

Abstract
Several biological processes in Trypanosoma brucei are affected by chromatin structure, including gene expression, cell cycle regulation, and lifecycle stage differentiation. In Saccharomyces cerevisiae and other organisms, chromatin structure is dependent upon posttranslational modifications of histones, which have been mapped in detail. The tails of the four core histones of T. brucei are highly diverged from those of mammals and yeasts, so sites of potential modification cannot be reliably inferred, and no cross-species antibodies are available to map the modifications. We therefore undertook an extensive survey to identify posttranslational modifications by Edman degradation and mass spectrometry. Edman analysis showed that the N-terminal alanine of H2A, H2B, and H4 can be monomethylated. We found that the histone H4 N-terminus is heavily modified, while, in contrast to other organisms, the histone H2A and H2B N-termini have relatively few modifications. Histone H3 appears to have a number of modifications at the N-terminus, but we were unable to assign many of these to a specific amino acid. Therefore, we focused our efforts on uncovering modification-states of H4. We discuss the potential relevance of these modifications.
1. Introduction
The basic unit of chromatin is the nucleosome, formed by 146 bp of DNA wrapped around an octamer containing two copies of each of the core histones, H2A, H2B, H3, and H4. The core histones contain many posttranslational modifications (PTMs), including phosphorylation, acetylation, methylation, and ubiquitination. These PTMs affect chromatin structure, which has been shown to have consequences for nucleosome assembly, DNA replication and repair, and, most notably, gene expression [1]. Histone acetylation, for example, is generally associated with transcriptionally permissive chromatin, or euchromatin [2]. N-terminal acetylation is thought to disrupt interactions that stabilize the nucleosome, making the DNA more accessible to transcription-promoting complexes [3]. Furthermore, transcription factors recruit histone acetyltransferases to the promoter region of target genes, and, accordingly, histone deacetylase activity has been detected in transcriptional co-repressor complexes [4,5]. Histone methylation also affects transcriptional activity of chromatin, but these effects are dependent upon the particular lysine that is methylated: H3 K9 and H4 K20 methylation are associated with silent chromatin, or heterochromatin, and H3 K4 and K79 methylation are associated with euchromatin [6]. In S. cerevisiae and other organisms, histone modifications clearly play a role in determining whether chromatin is in a transcriptionally permissive or restrictive configuration.
Trypanosoma brucei and its relatives are the causative agents of human Sleeping Sickness and widespread animal diseases in Africa and, to a lesser extent, other continents. This parasite evades the host immune response by spontaneously switching its Variant Surface Glycoprotein (VSG), in a process known as antigenic variation. It has been shown that chromatin structure at the subtelomeric VSG Expression Site (ES) may influence its transcriptional activity. The actively transcribed VSG demonstrates greater sensitivity to digestion with DNase I and single-strand-specific endonucleases than untranscribed VSGs, which suggested a more open chromatin conformation at the active ES [7,8]. Chromatin structure was further implicated in VSG silencing when it was shown that transcription was repressed from ES and rRNA promoters that were inserted in the silent ES, and that this effect was more pronounced as the promoters were placed closer to the telomere [9,10]. These studies suggest that VSG silencing is due to a repressive chromatin conformation.
Apart from VSG expression, several biological processes in trypanosomes involve histone PTMs. H3 K76 di- and trimethylation and the responsible histone methyltransferases, DOT1A and DOT1B, have been studied in detail in T. brucei [11]. Deletion of trypanosome DOT1 genes caused defects in cell cycle regulation and differentiation between lifecycle stages. Also, it was shown that overexpression of a histone deacetylase, T. brucei SIR2 related protein 1, resulted in increased sensitivity to DNA damaging agents [12].
Identifying histone PTMs in trypanosomes is an important first step towards studying the biological processes that are influenced by chromatin structure. Although histones are highly conserved in most organisms, the amino acid sequences of T. brucei histones are very different (Fig. 1), and we cannot infer what modifications will be present, based on sequence alignments. Commercial antibodies that recognize modifications in other organisms cannot be expected to specifically cross-react with trypanosome PTMs, and the most likely ones do not (unpublished data).
Fig. 1.

Representative sequence alignments of the N-termini of each core histone and the C-terminus of H2A. Identical residues are shaded. Important sites of homology, which are referred to in the text, are indicated by arrows. The sequence ruler, which is included only when the sequences are very concordant, is numbered according to the Homo sapiens sequence position. Residues found to be acetylated in T. brucei histones are indicated with an (*) and methylated residues with a (+). A putative site of ubiquitination at the H2A C-terminus is indicated with a (#). (A) H2A N-terminus. (B) H2A C-terminus. (C) H2B N-terminus. (D) H3 N-terminus. (E) H4 N-terminus.
Previous studies in Trypanosoma cruzi and Procyclic Form (PF) T. brucei, the insect (tsetse) midgut life-cycle stage, have identified some histone PTMs [13,14]. We add to this current knowledge by performing a detailed survey, using Edman degradation and mass spectrometry (MS), to identify as many as possible of the PTMs present on histones H2A, H2B, H3, and H4, which we purified from the Bloodstream Form (BF) of T. brucei strain Lister 427. Edman analysis provided us with quantitative data of PTMs found at the N-termini of H2A, H2B, and H4, demonstrating that H2A and H2B had relatively few modifications, while the H4 N-terminus possessed numerous modifications. Edman analysis also showed that H2A, H2B, and H4 can be monomethylated at A1, which is a histone modification that, to our knowledge, has only been found in T. brucei and T. cruzi [13,14]. Matrix-assisted laser-desorption/ionization time of flight mass spectrometry (MALDI-TOF MS) showed that the H3 and H4 N-terminal peptides are present in multiple modification-states. The H4 N-terminus was studied in greater detail by tandem MS. We conclude by comparing our findings to S. cerevisiae and other organisms.
2. Materials and Methods
2.1. Histone Purification
BF T. brucei cells of the Lister Strain 427 clone 221a were cultured in HMI-9 [15]. Approximately 3×105 cells were used to infect CD-1 female Swiss white mice. After 3 days, trypanosomes from the infected mice were used to inoculate Sprague-Dawley male rats (1–2×107 cells per rat). After a 3-day infection, trypanosomes were collected after exsanguination of the rats as previously described [16]. For purification of histones, 1×1010 cells were centrifuged at 1,800g for 10 min at 4 °C. Cells were resuspended in 20 ml 8% polyvinylpyrrolidone (containing 0.05% octyl glucoside) and lysed with a Polytron as described elsewhere [17]. Lysed cells were centrifuged at 16,000g for 5 min at 4 °C. The pellet was washed with 10 ml low salt buffer (10 mM Tris, pH 8.0; 75 mM NaCl; 0.05% octyl glucoside) and resuspended in 10 ml high salt buffer (10 mM Tris, pH 8.0; 2.0 M NaCl, 1% octyl glucoside). Octyl glucoside can be replaced by NP-40 for a better yield of histones, but NP-40 detergent contamination interfered with MS analysis. Nuclei were lysed with a Dounce homogenizer and centrifuged at 16,000g for 20 min at 4 °C in an SS-34 rotor (Sorvall). Histones from the supernatant were extracted in 0.4 M H2SO4 overnight at 4 °C. All solutions were treated with 1 mM DTT, 50 mM sodium butyrate, 0.5 mg/ml TLCK, 0.174 mg/ml PMSF, and a protease inhibitor cocktail (Sigma).
The main contaminant in this histone preparation was VSG, the most abundant protein in BF T. brucei. VSG was removed from the sample by passing it through a Concanavalin A (ConA) column. The sample was first dialyzed overnight at 4 °C in ConA equilibration buffer (20 mM Tris, pH 7.4; 0.5 M NaCl) and applied to a ConA column (Sigma). The flow-through was collected and lyophilized. The sample was fractionated by RP-HPLC on a C8 column using an acetonitrile gradient (0 to 60%) in 0.1% trifluoroacetic acid. Fractions were collected every minute for 70 minutes, lyophilized, then resuspended in 50 μl H2O. Fractions were analyzed by SDS-PAGE and MALDI-TOF.
2.2. Analysis of Histones by Edman Degradation and Mass Spectrometry
Edman degradation was performed using a Procise 494-HT with a PTH C18 HPLC column (Applied Biosystems). Modified PTH standards (acetyl or methyl lysines) were made by reacting free amino acids (Sigma) with PITC to create PTC-amino acids. These were then converted to PTH-amino acids by incubation with 25% trifluoroacetic acid.
Histones were digested with endoproteinases Glu-C, trypsin, and Asp-N, according to manufacturer’s instructions (Roche). To prevent trypsin cleavage at lysine, some histone preparations were reacted with 1% propionic anhydride (Sigma) in acetonitrile:50 mM NH4HCO3 (1:1 v/v) [18]. After propionylation, the products were dried and redissolved in water.
Digested histones were analyzed by a MALDI-TOF MS or ESI-LC-MS/MS. MALDI-TOF was conducted using a DE-STR mass spectrometer operating in delayed and reflector modes (Applied Biosystems). For ESI-LC-MS/MS, digests were injected on a 0.3 mm internal diameter by 65 mm long trapping column (PepMap; Dionex) at a flow rate of 20 μL/min (total loading time of 5 min). By switching the stream valve in the SWITCHOS (Dionex), the trapping column is back-flushed with a binary solvent gradient, which is started simultaneously with the injection cycle. The sample is thereby loaded onto a nanoscale RP C18 column (0.75 mm internal diameter by 150 mm long PepMapT column; LC Packings). Peptides were eluted from the stationary phase using a linear gradient from 0 to 75% solvent B (100% ACN in 0.1% formic acid) applied over a period of 45 min. The solvent delivery system was set at a constant flow of 20 μL/min and using a 1/1000 flow splitter, 200 nL/min of solvent was directed through the nanocolumn. The outlet of the nanocolumn was connected in-line with a distal metal-coated fused silica PicoTipT needle (PicoTipT FS360-20-10-D-C7, New Objective, Woburn, MA, USA) that is interfaced with a QSTAR XL QqTOF mass spectrometer (Applied Biosystems).
3. Results
3.1. Histone Purification
We first developed a method for purifying histones from BF T. brucei. Histones were previously purified from PF by isolating nuclei, then using acid extraction and RP-HPLC purification [14,17]. This method was attempted in BF trypanosomes, but the yield was extremely low (<1%). The greatest loss of material occurred during nuclei isolation, which we attribute to the increased fragility of the BF nucleus. Consequently, the protocol we developed (see Materials and Methods) did not involve the purification of nuclei as an intermediate step. Analysis of HPLC fractions by Coomassie staining of polyacrylamide gels and by MALDI-TOF MS demonstrated the high purity of the final histone preparations. When NP-40 was used in the protocol, our final yield of histones was approximately 30%. These samples were analyzed by Edman degradation, but NP-40 contamination of the purified histones prevented us from acquiring MS data. Substituting octyl glucoside for NP-40 prevented detergent contamination of the final samples, but resulted in a lower yield (~10%).
3.2. Edman Degradation and MS Analysis
A combination of MS techniques, including MALDI-TOF and MS/MS, and Edman degradation, were used to analyze T. brucei histones for PTMs. Edman degradation provided quantitative data for PTMs on the N-terminal 20–30 amino acids. For MS, histones were first digested with trypsin. Histones contain many arginine and lysine residues, resulting in excessive fragmentation by trypsin. In some experiments, therefore, histones were modified with propionic anhydride to prevent trypsin cleavage at lysine residues by propionylation (+56 Da) of unmodified (K) and monomethylated (Kme1) lysines, and the N-terminus. Dimethylated (Kme2), trimethylated (Kme3) and acetylated (Kac) lysines were not propionylated, but are also not cleaved by trypsin. Histone H3 was also digested with endoproteinases Asp-N and Glu-C and H4 was digested with Glu-C.
MALDI-TOF analysis provided the molecular mass of the digested peptides. Mass shifts of 14, 28, and 42 Da may represent peptides with a monomethyl, dimethyl, and trimethyl or acetyl group, respectively. The peptides were then sequenced by tandem MS to assign PTMs to their parent residue. Acetyl (42.0105 Da) and trimethyl (42.0468 Da) groups are difficult to distinguish by tandem MS. We relied on data generated by Edman degradation to help us assign mass shifts of 42 Da. Acetylated lysines also produce a characteristic immonium ion at m/z 126, and trimethylated lysines have a characteristic ion at MH+-59, both of which were used to make assignments [19].
3.3. H2A Modifications
Histones H2A and H3 co-eluted on the HPLC. We did not attempt to separate them because the presence of both histones in a single sample did not interfere with MS or Edman analysis. Only the H2A N-terminus was susceptible to Edman sequencing, because the H3 N-terminal Serine (S1) was blocked, as previously shown in PF [14]. Analysis of the first 22 amino acids of H2A demonstrated that 60% of A1 is monomethylated, and ~1% of K4 is acetylated (Table 3).
Table 3.
Summary of Histone Modifications in Bloodstream Forms
| Histone | N-terminus | Acetyl Lysine | Methyl Lysine |
|---|---|---|---|
| H2A | A1me1 (60%)a,b | K4 (1%)a | none |
| A1aca | K115b | ||
| K119b | |||
| K120b | |||
| K122b | |||
| K125b | |||
| K128b | |||
| H2B | A1me1 (60%)a,b | K4 (1%)a | none |
| A1me2b | K12b | ||
| K16b | |||
| H3 | S1acb | K23b | K32me3b |
| K76me1, K76me2, K76me3b | |||
| H4 | A1me1 (60%)a,b | K2 (2%)a,b | K2me2b |
| K4 (73%)a,b | K17me1 (4%), K17me2 (8%), K17me3 (9%)a,b | ||
| K5 (7%)a,b | |||
| K10 (7%)a,b | K18me1 (6%), K18me2 (4%), K18me3 (18%)a,b | ||
| K14 (1%)a |
PTMs observed by Edman degradation. The levels of acetylation and methylation, shown in parenthesis, were quantified by comparison to known standards.
PTMs observed by tandem MS
Kme1, Kme2, and Kme3 refer to mono-, di-, and tri-methyl lysine, respectively; Aac, Ame1, and Ame2 refer to acetyl, monomethyl, and dimethyl alanine, respectively; Sac refers to acetyl serine.
Tandem MS data were acquired for trypsin-digested H2A (Fig. 2A). Peptides that were analyzed are shown in red. Trypsin digestion produced several spectra representing modified C-terminal peptides, demonstrating that K115, K119, K120, K122, K125, and K128 can be acetylated (Table 1). We assigned these modifications as acetylation rather than trimethylation, because studies of PF histones showed that H2A exists in multiple acetylation states [14]. The presence of the immonium ion for acetylated lysine at m/z 126 supported this assignment. The MH+-59 ion was not present on these spectra. Multiple modifications of the H2A C-terminus have not been reported in other organisms.
Fig. 2.

T. brucei histone sequences with regions for which tandem MS data were acquired after endoproteinase digestion highlighted in red. (A) H2A trypsin digest. (B) H2A PA-trypsin digest. (C) H2B PA-trypsin digest. (D) H3 PA-trypsin digest. (E) H4 PA-trypsin digest.
Table 1.
Posttranslational modifications present on histones H2A, H2B, and H3
| Histone | Digest | Modified Peptide |
|---|---|---|
| H2A | Trypsin | GGVMPSLN115KacALAK (107–119) |
| GGVMPSLN115KacALA119Kac120KacQK (107–122) | ||
| ALA119Kac120KacQ122KacSGK (116–125) | ||
| SG125KacHA128KacATPSV (123–133) | ||
| HA128KacATPSV (126–133) | ||
| PA-Trypsin | 1AacTPKQAVKKASKGGSSR (1–17) | |
| 1Ame1TPKQAVKKASKGGSSR (1–17) | ||
| GGVMPSLN115KacALA119Kac120KacQKSGKHAKATPSV (107–133) | ||
| H2B | PA-Trypsin | 1Ame1TPKSTPAKTR (1–11) |
| 1Ame2TPKSTPAKTR (1–11) | ||
| 12KacEAKKTR (12–18) | ||
| KEAK16KacTR (12–18) | ||
| H3 | Trypsin | AS23KacGSDAASGVK (21–32) |
| GSDAASGV32Kme3TAQR (24–36) | ||
| PA-Trypsin | 1SacRTKETAR (1–8) | |
| EVSGAQ76Kme1EGLR (70–80) | ||
| EVSGAQ76Kme2EGLR (70–80) | ||
| EVSGAQ76Kme3EGLR (70–80) |
Histones, which in some cases were modified with propionic anhydride, were digested with trypsin. Tryptic peptides were sequenced by tandem MS, and peptides that have covalent modifications are listed here. Kac refers to acetyl lysine; Kme1, Kme2, and Kme3 refer to mono-, di-, and tri-methyl lysine, respectively; Aac, Ame1, and Ame2 refer to acetyl, monomethyl, and dimethyl alanine, respectively; Sac refers to acetyl serine.
Next, H2A was modified with propionic anhydride prior to trypsin digest (hereafter referred to as PA-trypsin digest) to acquire information about the N-terminus (Fig. 2B). MALDI-TOF analysis of the PA-trypsin-digested H2A demonstrated that the N-terminus is present in three modified states (Fig. 3A). Tandem MS sequencing of these species demonstrated that A1 can be monomethylated (Fig. 3B), acetylated, or unmodified (Table 1). A spectrum representing peptide 107–133 confirmed acetylation of K115, K119, and K120 (Table 1).
Fig. 3.
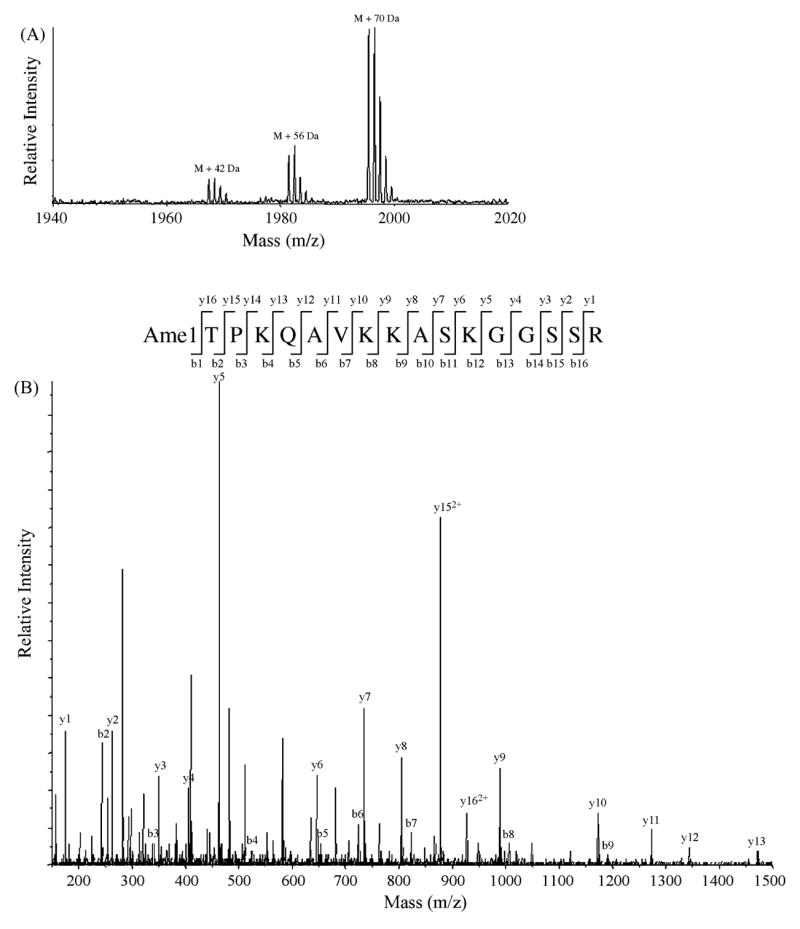
Histone H2A alanine 1 can be unmodified, acetylated, or monomethylated. H2A was propionylated and digested with trypsin. Peptide 1–17 (ATPKQAVKKASKGGSSR) was subjected to MALDI-TOF analysis and sequencing by tandem MS. (A) MALDI-TOF shows that peptide 1–17 is present in three modified states. M is the molecular mass ion of peptide 1–17 with all lysines propionylated (1924 Da). Tandem MS sequencing of peaks labeled M + 42 Da, M + 56 Da, and M + 70 Da species indicate that H2A A1 can be acetylated, propionylated (meaning it is unmodified), or both propionylated and monomethylated, respectively. Multiple peaks representing each modified peptide show the isotope pattern, with the first peak representing the monoisotopic m/z. (B) A representative tandem MS showing that H2A A1 is monomethylated. All of the b-series ions are shifted +70 Da relative to the unmodified species. Also, the difference between the MH+ of the spectrum (m/z 1995) and y16 provides the mass of the modified A1. y152+ and y162+ represent doubly charged ions [M + 2H]/[2+].
In other organisms, the C-termini of H2A and H2B are ubiquitinated [20,21]. Alignment of the H2A C-terminus (Fig. 1) suggests that T. brucei H2A K122 might be the homologue of the ubiquitinated H2A K119 in other organisms. We therefore performed a directed search for ubiquitinated H2A K122. MS can identify sites of ubiquitination by looking for a GG dipeptide linkage (114 Da) to lysine, which results from tryptic cleavage of covalently linked ubiquitin [22]. The tandem MS spectrum of a peptide with m/z 1029.263+ displayed partial y-series (y1–y6, y8, y15) and b-series (b3, b12, b22-24) ions, demonstrating that the spectrum represents propionylated H2A peptide 107–133 (data not shown). The molecular weight of this species and the y- and b-ions indicate that K115 and K128 are propionylated, and, of the remaining 4 lysines (K119, K120, K122, and K125), one may be ubiquitinated and three are acetylated.
3.4. H2B Modifications
Edman degradation of the first 25 amino acids of H2B demonstrated that 60% of A1 is monomethylated, and ~1% of K4 is acetylated (Table 3). Both H2A and H2B begin with the sequence ATPK, and both are modified at A1 and K4 to the same degree. PA-trypsin digestion of H2B provided MS/MS data for the lysine-rich N-terminus (Fig. 2C). Three spectra representing peptide 1–11 showed that A1 can be unmodified, mono-, or di-methylated (Table 1). One spectrum was obtained representing two species of peptide 12–18, which showed that K12 or K16 is acetylated (Fig. 4). These PTMs were designated as acetylated lysines because the Kac immonium ion was present at m/z 126. These marks are presumably minor, because they were not observed by Edman analysis.
Fig. 4.

Histone H2B lysines 12 and 16 are acetylated. H2B was propionylated and digested by trypsin. Peptide 12–18 (KEAKKTR) was sequenced by tandem MS. A single MS spectrum contains two modified species, demonstrating that either K12 (b2″–b3″ and y3″–y6″) or K16 (b1′–b3′ and y3′–y6′) are acetylated. An arrow points to the immonium ion of acetylated lysine (m/z 126). b72+ and y72+ represent to doubly charged ions.
3.5. H3 Modifications
Multiple attempts were made to acquire information on the modification state of H3. Many of the peptides produced by the PA-trypsin digest of H3 were sequenced by tandem MS (Fig. 2D). A spectrum corresponding to the N-terminus, peptide 1–8, showed that S1 can be acetylated (Table 1). Four spectra representing peptide 70–80 showed that K76 can be unmodified, mono-, di-, or tri-methylated (Table 1).
Quadrupole-quadrupole-TOF (QqTOF) analysis of the H3 PA-trypsin digest showed multiple modification states for the lysine-rich peptide 9–36 (Fig. 5A). Sequencing by tandem MS showed that all 8 lysines are propionylated in the species represented as M + 448 Da. Tandem MS of the three remaining peaks showed that they represent modified species of peptide 9–36, but modifications could not be assigned to specific lysines. The presence of these peaks suggests that several lysines at the H3 N-terminus can be modified.
Fig. 5.
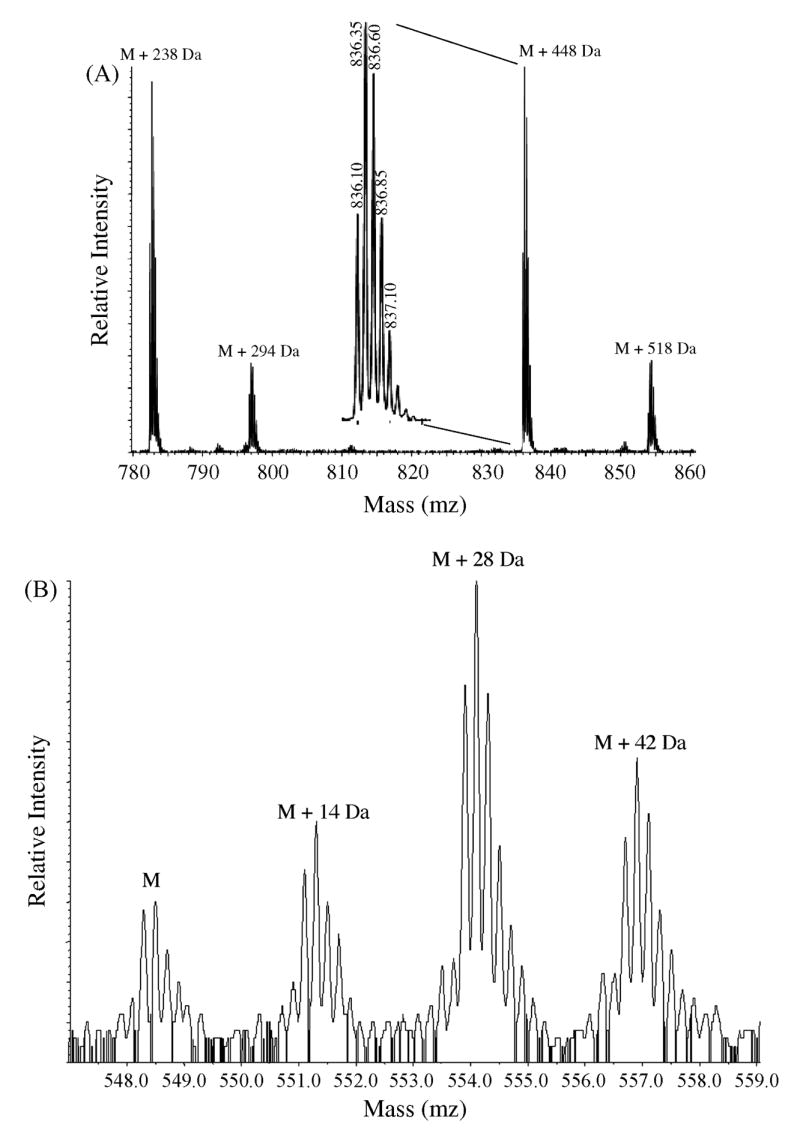
The N-terminus of histone H3 is present in multiple modification states. Quadrupole-quadrupole-TOF data were acquired for H3 digested with endoproteinases trypsin and Asp-N. M is the molecular ion mass of each peptide. (A) Propionylated H3 digested with trypsin produces peptide 9–36 (TKKTITSKKSKKASKGSDAASGVKTAQR), which was observed as [MH4]4+(m/z 723.9). Sequencing by tandem MS of the peptide labeled M + 448 Da shows that this is the species for which all 8 lysines are propionylated. Magnification of peak M + 448 Da (inset) shows the isotope pattern of this peptide, which enabled us to determine that this is a +4 species. Partial sequencing by tandem MS of the remaining three peaks confirms that they correspond to peptide 9–36, but modifications could not be assigned to specific parent residues. Based on the possible combinations allowed by the mass of the modified peptide, we speculate that M + 238 Da is a species that possesses 1 trimethyl (or acetyl) and 7 dimethyl lysines. We also speculate that M + 518 Da is a species that possesses either 5 monomethyl lysines or 1 trimethyl (or acetyl) and 6 monomethyl lysines. (B) Asp-N-digested H3 produces peptide 1–25 (SRTKETARTKKTITSKKSKKASKGS), which was observed at m/z 548.3 for [MH5]5+. Sequencing by tandem MS confirms that the peak labeled M is unmodified peptide 1–25. Partial sequencing by tandem MS of the three modified species M + 14 Da, M + 28 Da, and M + 42 Da confirms that they correspond to peptide 1–25, but modifications could not be assigned to specific parent residues.
Although the N-terminus of H3 was blocked to analysis by Edman degradation, we attempted to acquire Edman degradation data by separating the peptides from the PA-trypsin digest by HPLC and sequencing peptide 9–36. A weak profile was obtained, but no PTMs were detectable.
Tandem MS sequencing of trypsin-digested H3 uncovered two modifications at the N-terminus — K23ac and K32me3 (Table 1). K23 was shown to be acetylated by the presence of the immonium ion at m/z 126. K32 was shown to be methylated by the presence of the MH+-59 ion. Finally, we attempted MS analysis of Asp-N- and Glu-C-digested H3. No interpretable tandem MS data were acquired, although Qq-TOF analysis of Asp-N-digested H3 confirmed that multiple modification-states exist for N-terminal peptide 1–25 (Fig. 5B). We concluded our study of H3 with the knowledge that PTMs were present at the N-terminus that could not be assigned to specific amino acids.
3.6. H4 Modifications
Edman degradation of the first 22 amino acids showed that, unlike H2A and H2B, the H4 N-terminus has numerous PTMs (Table 3). K2, K5, K10, and K14, are acetylated in less than 10% of H4. K4ac is an abundant mark that is found in 73% of H4. Both K17 and K18 can be mono-, di-, or tri-methylated and 60% of A1 is monomethylated.
We examined H4 in more detail by MS, to confirm the PTMs observed by Edman as well as to determine whether less abundant PTMs were present. H4 Glu-C, trypsin-, and PA-trypsin-digested peptides were fractionated by HPLC, and individual fractions were analysed by MALDI-TOF. Peptides were then sequenced by tandem MS.
PA-trypsin-digested H4 had extensive sequence coverage by tandem MS (Fig. 2F). The data did not suggest the presence of additional PTMs in the core domain or C-terminus of H4. MALDI-TOF demonstrated multiple modification states for peptides 1–15 and 16–21 (Table 2 and data not shown). Sequencing of peptide 1–15 by tandem MS revealed a modification, K2me2, that was not observed by Edman degradation. We also confirmed that K2, K4, and K10 can be acetylated. Tandem MS analysis of peptide 16–21 demonstrated that K17 is mono- and tri-methylated, and K18 is mono-, di-, and tri-methylated (Table 2). Tandem MS spectra representing peptide 16–21 were initially difficult to interpret because the mass of the peptide was 17 Da less than the theoretical mass. Closer examination of these spectra demonstrate that glutamine 16 (Q16) forms a cyclic compound between its free amine and amide carbon, resulting in the loss of ammonia and consequent molecular weight decrease of 17 Da. The cyclization of glutamine to form pyroglutamic acid has been characterized previously by MS [23].
Table 2.
Posttranslational modifications are present at the N-terminus of Histone H4
| Digest | Modified Peptide |
|---|---|
| Glu-C | 1Ame1KG4KacKSGE (1–8) |
| A10KacGSQKRQKKVLRE (9–22) | |
| AKGSQKRQ17Kme3KVLRE (9–22) | |
| A10KacGSQKRQK18Kme1VLRE (9–22) | |
| AKGSQKRQ17Kme318Kme2VLRE (9–22) | |
| AKGSQKRQ17Kme318Kme3VLRE (9–22) | |
| A10KacGSQKRQ17Kme318Kme3VLRE (9–22) | |
| Trypsin | 5KacSGEA10KacGSQK (5–14) |
| PA-Trypsin | AKG4KacKSGEAKGSQKR (1–15) |
| 1Ame1KGKKSGEAKGSQKR (1–15) | |
| 1Ame1KG4KacKSGEAKGSQKR (1–15) | |
| 1Ame12KacG4KacKSGEAKGSQKR (1–15) | |
| 1Ame12KacG4KacKSGEA10KacGSQKR (1–15) | |
| 1Ame12Kme2G4KacKSGEAKGSQKR (1–15) | |
| Q17Kme1KVLR (16–21) | |
| QK18Kme1VLR (16–21) | |
| QK18Kme2VLR (16–21) | |
| Q17Kme3KVLR (16–21) | |
| QK18Kme3VLR (16–21) | |
| Q17Kme118Kme1VLR (16–21) |
N-terminal peptides resulting from each digest were sequenced by tandem MS. Peptides that have covalent modifications are listed here. Kac refers to acetyl lysine; Kme1, Kme2, and Kme3 refer to mono-, di-, and tri-methyl lysine, respectively; Ame1 refers to monomethyl alanine.
Sequencing by tandem MS of trypsin-digested H4 provided minimal information due to excessive fragmentation of the N-terminus. We discovered one modified species at the H4 N-terminus, demonstrating that K5 and K10 are acetylated (Table 2).
MS analysis of Glu-C-digested H4 was focused on N-terminal peptides 1–8 and 9–22. We were unable to acquire MALDI-TOF data for peptide 1–8 and found only one modified species by tandem MS, demonstrating that A1 is monomethylated and K4 is acetylated (Table 2). Edman degradation showed that this is the most abundant species of peptide 1–8. MALDI-TOF analysis of the fractionated Glu-C digest showed that peptide 9–22 is present in multiple modification-states (Fig. 6). Tandem MS sequencing of peptide 9–22 illustrated the combinations in which K10ac, K17me, and K18me may be present (Table 2). We were unable to sequence all of the modified species of peptide 9–22, so PTMs for the following species were not assigned: M + 14 Da, M + 28 Da, M + 98 Da, and M + 112 Da. The significance of these PTMs and the combinations in which they are present is currently unknown, but functional studies of individual marks and their corresponding histone-modifying enzymes will enable us to interpret this data in a meaningful way in the future.
Fig. 6.

Histone H4 peptide 9–22 is present in multiple modification states. H4 digested by endoproteinase Glu-C was fractionated by HPLC. MALDI-TOF data were acquired for 5 consecutive HPLC fractions. Sequencing by tandem MS confirmed that these peaks correspond to peptide 9–22 (AKGSQKRQKKVLRE). Peaks are separated by 14 Da, the molecular mass of a methyl group. M is the molecular ion mass of peptide 9–22 (1655 Da).
4. Discussion
In this study, we attempted to identify the major covalent modifications present on the four core histones of BF T. brucei. All of the identified PTMs and the methods used to discover them are summarized in Table 3. The N-terminus of H3 was blocked to Edman degradation, and minimal tandem MS data were acquired for modified N-terminal peptides. However, Qq-TOF data of the H3 PA-trypsin and Asp-N digests suggested that several lysines at the H3 N-terminus can be modified. MALDI-TOF data of the PF H3 N-terminus also suggested that a number of PTMs were present, although no tandem MS data were acquired to assign PTMs to their specific amino acids. Sequencing of trypsin-digested H3 by tandem MS showed that K23 is acetylated and K32 is trimethylated. T. brucei K23 and K32 are the possible homologues of K27 and K36 in other organisms (Fig. 1D), where their modifications are important for gene regulation. There are also predicted homologues of modified lysines 18, 23, 27, and 36 (T. brucei K16, K19, K23, and K32), indicating several possible marks for future study.
Next we investigated the PTMs on T. brucei histone H4. It has been shown that the acetylation of the S. cerevisiae H4 N-terminus at K5, K8, K12, and K16 is classically associated with transcriptional activation [2]. According to our alignment, S. cerevisiae K5, K12, and K16 could correspond to T. brucei K4, K10, and K14, all of which are acetylated (Fig. 1E). Although the T. brucei H4 sequence diverges considerably from the extremely conserved H4 N-terminus of other organisms, 3 of the 4 acetylated lysines appear to be conserved.
Importantly, in addition to the conserved acetylated lysines found at the H4 N-terminus, there is an apparent homologue of the methylated H4 K20, T. brucei H4 K18 (Fig. 1E). Trimethyl H4 K20 is a marker of constitutive and facultative heterochromatin and is in competition with the hyperacetylated H4 N-terminus [24]. Monomethyl H4 K20 is present at active gene promoters in association with H4 hyperacetylation, which demonstrates that the degree of methylation may be associated with distinct transcriptional states [25]. We found that T. brucei H4 K18 and the adjacent K17 may both be mono-, di-, or tri-methylated. K17 and K18 exist together in several modification states (Table 3). It is unlikely that these different methylation levels are created by different methyltransferases because there are only three identifiable putative SET domain-containing methyltransferases in the T. brucei genome [26]. Instead, it is more likely that a single methyltransferase acts on both K17 and K18, and the degree of methylation is determined by cofactors acting on the methyltransferase. We cannot predict whether the degree of methylation or the combination of modification states in which K17 and K18 co-exist will result in distinct biological readouts. If so, the amount of variability would allow for intricately regulated downstream effects.
Two modified lysines in the T. brucei H4 N-terminus, K2 and K5, did not have homologues in other organisms. We have shown that H4 K2 can be either acetylated or dimethylated. Histones from other organisms possess a number of lysines that may be either acetylated or methylated as well. Chicken H3 K9, for example, is subject to both types of covalent modification: H3K9me3 is associated with inactive chromatin, whereas H3K9ac is found at the boundaries of heterochromatin [27]. These marks have antagonistic roles in the establishment of different chromatin states. Whether T. brucei H4 K2 is involved in determining chromatin structure is unknown, but we propose that the dual modification status of this lysine makes it an interesting target for further characterization.
The H2A C-terminus contains 6 acetylated lysines (K115, K119, K120, K122, K125, and K128). Three of these, K120, K122, and K128, are probable homologues of lysines in other organisms, but the homologues of T. brucei K120 and K128 do not appear to be modified (Fig. 1B). T. brucei H2A K122 aligns with human H2A K119, the site of ubiquitination on this histone [21]. In other organisms, H2A ubiquitination is associated with transcriptional silencing [28,29]. We found evidence that T. brucei H2A C-terminus is ubiquitinated — the molecular weight of an H2A peptide 107–133 corresponds to a modified species that has one ubiquitinated and three acetylated lysines. Interestingly, T. brucei K122 is the only lysine at the H2A C-terminus that is adjacent to a possible phosphorylation site, S123. In other organisms, ubiquitinated lysines at the H2A and H2B C-termini are adjacent to phospho-acceptor sites. It has been suggested that phosphorylation influences ubiquitination and de-ubiquitination of its neighboring lysine and also influences whether effector proteins bind ubiquitinated lysine [30]. Based on our data and the alignment of the H2A C-terminus with the C-terminus of H2A from other organisms, we propose that T. brucei H2A K122 can be ubiquitinated.
Two recent studies identifying covalent histone modifications in T. cruzi and PF T. brucei were published recently [13,14]. Both studies uncovered H4 acetylation at K4 and K10 (and K14 in T. cruzi) and monomethylation at K18 (and mono- and dimethylation at K17 and trimethylation at K18 in PF T. brucei). Additionally, our study detected H4 acetylation at K2 and K5, dimethylation at K2, trimethylation at K17, and dimethylation at K18. Perhaps these marks reflect lifecycle stage variation in BF T. brucei. The same may be said for the acetylated lysines found in BF H2A (K4 and K122) and H2B (K4, K12, and K16), which were not detected in PF. Finally, the T. cruzi H4 study detected K57 acetylation and R53 dimethylation. After a thorough analysis of T. brucei H4, we conclude that these marks are not present and may reflect species-specific variation of histone PTMs.
In higher eukaryotes, histones are highly conserved proteins. The extreme sequence divergence of T. brucei histones makes them interesting targets for phylogenetic comparisons. We demonstrate that, despite the sequence divergence, the locations of several modified residues are similar. The information compiled here provides a number of starting points for functional studies, some of which we are beginning to undertake. Future work will focus on identifying the modifications that play key roles in transcriptional regulation in trypanosomes.
Acknowledgments
We are grateful to several members of the Cross lab, especially Luisa Figueredo, Nicolai Siegel, and Eiji Okubo, for editorial comments on the manuscript, and Jenny Li for technical assistance. We thank David Allis and Brian Chait (Rockefeller University), and Kirk Deitsch (Weill Medical College of Cornell University), for their advice and discussions. This work was supported by NIH grants AI21729 and GM07739.
Abbreviations
- BF
bloodstream-form
- ES
expression site
- MALDI-TOF
matrix-assisted laser-desorption/ionization time of flight
- MS
mass spectrometry
- PF
procyclic form
- PTM
posttranslational modification
- VSG
variant surface glycoprotein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fischle W, Wang Y, Allis CD. Histone and chromatin cross-talk. Curr Opin Cell Biol. 2003;15:172–83. doi: 10.1016/s0955-0674(03)00013-9. [DOI] [PubMed] [Google Scholar]
- 2.Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997;389:349–52. doi: 10.1038/38664. [DOI] [PubMed] [Google Scholar]
- 3.Brower-Toland B, Wacker DA, Fulbright RM, Lis JT, Kraus WL, Wang MD. Specific contributions of histone tails and their acetylation to the mechanical stability of nucleosomes. J Mol Biol. 2005;346:135–46. doi: 10.1016/j.jmb.2004.11.056. [DOI] [PubMed] [Google Scholar]
- 4.Kuo MH, vom Baur E, Struhl K, Allis CD. Gcn4 activator targets Gcn5 histone acetyltransferase to specific promoters independently of transcription. Mol Cell. 2000;6:1309–20. doi: 10.1016/s1097-2765(00)00129-5. [DOI] [PubMed] [Google Scholar]
- 5.Kurdistani SK, Grunstein M. Histone acetylation and deacetylation in yeast. Nat Rev Mol Cell Biol. 2003;4:276–84. doi: 10.1038/nrm1075. [DOI] [PubMed] [Google Scholar]
- 6.Sims RJ, 3rd, Nishioka K, Reinberg D. Histone lysine methylation: a signature for chromatin function. Trends Genet. 2003;19:629–39. doi: 10.1016/j.tig.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 7.Pays E, Lheureux M, Steinert M. The expression-linked copy of a surface antigen gene in Trypanosoma brucei is probably the one transcribed. Nature. 1981;292:265–7. doi: 10.1038/292265a0. [DOI] [PubMed] [Google Scholar]
- 8.Greaves DR, Borst P. Trypanosoma brucei variant-specific glycoprotein gene chromatin is sensitive to single-strand-specific endonuclease digestion. J Mol Biol. 1987;197:471–83. doi: 10.1016/0022-2836(87)90559-6. [DOI] [PubMed] [Google Scholar]
- 9.Horn D, Cross GAM. A developmentally regulated position effect at a telomeric locus in Trypanosoma brucei. Cell. 1995;83:555–61. doi: 10.1016/0092-8674(95)90095-0. [DOI] [PubMed] [Google Scholar]
- 10.Horn D, Cross GAM. Position-dependent and promoter-specific regulation of gene expression in Trypanosoma brucei. EMBO J. 1997;16:7422–31. doi: 10.1093/emboj/16.24.7422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Janzen CJ, Hake SB, Lowell JE, Cross GAM. Selective di- or trimethylation of histone H3 lysine 76 by two DOT1 homologs is important for cell cycle regulation in Trypanosoma brucei. Mol Cell. 2006;23:497–507. doi: 10.1016/j.molcel.2006.06.027. [DOI] [PubMed] [Google Scholar]
- 12.Garcia-Salcedo JA, Gijon P, Nolan DP, Tebabi P, Pays E. A chromosomal SIR2 homologue with both histone NAD-dependent ADP-ribosyltransferase and deacetylase activities is involved in DNA repair in Trypanosoma brucei. EMBO J. 2003;22:5851–62. doi: 10.1093/emboj/cdg553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.da Cunha JP, Nakayasu ES, de Almeida IC, Schenkman S. Post-translational modifications of Trypanosoma cruzi histone H4. Mol Biochem Parasitol. 2006;150:268–77. doi: 10.1016/j.molbiopara.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 14.Janzen CJ, Fernandez JP, Deng H, Diaz R, Hake SB, Cross GAM. Unusual histone modifications in Trypanosoma brucei. FEBS Lett. 2006;580:2306–10. doi: 10.1016/j.febslet.2006.03.044. [DOI] [PubMed] [Google Scholar]
- 15.Hirumi H, Hirumi K. Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J Parasitol. 1989;75:985–9. [PubMed] [Google Scholar]
- 16.Taylor AE, Lanham SM, Williams JE. Influence of methods of preparation on the infectivity, agglutination, activity, and ultrastructure of bloodstream trypanosomes. Exp Parasitol. 1974;35:196–208. doi: 10.1016/0014-4894(74)90023-x. [DOI] [PubMed] [Google Scholar]
- 17.Rout MP, Field MC. Isolation and characterization of subnuclear compartments from Trypanosoma brucei. Identification of a major repetitive nuclear lamina component. J Biol Chem. 2001;276:38261–71. doi: 10.1074/jbc.M104024200. [DOI] [PubMed] [Google Scholar]
- 18.Jin M, Bateup H, Padovan JC, Greengard P, Nairn AC, Chait BT. Quantitative analysis of protein phosphorylation in mouse brain by hypothesis-driven multistage mass spectrometry. Anal Chem. 2005;77:7845–51. doi: 10.1021/ac051519m. [DOI] [PubMed] [Google Scholar]
- 19.Zhang K, Yau PM, Chandrasekhar B, New R, Kondrat R, Imai BS, Bradbury ME. Differentiation between peptides containing acetylated or tri-methylated lysines by mass spectrometry: an application for determining lysine 9 acetylation and methylation of histone H3. Proteomics. 2004;4:1–10. doi: 10.1002/pmic.200300503. [DOI] [PubMed] [Google Scholar]
- 20.West MH, Bonner WM. Histone 2B can be modified by the attachment of ubiquitin. Nucleic Acids Res. 1980;8:4671–80. doi: 10.1093/nar/8.20.4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goldknopf IL, Busch H. Isopeptide linkage between nonhistone and histone 2A polypeptides of chromosomal conjugate-protein A24. Proc Natl Acad Sci U S A. 1977;74:864–8. doi: 10.1073/pnas.74.3.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chu F, Nusinow DA, Chalkley RJ, Plath K, Panning B, Burlingame AL. Mapping post-translational modifications of the histone variant MacroH2A1 using tandem mass spectrometry. Mol Cell Proteomics. 2006;5:194–203. doi: 10.1074/mcp.M500285-MCP200. [DOI] [PubMed] [Google Scholar]
- 23.Khandke KM, Fairwell T, Chait BT, Manjula BN. Influence of ions on cyclization of the amino terminal glutamine residues of tryptic peptides of streptococcal PepM49 protein. Resolution of cyclized peptides by HPLC and characterization by mass spectrometry. Int J Pept Protein Res. 1989;34:118–23. doi: 10.1111/j.1399-3011.1989.tb01499.x. [DOI] [PubMed] [Google Scholar]
- 24.Sarg B, Helliger W, Talasz H, Koutzamani E, Lindner HH. Histone H4 hyperacetylation precludes histone H4 lysine 20 trimethylation. J Biol Chem. 2004;279:53458–64. doi: 10.1074/jbc.M409099200. [DOI] [PubMed] [Google Scholar]
- 25.Talasz H, Lindner HH, Sarg B, Helliger W. Histone H4-lysine 20 monomethylation is increased in promoter and coding regions of active genes and correlates with hyperacetylation. J Biol Chem. 2005;280:38814–22. doi: 10.1074/jbc.M505563200. [DOI] [PubMed] [Google Scholar]
- 26.Ivens AC, Peacock CS, Worthey EA, Murphy L, Aggarwal G, Berriman M, Sisk E, Rajandream MA, Adlem E, Aert R, Anupama A, Apostolou Z, Attipoe P, Bason N, Bauser C, Beck A, Beverley SM, Bianchettin G, Borzym K, Bothe G, Bruschi CV, Collins M, Cadag E, Ciarloni L, Clayton C, Coulson RM, Cronin A, Cruz AK, Davies RM, De Gaudenzi J, Dobson DE, Duesterhoeft A, Fazelina G, Fosker N, Frasch AC, Fraser A, Fuchs M, Gabel C, Goble A, Goffeau A, Harris D, Hertz-Fowler C, Hilbert H, Horn D, Huang Y, Klages S, Knights A, Kube M, Larke N, Litvin L, Lord A, Louie T, Marra M, Masuy D, Matthews K, Michaeli S, Mottram JC, Muller-Auer S, Munden H, Nelson S, Norbertczak H, Oliver K, O’Neil S, Pentony M, Pohl TM, Price C, Purnelle B, Quail MA, Rabbinowitsch E, Reinhardt R, Rieger M, Rinta J, Robben J, Robertson L, Ruiz JC, Rutter S, Saunders D, Schafer M, Schein J, Schwartz DC, Seeger K, Seyler A, Sharp S, Shin H, Sivam D, Squares R, Squares S, Tosato V, Vogt C, Volckaert G, Wambutt R, Warren T, Wedler H, Woodward J, Zhou S, Zimmermann W, Smith DF, Blackwell JM, Stuart KD, Barrell B, Myler PJ. The genome of the kinetoplastid parasite, Leishmania major. Science. 2005;309:436–42. doi: 10.1126/science.1112680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Litt MD, Simpson M, Gaszner M, Allis CD, Felsenfeld G. Correlation between histone lysine methylation and developmental changes at the chicken beta-globin locus. Science. 2001;293:2453–5. doi: 10.1126/science.1064413. [DOI] [PubMed] [Google Scholar]
- 28.Baarends WM, Hoogerbrugge JW, Roest HP, Ooms M, Vreeburg J, Hoeijmakers JH, Grootegoed JA. Histone ubiquitination and chromatin remodeling in mouse spermatogenesis. Dev Biol. 1999;207:322–33. doi: 10.1006/dbio.1998.9155. [DOI] [PubMed] [Google Scholar]
- 29.Wang H, Wang L, Erdjument-Bromage H, Vidal M, Tempst P, Jones RS, Zhang Y. Role of histone H2A ubiquitination in Polycomb silencing. Nature. 2004;431:873–8. doi: 10.1038/nature02985. [DOI] [PubMed] [Google Scholar]
- 30.Fischle W, Wang Y, Allis CD. Binary switches and modification cassettes in histone biology and beyond. Nature. 2003;425:475–9. doi: 10.1038/nature02017. [DOI] [PubMed] [Google Scholar]