

Abstract
Our goal is to understand how enzymes adapt to utilize novel substrates. We and others have shown that directed evolution tends to generate enzyme variants with broadened substrate specificity. Broad-specificity enzymes are generally deleterious to living cells, so this observed trend might be an artifact of the most commonly employed high throughput screens. Here, we demonstrate a more natural and effective screening strategy for directed evolution. The gene encoding model enzyme HIV protease was randomly mutated, and the resulting library was expressed in Escherichia coli cells to eliminate cytotoxic broad-specificity variants. The surviving variants were screened for clones with activity against a reporter enzyme. The wild-type human immunodeficiency virus type I protease (HIV PR) is cytotoxic and exhibits no detectable activity in reactions with beta-galactosidase (BGAL). In contrast, the selected variants were nontoxic and exhibited greater activity and specificity against BGAL than did the wild-type HIV PR in reactions with any substrate. A single round of whole gene random mutagenesis and conventional high-throughput screening does not usually effect complete inversions of substrate specificity. This suggests that a combination of positive and purifying selection engenders more rapid adaptation than positive selection alone.
Keywords: directed evolution, random mutagenesis, high-throughput screening, aspartic protease
Introduction
Our goal is to understand how enzymes evolve to recognize novel substrates. Adaptive molecular evolution is worthy of study for several reasons. Metazoan speciation is manifested at the macroscopic level but is driven by microscopic changes in protein structure and function. Population genetic models are generally applicable to evolving populations of proteins as the same rules should apply to complex systems at all levels of biological order. Molecules are easier to manipulate and study than whole organisms and are therefore more amenable to experimental hypothesis testing. Finally, studies of molecular adaptation are very practical as they enable the artificial evolution of new enzymes with useful properties.
Phylogenetic analysis has proven successful at identifying instances of natural adaptive molecular evolution. This contribution is significant because neutral mutations are fixed at a relatively high constant rate due to genetic drift (Kimura 1983). The most common approach is to compare the rates of nonsynonymous (missense) and synonymous (silent) substitutions; unusually high ratios are taken to indicate adaptive evolution (Yang and Bielawski 2000; Swanson 2003). In certain cases, gene synthesis or site-directed mutagenesis has been applied to ‘‘resurrect’’ the ancestral genes predicted by phylogenetic inference algorithms. The subsequent expression, purification, and characterization of the corresponding proteins can support (or reject) predictions based on phylogenetic analyses (Thornton 2004).
Adaptive mutations that affect substrate specificity are particularly difficult to identify for several reasons. First, enzyme-substrate specificity is often conserved for billions of years (Golding and Dean 1998). The adaptive mutations that affect specificity have thus become obscured by the accumulation of subsequent neutral mutations. Second, the relationship between the structure and function of existing proteins is often unclear even when an X-ray crystal structure is available. The conformational dynamics of an enzyme is often an important determinant of its specificity, and mutations that affect this property can occur anywhere in the protein (Benkovic and Hammes-Schiffer 2003; James and Tawfik 2003). Third, the criteria that determine the overall fitness of any particular protein at any particular time remain incompletely understood. Phylogenetic approaches alone are thus insufficient to understand the rules that govern of adaptive molecular evolution.
We and many others (Stemmer 1994; Moore and Arnold 1996; Yano, Oue, and Kagamiyama 1998; Aharoni et al. 2005; Varadarajan et al. 2005) control the evolutionary process in the laboratory and observe populations of proteins as they adapt to new substrates. We can repeat informative experiments, varying population sizes, mutation rates, recombination rates, and the selection criteria. In contrast to those who evolve bacterial populations in vitro, it is easy for us to identify potentially beneficial mutations, infer their mechanistic effects (through computational molecular modeling), and to purify and characterize the ancestral and evolved protein variants. Neutral and slightly deleterious mutations are rarely fixed in any of our experiments, in contrast to the rates observed in natural gene families.
Laboratory evolution can provide insight into the mechanisms of adaptive molecular evolution. We (Matsumura and Ellington 2001) and others (Aharoni et al. 2005) have shown that directed evolution tends to produce enzyme variants with broadened substrate specificity. This trend suggests that enzymes fluctuate through ‘‘specialist’’ and ‘‘generalist’’ states as they adapt to new substrates. Laboratory evolution may allow the isolation of mutants that would not be discovered through natural evolution, and vice versa, due to operational differences. First, natural molecular populations are larger than those produced by the polymerase chain reaction (PCR) and cloning. Second, cells in nature are normally challenged with libraries of novel substrates. In contrast, directed evolution experiments are generally contrived with a wild-type enzyme that exhibits modest but detectable activity against a single novel substrate. Third, laboratory evolution is able to isolate certain generalist enzymes that may be deleterious to cells in nature. We are therefore motivated to devise more realistic and effective selection schemes.
The human immunodeficiency virus type I protease (HIV PR) is a good model protein. Its structure and function are well understood, and it is amenable to high throughput assays. HIV PR normally catalyzes the cleavage of nine divergent amino acid sequences within the HIV-1 Gag-Pol polyprotein, thereby activating viral proteins essential for replication (Kohl et al. 1988). The enzyme is thought to recognize a shape assumed by its polypeptide substrates, rather than a consensus amino acid sequence (Prabu-Jeyabalan, Nalivaika, and Schiffer 2002). The enzyme is specific in the sense that it does not recognize other sites within the HIV-1 proteome, but we consider it nonspecific because it recognizes essential proteins in Escherichia coli (Baum, Bebernitz, and Gluzman 1990b), yeast, and mammalian cells (Blanco, Carrasco, and Ventoso 2003) and is cytotoxic to all three species. HIV protease is normally expressed late in the viral life cycle, so the cleavage of host proteins imposes little or no fitness cost to the virus in its natural environment.
Here, we observe the divergence of HIV PR specificity in an artificial system designed to recapitulate the natural selection of a generic protease. Random mutations were introduced by mutagenic PCR. The toxicity of the wild-type enzyme was exploited to eliminate variants with broad specificity, reflecting the fitness costs of generalist enzymes upon living cells. The surviving variants were challenged with a de facto library of peptide substrates consisting of the ~1,023 amino acids of a modified form of the E. coli beta-galactosidase (BGAL). Several specific (nontoxic) variants emerged after a single round of random mutagenesis and screening, some of which exhibited wild-type–like levels of activity against novel substrates not recognized by the wild-type enzyme.
Experimental Procedures
Materials
The HIV PR/lacZ coexpression vector, p1 + IQ (Baum, Bebernitz, and Gluzman 1990a), was purchased from the American Type Culture Collection (Manassas, Va.). 6his-lacZ-pET28 (induction control E) was from Novagen (Madison, Wisc.), and E. coli strain DH5Δlac (DE3) was constructed as previously described (Matsumura and Ellington 2001). Escherichia coli strain InvαF′ was from Invitrogen (Carlsbad, Calif.). The GeneAmp XL PCR kit was from Perkin Elmer (Boston, Mass.). Molecular biology enzymes, including restriction enzymes, T4 DNA ligase and T4 DNA polymerase, were from New England Biolabs (Beverly, Mass.). All oligonucleotides were custom synthesized by IDT (Coralville, Iowa); rabbit polyclonal antibodies to BGAL and monoclonal anti-HIV antibodies were from Abcam (ab616); the IRD700 infrared–labeled goat anti-rabbit antibody was obtained from LI-COR (Lincoln, Nebr.). All other chemicals, including the fluorogenic peptide HIV PR substrate I (with the sequence Arg-Glu-(EDANS)-Ser-Gln-Asn-Tyr-Pro-Ile-Val-Gln-Lys-(DABCYL)-Arg), M9 salts, ampicillin, and para-nitrophenyl-beta-D-galactopyra-noside (pNP-gal) were from Sigma-Aldrich (St. Louis, Mo.).
Cloning
Vector p1 + IQ (Baum, Bebernitz, and Gluzman 1990a) was used to coexpress the HIV PR and BGAL variants in E. coli. The p6 site in lacZ (after codon 80) was present in the p1 + IQ vector. The TNF-α peptide sequence (NRPDYLLFAE) was cloned into this vector, replacing the p6, by whole plasmid PCR and self-ligation (Geddie and Matsumura 2004) as follows. The p1 + IQ plasmid was amplified using primers: 5′-CTTCTTTTCG-CCGAGGAGGCCGATACTGTCGTCGTCCCC-3′ and 5′-GTAATCGGGTCGGTTAGGAAGATCGCACTCC-AGCCAGCTT-3′ (bold sequence encodes TNF-α peptide and underlined sequence complements lacZ). The presence of the TNF-α site was verified with DNA sequencing. Random mutations were introduced into the HIV PR gene in a error-prone PCR (Cadwell and Joyce 1992) using the extragenic primers 5′-CTACCATTACCAGTTGGTC-TGGTGTCA-3′ and 5′-CGTTTCACTTCTGAGTTCGG-CATG-3′. In short, the mutations were introduced in an error-prone PCR containing 5 microliters of a 10× mutagenic PCR mix (8 mM dTTP, 8 mM dCTP, 48 mM MgCl2, 5 mM MnCl2) in 50 microliters (total volume). The PCR was cycled 25 times between 94°C for 30 seconds, 65°C for 30 seconds and 72° C for 1 minute. The addition of MnCl2 to the reaction reduced the fidelity of the polymerase and the excess of dTTP and dCTP offset the balance of dNTPs. Adding MnCl2 to the reaction reduces the fidelity of the polymerase, and the excess of dTTP and dCTP offsets the balance of deoxynucleoside triphosphates. The library of mutant HIV PR alleles was cloned into a TNF-α/BGAL variant of the p1 + IQ coexpression vector using the enzymes BsmI and PstI and electroporated (Dower, Miller, and Ragsdale 1988) into E. coli W2244 (lacZ−, lacY+ ) (Cook and Lederberg 1962) to produce a library containing 500,000 random mutants.
Western Blot Analysis
p1 + IQ vectors containing various combinations of HIV PR (wild type, D25N, P9S/I50L) and lacZ (wild type, p6, and TNF-α) alleles were propagated in LB-ampicillin media to midlogarithmic phase and induced with 1 mM iso-propyl-beta-D-thiogalactopyranoside (IPTG) for 1 h at 37°C. Cells were harvested by micro-centrifugation in microfuge at 3,000 rpm for 10 min and resuspended in sodium dodecyl sulfate (SDS)/beta-mercaptoethanol buffer (Sambrook and Russell 2001). The proteins in these extracts were electrophoretically separated in a 6% polyacrylamide gel and transferred to a nitrocellulose membrane. The membrane was blocked with 4% nonfat milk, reacted with polyclonal anti-BGAL antibodies and IRD700-labeled secondary antibodies. The immobilized antibody-antigen complexes were visualized and quantified on a LI-COR Odyssey infrared imager.
In Vivo BGAL Activity
P1 + IQ-type vectors for the coexpression of HIV PR (wild type, D25N, or P9S/I50L) and BGAL (wild type, native p6, or novel TNF-α) were constructed. Escherichia coli strain W2244 (lacZ−, lacY+ ) was transformed with each of the nine constructs and separately propagated on LB-ampicillin agar plates. The resulting colonies were scraped off of each plate and resuspended in liquid LB-ampicillin. The OD600 of each culture was measured, andthe cells were diluted to the same density. Twenty micro-liters of each culture (in triplicate) were reacted with 2 mM pNP-gal in 50 mM Tris-HCl/10 mM MgCl2 (pH 7.6). The OD405 was then monitored for 5 h in a Thermo Lab- systems Multiskan Ascent microplate spectrophotometer.
Protein Purification
The HIV PR alleles from p1 + IQ were amplified with 5′-NdeI HIV PR (5′-GGAATTCCATATGCCTCAGAT-CACTCTTTGGCAACGACC-3′), where the bold sequence is an NdeI restriction site and the underlined sequence complements the HIV PR gene and the extragenic pUC-600 (5′-GAAAACGTTCTTCGGGGCGAA-3′). The PCR products were purified and cloned into the pET28+ vector (Novagen) using restriction enzymes NdeI and HindIII. This fused DNA encoding an N-terminal histidine tag and thrombin cleavage site to the 5′ end of the HIV PR gene. The clone was sequenced to verify the absence of undesired mutations from the PCR procedure. The protease was purified according to an established protocol (Vickrey et al. 2003) that describes the purification of the protease from inclusion bodies. Briefly, a 10-ml culture of the his-tagged protease was grown to saturation in LB media supplemented with 25 mg/ml kanamycin at 37°C (LB-kan). The culture was diluted 1:100 in 1 liter LB-kan, grown to mid-logarithmic phase, and induced for 3 h with 1 mM IPTG. The cells were harvested by centrifugation, and the pellet was frozen at −80°C.
The cells were resuspended in lysis buffer (50 mM Tris, pH 7.5, 200 mM sodium chloride, and 2 mM ethyl-enediaminetetraacetic acid [EDTA]) and lysed by the application of 20,000 PSI of force in two passages through a French pressure cell. The inclusion bodies were separated from the soluble protein fraction by centrifugation, resuspended in 6 M urea, and applied to a nickel affinity column equilibrated with lysis buffer and 6 M urea. The column was sequentially washed with three lysis buffers containing 6 M urea and 10, 25, and 50 mM imidazole, respectively. HIV PR was step eluted with 200 mM and 1 M imidazole. The denatured protease was placed in dialysis tubing and dialyzed initially against buffer (20 mM sodium acetate, pH 5, 1 mM dithiothreitol DTT), plus 3 M urea and then the same buffer with 1 M urea and finally two exchanges of buffer without urea. The protein was concentrated using a Centriprep YM-3 concentrator (Millipore, Bedford, Mass.). All HIV PR protein was concentrated to > 1 microgram/ microliter, as quantified by a Bradford assay (Bradford 1976). The proteins were stored at −20°C in 20 mM sodium acetate, pH 5, 1 mM DTT, and 50% glycerol and were stable for over 2 months. The protease was purified to greater than 95% purity, as judged by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) analysis (Sambrook and Russell 2001).
Enzyme Assays with Fluorogenic Oligopeptide Substrates
The protease assays were initiated by first adding varying amounts of HIV protease substrate I (1.45–13.85 μM) to HIV PR assay buffer (0.1 M sodium acetate, 1.0 M sodium chloride, 1.0 mM EDTA, 1.0 mM DTT, 1mg/ml BSA, pH 4.7) and then adding a constant amount of purified protease (191.5 nM) to a total reaction volume of 130 μl. The enzymes and substrates were reacted for 20 min at 25°C on an SLM-Aminco Bowman Series 2 spec-trofluorimeter. The assays were performed in triplicate at each substrate concentration. The purified, inactive D25N variant did not react with either peptide substrate (negative control, data not shown). To convert fluorescence units per time to molarity per time, a known amount of the 5-(2-aminoethylamino)-1-naphthalene sulfonate (EDANS) fluorophore was used as a reference compound, and its absorbance was recorded. The equation Vc = Vi(Cref/Iref), where Vc is the concentration of product per unit time, Vi is the fluorescence per unit time, Cref is the known concentration of EDANS, and Iref is the fluorescence intensity of EDANS for the given concentration. The values were also corrected for the differences of the intensities of equimolar quantities of reference compound to that of the free cleavage product by multiplying by this ratio (Matayoshi et al. 1990).
Results
High-Throughput HIV PR Screen
Our objective is to study adaptive enzyme evolution under conditions that simulate the natural selection of a generic protease. Previous workers have found that many high-throughput screens (those divorced from organismal fitness) tend to favor the selection of generalist variants with broad specificity, at least as an intermediate. Here, we describe a system that discriminates against such generalists and challenges each randomly mutated enzyme with a library of possible substrates. It has been shown that HIV PR catalyzes the cleavage of the native p6 peptide (VSFNF↓PQITL) in the context of a permissive surface loop (amino acid position 80) of the E. coli BGAL (p6/BGAL). The cleavage reaction occurs in vivo when the HIV PR and p6/BGAL proteins are coexpressed in E.coli, enablinghigh-throughputassaysbasedoninactivation of BGAL activity (Baum, Bebernitz, and Gluzman 1990a).
We arbitrarily chose a relatively hydrophobic 10-amino acid peptide (NRPDY↓LLFAE) as a novel HIV PR substrate; this sequence is normally displayed on a surface loop of TNF-α (Eck and Sprang 1989), a drug target that causes inflammatory diseases (Beutler and Cerami 1986). We inserted the TNF-α peptide into lacZ after codon 80 (TNF-α/BGAL) by site-directed insertion mutagenesis. Coexpression of TNF-α/BGAL with either the wild-type or the catalytically inactive (negative control) D25N HIV PR mutant demonstrated that the wild-type protease exhibited detectable activity against the novel substrate (table 1), normally a prerequisite for in vitro evolution. The wild-type HIV PR thus strongly prefers the TNF-α peptide over all the other peptides of TNF-α/BGAL.
Table 1.
In Vivo Coupled Assays of HIV PR Activity
| Wild-Type BGAL | p6/BGAL | TNF-α/BGAL | |
|---|---|---|---|
| Wild-type HIV PR | 1.966 ± 0.03a | 0.834 ± 0.01 | 1.053 ± 0.06 |
| D25N HIV PRb | 2.063 ± 0.09 | 1.060 ± 0.04 | 1.384 ± 0.03 |
| P9S/I50L HIV PR | 1.884 ± 0.04 | 1.000 ± 0.08 | 0.687 ± 0.02 |
The OD405 was monitored for 5 h in a microplate spectrophotometer, and the activities (in units of change in mAbs405 per hour per cell) are reported.
All values should be compared to those of the catalytically inactive D25N (negative control), which should have little effect upon BGAL activity.
Random Mutagenesis and Selection
Random mutations were introduced into the HIV PR gene in a error-prone PCR; the cumulative mutation rate of this procedure is about 0.66 ± 0.13% per position (Cadwell and Joyce 1992). The resulting library of mutant HIV PR alleles was cloned into a TNF-α/BGAL coexpression vector and electroporated into E. coli W2244 (lacZ−, lacY+). Sixty-thousand transformed cells were propagated in LB-ampicillin media to midlogarithmic phase; expression of the HIV PR library and TNF-α/BGAL proteins were induced with IPTG. The wild-type HIV PR catalyzes the cleavage of essential E. coli proteins and is therefore cyto-toxic (Baum, Bebernitz, and Gluzman 1990b); this initial selection eliminated active variants with broad specificity. After 2 h of induction, the surviving ~15,000 cells were spread onto plates containing 100 μg/μl ampicillin at a density of ~500 colony-forming units per plate and grown overnight at 37°C.
The transformed colonies were adsorbed to nitrocellulose filters and transferred (colony side up) onto plates containing ampicillin and 0.08 mg/ml 5-bromo-4-chloro-3-indolyl-beta-D-galactopyranoside (X-gal), a histochemical marker of BGAL activity. Control colonies expressing the wild-type HIV PR and TNF-α/BGAL were a lighter blue, indicating protease activity, compared with those expressing with D25N variant under these conditions, indicating protease activity. We identified 27 light blue colonies (presumably expressing HIV PR variants with altered specificity) and restreaked these onto LB-ampicillin–X-gal plates to verify the decrease in BGAL activity. Fewer than half (12 clones) retained the light blue phenotype, and seven of these were discarded after restriction mapping analysis indicatedlargedeletionswithinthep1+ IQplasmid. In summary, we identified five protease variants (out of 60,000 clones) that were less toxic than the wild type but exhibited reproducibly greater activity against the TNF-α/ BGAL protein.
Sequences of the Evolved HIV PR Variants
The selected HIV PR genes were sequenced (both strands) in order to determine the mutations that altered specificity. The following amino acid replacements were observed: P9S/I50L, Q7L (twice), W6R/F53S/N83I/I93N, L5F. We mapped them upon the crystal structure of the enzyme-substrate complex (Prabu-Jeyabalan, Nalivaika, and Schiffer 2002) and found that the mutations occurred in residues either in the substrate-binding cleft or at the dimer interface (fig. 1). The possible adaptive mechanisms of these mutations are described in Discussion. We studied the behavior of these mutants in vivo (within E. coli) and in vitro to learn how the amino acid changes altered substrate specificity.
Fig. 1.

Models of the artificially evolved HIV PR variants. The cocrystal structure of the D25N HIV PR/p6 peptide complex (Prabu-Jeyabalan, Nalivaika, and Schiffer 2002) was modified by molecular modeling (using the SYBYL software package) as follows. The ‘‘native’’ p6 peptide was mutated into the novel NRPDYLLFAE peptide substrate (derived from TNF-α); the P9S and I50L mutations were made (A). The new complex was energy minimized. The Q7L (B), W6R, F53S, N83I, I93N (C), and L5F (D) mutations, which are also associated with changes in substrate specificity, are modeled on the unaltered D25N HIV PR/p6 structure.
In Vivo Behavior
In Vivo Activity
Amongst the four unique HIV PR variants (P9S/ I50L, Q7L, W6R/F53S/N83I/I93N, L5F), P9S/I50L was the sole protease that exhibited improved specificity for the TNF-α/BGAL substrate. The three other mutations showed decreased BGAL activity, regardless of the sequence of the inserted peptide. To quantify the inversion of specificity exhibited by the P9S/I50L variant, we co-expressed three HIV PR variants (wild type, D25N, and P9S/I50L) with each of the three BGAL variants (containing the wild type, p6, and TNF-α peptides at amino acid 80) in liquid culture and reacted each culture with pNP-gal (table 1). Increased protease activity is revealed by diminished BGAL activity (relative to the catalytically inactive D25N control). The wild-type HIV PR exhibited similar activity in reactions with p6/BGAL (~21% less BGAL activity than corresponding D25N control) and TNF-α/BGAL (~25% less BGAL activity than D25N). In contrast, the P9S/I50L variant exhibited little activity against p6/BGAL (~6% less than D25N) but significantly improved activity against TNF-α/BGAL (~50% less BGAL activity than D25N). These results suggest that the P9S/I50L mutations confer specificity for the TNF-α peptide.
Toxicity
The heterologous expression of the wild-type protease is toxic to E. coli due to nonspecific cleavage of essential cellular proteins (Baum, Bebernitz, and Gluzman 1990b). Escherichia coli transformed with HIV PR/BGAL coexpression vectors were grown to midlogarithmic phase. The HIV PRs (wild type, D25N, P9S/I50L, Q7L, or W6R/ F53S/N83I/I93N, L5F) and BGAL (p6/BGAL) were induced with IPTG. The cultures were diluted 105-fold and plated at time points 0, 1, and 2 h. After overnight incubation at 37°C, the colonies were counted and the viability of each culture was calculated. This survival assay confirmed that the wild-type HIV PR was toxic to E. coli, but the catalytically inactive D25N and artificially selected HIV PR variants were not (Figure 2).
Fig. 2.

Toxicity of HIV PR variants. Two milliliters of Escherichia coli transformed with HIV PR/BGAL coexpression vectors were grown to midlogarithmic phase in LB-ampicillin media at 37°C. The HIV PRs and BGAL were induced with 1 mM isopropyl-beta-D-thiogalactoside. Before induction and at the 1- and 2-h time points, cultures were diluted 105-fold and 100 μl was plated onto LB-ampicillin plates. After overnight incubation at 37°C, the colonies were counted and the viability of each culture was calculated.
Western Blot
Anti-BGAL polyclonal antibodies were employed to visualize the cleavage products of the PRs within cell extracts. Cells transformed with variants of the p1 + IQ plasmid (wild type, D25N or P9S/I50L HIV PR plus wild type, p6/BGAL or TNF-a/BGAL) were propagated to midlogarithmic phase and induced with IPTG. Extracts from these cells were visualized by SDS-PAGE and immunoblotting (fig. 3). The expected fragments of BGAL when cleaved at the engineered site are approximately 17 and 108 kDa. The western blot clearly shows cleavage of the p6/BGAL by wild-type protease (lane 2) but not by D25N (lane 3); a modest product can be seen when coexpressed with P9S, I50L (lane 4). Similarly, the TNF-α site within TNF-α/BGAL is cleaved (albeit incompletely) by the selected P9S/I50L variant (lane 7), but not by D25N (lane 6), and to a lesser degree by the wild-type protease (lane 5). A lower band present in all lanes, including those containing the inactive D25N HIV PR mutant, is probably either a nonspecific cross-reactant with the BGAL antibody or the product of some endogenous protease. These results suggest that the P9S/I50L HIV PR variant recognizes the novel TNF-α peptide rather than some other part of BGAL. To determine if the P9S/I50L mutation affected protein expression levels, soluble and whole cell extracts were analyzed by Western blotting analysis using a monoclonal anti-HIV PR antibody (Figure 4). The levels of expression differed only modestly. The increased activity of the P9S/ I50L HIV protease against the TNF-α/BGAL reporter is therefore not likely due to increased expression levels.
Fig. 3.

The P9S/I50L HIV PR variant cleaves the TNF-α/BGAL in vivo. Escherichia coli were transformed with various HIV PR/BGAL co-expression vectors, propagated to midlogarithmic stage, and induced with 1 mM IPTG for 30 min. The proteins were subsequently harvested by centrifugation, resuspended in denaturing buffer, and separated by 6% SDS-PAGE. The proteins were transferred to nitrocellulose and probed with an anti-BGAL polyclonal antibody. The antibody-BGAL complexes were detected as described for figure 3. Lanes: (1) molecular weight marker; (2) wild-type (WT) HIV PR, p6/BGAL; (3) D25N HIV PR, p6/BGAL; (4) P9S/I50L HIV PR, p6/BGAL; (5) WT HIV PR, TNF-α/BGAL; (6); D25N HIV PR, TNF-α/BGAL; and (7) P9S/I50L HIV PR, TNF-α/BGAL.
Fig. 4.
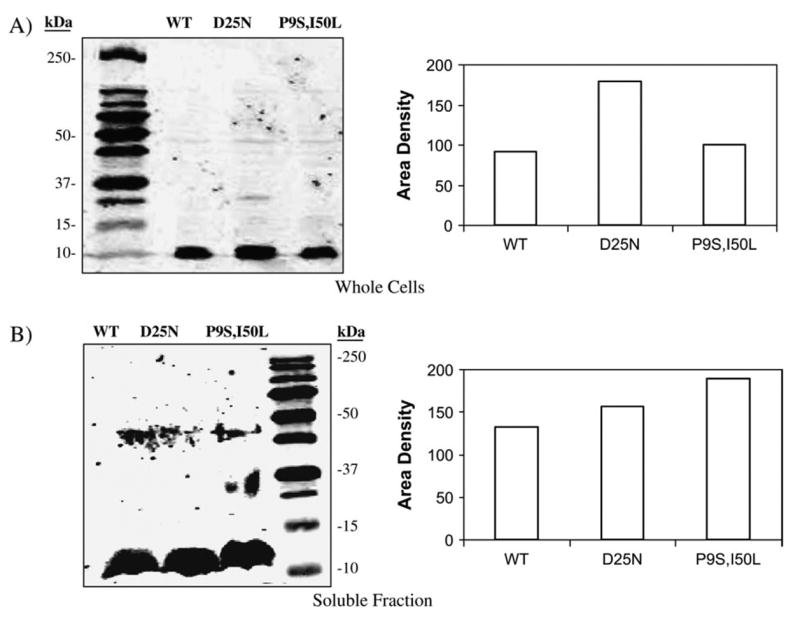
Expression levels of HIV PRs. E. coli transformed with WT, D25N or P9S/I50L HIV PR expression vectors were propagated to mid logarithmic phase in LB-ampicillin media. Gene expression was induced with 1 mM IPTG for 30 minutes. A) Whole cells were harvested by centrifugation, resuspended in 1 ml 1× SDS-PAGE running buffer, and analyzed by western blot analysis using an anti-HIV PR antibody. An IRD800 labeled secondary antibody and a LICOR Odyssey scanner were used to quantify the protein in each band. B) Cells were lysed, and the soluble fraction was removed and analyzed by western blot analysis.
In Vitro Behavior
Synthetic Substrate
The wild-type, D25N, P9S/I50L, W6R/F53S/N83I/ I93N, Q7L HIV PRs were purified and refolded by an established protocol (Vickrey et al. 2003). The purified wild-type and P9S/I50L HIV PRs were separately reacted with a commercially available peptide (HIV protease substrate I). This synthetic peptide contains an EDANS (5-(2-amino-ethylamino)-1-naphthalene sulfonate) molecule at the C-terminus and a 4′-dimethylaminoazobenzene-4-carboxylate (DABCYL) molecule at the N-terminus, which quenches the fluorophore when in close proximity (Matayoshi et al. 1990). Upon cleavage, the DABCYL group is separated from the fluorophore, leading to emission at 490 nm. The activity of the purified wild type is comparable to the value in the literature (fig. 5). In contrast, the P9S/I50L HIV PR exhibits only ~3% wild-type activity. These results are consistent with the in vivo assays above, which show that P9S/I50L HIV PR is relatively inactive against the p6/BGAL protein.
Fig. 5.
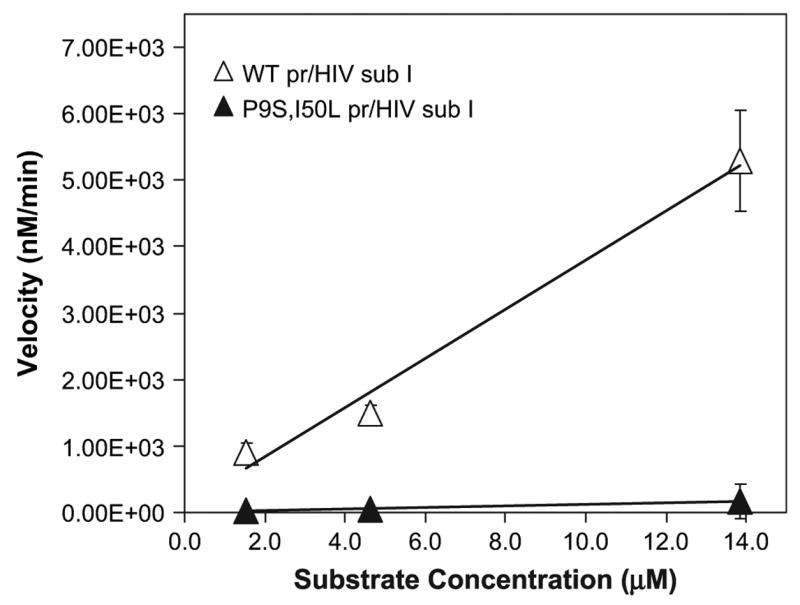
Activity of purified proteases against fluorogenic peptide substrates. The wild-type (WT) PR was purified, and equimolar concentrations (191 nM) were separately reacted with 1.45–13.85 μM native HIV PR substrate I. The initial velocities of product formation were recorded for up to 20 minutes in a fluorescence spectrophotometer. These rates were then converted to velocity in units of concentration per minute and plotted against substrate concentration.
BGAL Substrate
We next showed that the purified HIV PR variants could cleave the purified BGAL variants in vitro. The his-tagged wild-type, p6- and TNF-α/ BGAL proteins were purified by immobilized metal affinity chromatography. The HIV PRs were reacted with the BGAL variants in phosphate buffer (25 mM Na2HPO4, pH 7.0; 25 mM NaCl; 1 mM DTT, 30°C for 1 h). The cleavage products were then separated on 6% polyacrylamide gels and stained with SYPRO dye (Invitrogen). Figure 6A shows a representative gel of the degradation of p6/BGAL by HIV PR. Figure 6B shows the quantification of the upper band (full-length BGAL) at each time point. We chose to quantify the band representing the full-length BGAL in each experiment because it is evident that the 100-kDa product is subject to further degradation, possibly because this product is conformationally unstable. The purified P9S/I50L PR is ~2.5-fold more active than the wild-type enzyme in reactions with the TNF-α/BGAL. Neither version of BGAL is cleaved by D25N PR; the wild-type BGAL is not cleaved by the wild type, D25N, or P9S/I50L (data not shown). These results suggest that P9S/I50L is specific for the TNF-α peptide within the TNF-α/BGAL reporter.
Fig. 6.
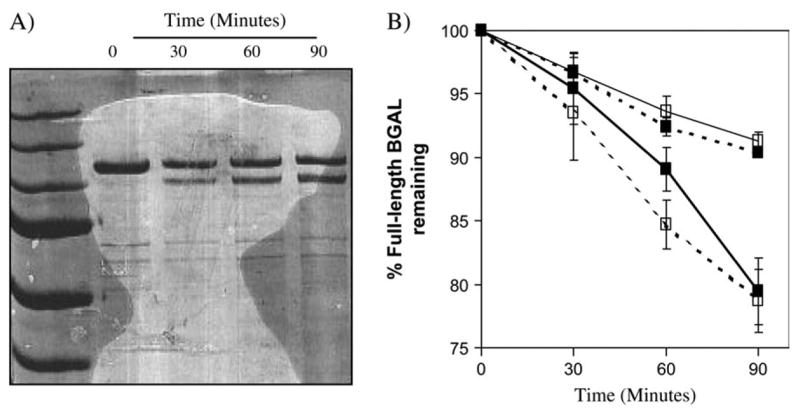
The purified P9S/I50L HIV PR variant cleaves the purified TNF-α/BGAL in vitro. The wild-type (WT), D25N (negative control), and P9S/ I50L (evolved) HIV PR alleles and the p6/and TNF-α/BGAL variants were cloned into expression vector pET28. This cloning step fused each gene to sequence encoding an N-terminal hexa-histidine tag. The protease and reporter proteins were purified and reacted with each other to give the corresponding graphs. For each experiment, 1.6 μM BGAL protein was reacted with 0.26 μM protease at 30°C, and time points were taken at 0, 30, 60, and 90 min. The reactions were stopped by the addition of SDS and beta-mercaptoethanol. The samples were analyzed by SDS-PAGE using the same molecular weight standard as shown in fig. 3. (A) Wild-type protease cleaves p6/BGAL in a linear fashion over a period of 90 min. (B) A graph indicating the percent of full-length BGAL remaining after specified times. ■ = wild-type HIV PR; □ = P9S/I50L PR; — = p6/BGAL; - - - - = TNF-α/BGAL.
The purified W6R/F53S/N83I/I93N and Q7L HIV PRs exhibited activity in reactions with the TNF-α/BGAL both in vivo and in vitro (fig. 7). The HIV PR variants were cloned into pET28 and transformed into the strain BL21(DE3), which is lacZ+ . Cultures transformed with various proteases show a profound decrease in BGAL activity (about 10-fold) when activity was measured with a pNP-gal assay (fig. 7A). In the in vitro assays, the evolved HIV PR variants caused the disappearance of the 116 kDa beta-galactosidase band, but not the appearance of the 100 kDa digestion product (fig. 7b). This result suggests that these mutants evolved to recognize sites within BGAL other than the TNF-α site. The wild-type HIV PR has no detectable activity against the wild-type BGAL either in vivo (fig. 7A) or in vitro (fig. 7B). In contrast, the purified W6R/ F53S/N83I/I93N and Q7L HIV PR variants appear to be even more active against wild-type BGAL than is the wild-type HIV PR is against p6/BGAL. We cannot pinpoint the site of cleavage due to the smearing produced by the protease, but it is clear that these HIV PR variants have evolved to recognize sites within BGAL not susceptible to the wild-type protease.
Fig. 7.
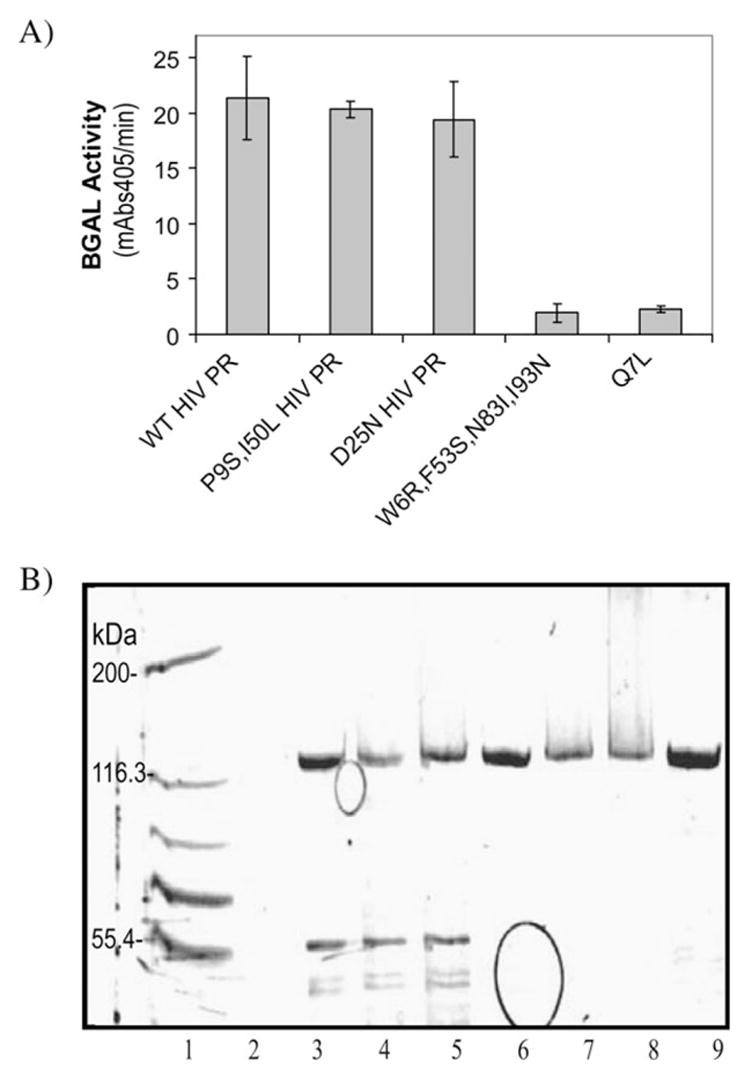
The Q7L and W6R/F53S/N83I/I93N PRs are active against the wild-type BGAL both in vivo and in vitro. A.) The wild-type (WT), D25N, P9S/I50L, W6R/F53S/N83I/I93N and Q7L HIV proteases were separately expressed in E. coli strain BL21(DE3), which is lacZ+ . The BGAL activity of each culture was measured by reacting it with para-nitro-phenyl-beta-D-galactopyranoside and monitoring the formation of the pNP product (Abs405) for 30 minutes. Decreased BGAL activity corresponds to increased HIV PR activity (B) The his-tagged HIV PR and BGAL variant proteins were purified, and 0.26 micromolar of each protease was reacted with 1.6 micromolar TNF-α/BGAL or wild-type BGAL for one hour at 30° C. The hydrolysis of the BGAL substrate (~116 kDa band) was visualized by SDS-PAGE. Lanes: 1) molecular weight markers representing 200, 116.3, 97.4, 66.3, and 55.4 kDa bands; 2) blank; 3) TNF-α/BGAL alone; 4) Q7L HIV PR + TNF-α/BGAL; 5) W6R/F53S/N83I/I93N HIV PR + TNF-α/BGAL; 6) WT BGAL alone; 7) Q7L HIV PR + WT BGAL; 8) W6R/F53S/N83I/I93N HIV PR + WT BGAL; 9) WT PR + WT BGAL.
Discussion
Positive and Purifying Selection
Laboratory evolution experiments can reveal possible mechanisms of adaptive molecular evolution, but their relevance to real-world populations is predicated upon the nature of the high-throughput screen. Protein engineers typically begin with a wild-type enzyme with modest but detectable catalytic activity in reactions with a single novel substrate. In contrast, natural selection adapts entire proteomes to libraries of novel substrates. The simplest high-throughput screens tend to favor enzyme variants with broadened substrate specificity (Aharoni et al. 2005). This trend suggests that proteins have the structural potential to evolve in this way, but it seems to us that most broad-specificity enzymes would be toxic to contemporary cells.
Our selection system discriminates against protease variants with broad substrate specificity. It also enabled the identification of variants that utilize any part of the 1023 amino acid BGAL protein as a substrate. These features apparently favored variants more specific than those commonly observed in directed evolution experiments. The P9S/I50L PR exhibits inverted specificity toward the TNF-α site within BGAL and is as active and specific in reactions with TNF-α/BGAL as the wild-type HIV PR is against p6/ BGAL. The W6R/F53S/N83I/I93N and Q7L HIV PR variants exhibit inverted specificity against unknown sites within the wild-type BGAL protein, even though the wild-type HIV PR shows no detectable activity against this substrate. None of the evolved protease variants are as toxic to E. coli cells as the wild-type enzyme. It is unusual to observe such inversions in substrate specificity after a single round of whole gene mutagenesis and screening. A recent study that utilizes bacterial display and flow cytometry (Varadarajan et al. 2005) applied positive and negative selection as well. Through this screening procedure, these investigators were able to isolate mutants of the E. coli endopeptidase OmpT that exhibited greater than three million-fold specificity toward a novel substrate over the native substrate. These results further validate this purifying selection strategy.
Structure and Function
We randomly mutated the HIV PR gene and identified a single clone (P9S/I50L) with improved activity against a designated target substrate (TNF-α peptide) in a high-throughput screen. Characterization of the mutant enzyme within the artificial E. coli system and in vitro showed that it had reduced activity against its native substrate and increased activity toward this novel substrate. To rationalize the effect of the P9S and I50L amino acid alterations on the overall structure and activity of the protease, we modeled the TNF-α substrate within the active site of HIV PR (fig. 1). I50 is part of the flap region, which in the closed conformation interacts with the substrate and is involved in hydrogen bonding within the protease (Hoog et al. 1996). The mutation of this isoleucine to leucine may alter hydrophobic packing within the enzyme-substrate complex. Interestingly, a mutation in I50 has also been identified in a selection searching for mutations in the protease that alleviate its toxicity (Baum, Bebernitz, and Gluzman 1990b). The effect of the P9S substitution is not obvious, but the proline residue is present in the dimer interface of the protease. This region of the protease is primarily hydrophobic, and a substitution from proline to serine may alter this hydrophobicity and thus the dimer interaction. Because dimerization influences the shape of the active site pocket, an alteration may allow the novel peptide substrate to bind (Prabu-Jeyabalan, Nalivaika, and Schiffer 2000). The substrate preference of other mutations (Q7L, W6R, F53S, N83I, I93N, and L5F) remains unknown, but each clone contains mutations also in the dimerization interface.
Acknowledgments
D.N.G. first expressed and purified the hexa-histidine tagged HIV PR, although she did not refold it. We are grateful to Lauren Ancel-Meyers, Stefan Lutz, Wayne Patrick, and Jennifer Stratton for their critical readings of the manuscript. We thank the National Institutes of Health/National Institute of Allergy and Infectious Diseases (1 R21AI054602-01) and National Institute of General Medical Services (1 R01 GM074264-01) for support.
Footnotes
Claudia Schmidt-Dannert, Associate Editor
Literature Cited
- Aharoni A, Gaidukov L, Khersonsky O, Mc QGS, Roodveldt C, Tawfik DS. The ‘evolvability’ of promiscuous protein functions. Nat Genet. 2005;37:73–76. doi: 10.1038/ng1482. [DOI] [PubMed] [Google Scholar]
- Baum EZ, Bebernitz GA, Gluzman Y. Beta-galactosidase containing a human immunodeficiency virus protease cleavage site is cleaved and inactivated by human immunodeficiency virus protease. Proc Natl Acad Sci USA. 1990a;87:10023–10027. doi: 10.1073/pnas.87.24.10023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum EZ, Bebernitz GA, Gluzman Y. Isolation of mutants of human immunodeficiency virus protease based on the toxicity of the enzyme in Escherichia coli. Proc Natl Acad Sci USA. 1990b;87:5573–5577. doi: 10.1073/pnas.87.14.5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkovic SJ, Hammes-Schiffer S. A perspective on enzyme catalysis. Science. 2003;301:1196–1202. doi: 10.1126/science.1085515. [DOI] [PubMed] [Google Scholar]
- Beutler B, Cerami A. Cachectin/tumor necrosis factor: an endogenous mediator of shock and inflammation. Immunol Res. 1986;5:281–293. doi: 10.1007/BF02935501. [DOI] [PubMed] [Google Scholar]
- Blanco R, Carrasco L, Ventoso I. Cell killing by HIV-1 protease. J Biol Chem. 2003;278:1086–1093. doi: 10.1074/jbc.M205636200. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Cadwell RC, Joyce GF. Randomization of genes by PCR mutagenesis. PCR Methods Appl. 1992;2:28–33. doi: 10.1101/gr.2.1.28. [DOI] [PubMed] [Google Scholar]
- Cook A, Lederberg J. Recombination studies of lactose nonfermenting mutants of Escherichia coli K-12. Genetics. 1962;47:1335–1353. doi: 10.1093/genetics/47.10.1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dower WJ, Miller JF, Ragsdale CW. High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res. 1988;16:6127–6145. doi: 10.1093/nar/16.13.6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eck MJ, Sprang SR. The structure of tumor necrosis factor-alpha at 2.6 A resolution. Implications for receptor binding. J Biol Chem. 1989;264:17595–17605. doi: 10.2210/pdb1tnf/pdb. [DOI] [PubMed] [Google Scholar]
- Geddie ML, Matsumura I. Rapid evolution of beta-glucuronidase specificity by saturation mutagenesis of an active site loop. J Biol Chem. 2004;279:26462–26468. doi: 10.1074/jbc.M401447200. [DOI] [PubMed] [Google Scholar]
- Golding GB, Dean AM. The structural basis of molecular adaptation. Mol Biol Evol. 1998;15:355–369. doi: 10.1093/oxfordjournals.molbev.a025932. [DOI] [PubMed] [Google Scholar]
- Hoog SS, Towler EM, Zhao B, Doyle ML, Debouck C, Abdel-Meguid SS. Human immunodeficiency virus protease ligand specificity conferred by residues outside of the active site cavity. Biochemistry. 1996;35:10279–10286. doi: 10.1021/bi960179j. [DOI] [PubMed] [Google Scholar]
- James LC, Tawfik DS. Conformational diversity and protein evolution—a 60-year-old hypothesis revisited. Trends Biochem Sci. 2003;28:361–368. doi: 10.1016/S0968-0004(03)00135-X. [DOI] [PubMed] [Google Scholar]
- Kimura M. The neutral theory of molecular evolution. Cambridge University Press; Cambridge: 1983. [Google Scholar]
- Kohl NE, Emini EA, Schleif WA, Davis LJ, Heimbach JC, Dixon RA, Scolnick EM, Sigal IS. Active human immunodeficiency virus protease is required for viral infectivity. Proc Natl Acad Sci USA. 1988;85:4686–4690. doi: 10.1073/pnas.85.13.4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matayoshi ED, Wang GT, Krafft GA, Erickson J. Novel fluorogenic substrates for assaying retroviral proteases by resonance energy transfer. Science. 1990;247:954–958. doi: 10.1126/science.2106161. [DOI] [PubMed] [Google Scholar]
- Matsumura I, Ellington AD. In vitro evolution of beta-glucuronidase into a beta-galactosidase proceeds through non-specific intermediates. J Mol Biol. 2001;305:331–339. doi: 10.1006/jmbi.2000.4259. [DOI] [PubMed] [Google Scholar]
- Moore JC, Arnold FH. Directed evolution of a para-nitrobenzyl esterase for aqueous-organic solvents. Nat Biotechnol. 1996;14:458–467. doi: 10.1038/nbt0496-458. [DOI] [PubMed] [Google Scholar]
- Prabu-Jeyabalan M, Nalivaika E, Schiffer CA. How does a symmetric dimer recognize an asymmetric substrate? A substrate complex of HIV-1 protease. J Mol Biol. 2000;301:1207–1220. doi: 10.1006/jmbi.2000.4018. [DOI] [PubMed] [Google Scholar]
- Prabu-Jeyabalan M, Nalivaika E, Schiffer CA. Substrate shape determines specificity of recognition for HIV-1 protease: analysis of crystal structures of six substrate complexes. Structure (Camb) 2002;10:369–381. doi: 10.1016/s0969-2126(02)00720-7. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. Molecular cloning: a laboratory manual. 3. Vol. 1. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, N.Y: 2001. [Google Scholar]
- Stemmer WP. Rapid evolution of a protein in vitro by DNA shuffling. Nature. 1994;370:389–391. doi: 10.1038/370389a0. [DOI] [PubMed] [Google Scholar]
- Swanson WJ. Adaptive evolution of genes and gene families. Curr Opin Genet Dev. 2003;13:617–622. doi: 10.1016/j.gde.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Thornton JW. Resurrecting ancient genes: experimental analysis of extinct molecules. Nat Rev Genet. 2004;5:366–375. doi: 10.1038/nrg1324. [DOI] [PubMed] [Google Scholar]
- Varadarajan N, Gam J, Olsen MJ, Georgiou G, Iverson BL. Engineering of protease variants exhibiting high catalytic activity and exquisite substrate selectivity. Proc Natl Acad Sci USA. 2005;102:6855–6860. doi: 10.1073/pnas.0500063102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickrey JF, Logsdon BC, Proteasa G, Palmer S, Winters MA, Merigan TC, Kovari LC. HIV-1 protease variants from 100-fold drug resistant clinical isolates: expression, purification, and crystallization. Protein Expr Purif. 2003;28:165–172. doi: 10.1016/s1046-5928(02)00650-2. [DOI] [PubMed] [Google Scholar]
- Yang Z, Bielawski JP. Statistical methods for detecting molecular adaptation. Trends Ecol Evol. 2000;15:496–503. doi: 10.1016/S0169-5347(00)01994-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano T, Oue S, Kagamiyama H. Directed evolution of an aspartate aminotransferase with new substrate specificities. Proc Natl Acad Sci USA. 1998;95:5511–5515. doi: 10.1073/pnas.95.10.5511. [DOI] [PMC free article] [PubMed] [Google Scholar]