pp. 245–284. [Google Scholar]
- Mann CC. Science. 1999;283:314–316. doi: 10.1126/science.283.5400.314. [DOI] [PubMed] [Google Scholar]
- Matsumura I, Wallingford JB, Surana NK, Vize PD, Ellington AD. Nat Biotechnol. 1999;17:696–701. doi: 10.1038/10910. [DOI] [PubMed] [Google Scholar]
- Ness JE, Kim S, Gottman A, Pak R, Krebber A, Borchert TV, Govindarajan S, Mundorff EC, Minshull J. Nat Biotechnol. 2002;20:1251–1255. doi: 10.1038/nbt754. [DOI] [PubMed] [Google Scholar]
- Newman J, Gutteridge S. J Biol Chem. 1993;268:25876–25886. [PubMed] [Google Scholar]
- Parry MA, Andralojc PJ, Mitchell RA, Madgwick PJ, Keys AJ. J Exp Bot. 2003;54:1321–1333. doi: 10.1093/jxb/erg141. [DOI] [PubMed] [Google Scholar]
- Pellicer MT, Nunez MF, Aguilar J, Badia J, Baldoma L. J Bacteriol. 2003;185:5815–5821. doi: 10.1128/JB.185.19.5815-5821.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce J, Carlson TJ, Williams JG. Proc Natl Acad Sci USA. 1989;86:5753–5757. doi: 10.1073/pnas.86.15.5753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sage RF. In: C4 Plant Biology. Sage RF, Monson RK, editors. Academic Press; NY: 1999. pp. 3–17. [Google Scholar]
- Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. 3. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2001. [Google Scholar]
- Shively JM, van Keulen G, Meijer WG. Annu Rev Microbiol. 1998;52:191–230. doi: 10.1146/annurev.micro.52.1.191. [DOI] [PubMed] [Google Scholar]
- Smith SA, Tabita FR. J Mol Biol. 2003;331:557–569. doi: 10.1016/s0022-2836(03)00786-1. [DOI] [PubMed] [Google Scholar]
- Spreitzer RJ, Salvucci ME. Annu Rev Plant Biol. 2002;53:449–475. doi: 10.1146/annurev.arplant.53.100301.135233. [DOI] [PubMed] [Google Scholar]
- Spreitzer RJ, Esquivel MG, Du YC, McLaughlin PD. Biochemistry. 2001;40:5615–5621. doi: 10.1021/bi002943e. [DOI] [PubMed] [Google Scholar]
- Watson GM, Tabita FR. FEMS Microbiol Lett. 1997;146:13–22. doi: 10.1111/j.1574-6968.1997.tb10165.x. [DOI] [PubMed] [Google Scholar]
- Zhao G, Winkler ME. J Bacteriol. 1994;176:6134–6138. doi: 10.1128/jb.176.19.6134-6138.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]