

Abstract
We develop the theory for computing the joint frequency spectra of alleles in two closely related species. We allow for arbitrary population growth in both species after they had a common ancestor. We focus on the case in which a single chromosome is sequenced from one of the species. We use classical diffusion theory to show that, if the ancestral species was at equilibrium under mutation and drift and a chromosome from one of the descendant species carries the derived allele, the frequency spectrum in the other species is uniform, independently of the demographic history of both species. We also predict the expected densities of segregating and fixed sites when the chromosome from the other species carries the ancestral allele. We compare the predictions of our model with the site-frequency spectra of SNPs in the four HapMap populations of humans when the nucleotide present in the Neanderthal DNA sequence is ancestral or derived, using the chimp genome as the outgroup.
RECENTLY separated species may share alleles that were present in their common ancestor. If trans-species polymorphism is likely, then allele frequencies in the two species are not independent. Instead, they are correlated because of alleles that arose in the common ancestor. In this article we develop the theory of the joint frequency spectra in two species, focusing on the case of neutral alleles when a single chromosome is sampled from one of the species. We compare the predictions of our theory to data from the HapMap project and from the Neanderthal genomic sequence published recently by Green et al. (2006).
The allele-frequency or site-frequency spectrum, hereafter called the frequency spectrum, is being used increasingly for the analysis of genomic data. We follow tradition and use the term allele when developing the theory. When discussing the Neanderthal data, each polymorphic site in humans is regarded as a locus and each polymorphic nucleotide as an allele. The underlying assumption of the frequency spectrum is that mutation is irreversible. Alleles that are polymorphic are only transiently so and hence the frequency distribution of any single allele cannot reach an equilibrium. However, the ensemble of polymorphic loci together can be characterized by a frequency spectrum, defined to be the number of polymorphic loci among all loci sampled at which alleles are found at a specified frequency or within a specified frequency range. In a population of constant size and with constant selection coefficients, the frequency spectrum can attain an equilibrium.
The frequency spectrum is of importance because it provides a way to combine information across a large number of loci. The frequency spectrum when n chromosomes are sampled is a set of n − 1 summary statistics for which considerable population genetics theory is available. The frequency spectrum does not make use of information about haplotype structure or linkage disequilibrium because loci are treated as being unlinked. The frequency spectrum of all loci together allows detailed examination of the effects of demographic history, while considering subsets of loci allows testing for selection on those subsets.
The theory of the equilibrium frequency spectrum traces to classical articles by Fisher (1930), Wright (1938), and Kimura (1964, 1969). Sawyer and Hartl (1992) developed a method based on Poisson random fields for the purpose of estimating selection intensities and mutation rates from observed frequency spectra. Bustamante et al. (2001) tested the efficacy of the Sawyer–Hartl method when sites are closely linked. Griffiths (2003) summarized the equilibrium theory of the frequency spectrum and extended it to allow for arbitrary fluctuations in population size in the case of neutral alleles and in special cases of selected nucleotides. Williamson et al. (2005) generalized the Sawyer–Hartl method to allow for stepwise changes in population size and applied their method to a large human data set. Williamson et al. (2005) allowed for past population growth by comparing neutral sites with other classes of sites and inferred which loci have been subject to recent selection in modern humans. Evans et al. (2007) extended classical diffusion theory to allow for arbitrary variation in population size and selection intensity with time.
One potential problem with using observed frequency spectra in humans to estimate population genetic parameters is that most data currently available are subject to ascertainment bias of an unknown extent. Wakeley et al. (2001), Clark et al. (2005), and others have suggested ways to take ascertainment into account when estimating parameters. Both DNA sequencing error and sequence changes resulting from degradation of ancient DNA can also affect parameter estimation. Johnson and Slatkin (2006) developed a likelihood method for allowing for sequencing error when using the frequency spectrum to estimate mutation and population growth rates.
In this article, we explore the effect on the frequency spectrum of having additional information available, namely that one chromosome from a closely related species and one from an outgroup are sampled. The outgroup chromosome allows us to infer which allele is ancestral, while the chromosome from the more closely related species allows us to understand recent changes in allele frequency. We develop the basic theory here in as simple a context as possible to demonstrate that additional information is available when even a single chromosome from a closely related species is sampled. We are not concerned here with the inference of population genetic parameters.
The theory presented here is based on classical diffusion theory and the extension to it by Evans et al. (2007). The joint frequency spectra of neutral alleles could also be obtained from the coalescent model of Wakeley and Hey (1997) or by Monte Carlo simulation (Hudson 2002). The analysis in terms of diffusion theory is simpler mathematically and can incorporate natural selection with only minor modification.
Our theory was motivated by the recent publication of nuclear sequences from a Neanderthal (Green et al. 2006; Noonan et al. 2006). Mitochondrial DNA sequences (mtDNA) from several Neanderthals lie outside the clade of modern human mtDNA sequences. Green et al. (2006) concluded that the mtDNA from the bone they analyzed, which provides the most extensive Neanderthal mtDNA sequence available, had a most recent common ancestor with modern humans between 416,000 and 825,000 years ago.
The nuclear DNA sequence data from Neanderthals raise many questions. Have differences between humans and chimpanzees arisen before or after the modern human lineage separated from the lineage leading to Neanderthals? Is there evidence of secondary contact of Neanderthals and modern humans during the 70,000 years they coexisted in Europe? Can knowledge of the Neanderthal genome help us understand the history of population growth and population subdivision of modern humans since divergence of the Neanderthal lineage? To answer these and other questions new theory will be needed. The theory developed here provides an analytic basis for studying the joint frequency spectrum and allows the easy exploration of the range of possibilities consistent with the recent evolution of closely related species. Although as presented it does not directly permit hypothesis testing and parameter estimation, it can serve as the basis for developing that theory.
THEORY
Joint spectra:
We assume data are available from three species. The model is tailored to the problem of interpreting data from modern humans (species 1), Neanderthals (species 2), and chimpanzees (species 3), as illustrated in Figure 1. We assume species 1 and 2 diverged recently enough that neutral loci in both species have a significant chance of being polymorphic for alleles that were present in their most recent common ancestor (node 4 in Figure 1). We assume the common ancestor of species 1 and 2 diverged from the ancestor of species 3 long enough ago in the past that neutral alleles polymorphic in the common ancestor of all three species (node 5 in Figure 1) were lost or fixed before the divergence of species 1 and 2. In other words, trans-species polymorphism of neutral alleles is possible between species 1 and 2 but not possible between species 1 and 3 or 2 and 3.
Figure 1.—

Illustration of the three-species tree assumed and the two types of SNPs analyzed. Node 4 represents the most recent common ancestor of Neanderthals and modern humans. Node 5 represents the most recent common ancestor of modern humans, Neanderthals, and chimpanzees (and bonobos, which are not represented). The top SNP is 2-ancestral (N-ancestral) because G is assumed to be the ancestral nucleotide present at node 5. The bottom SNP is 2-derived (N-derived) because A is assumed to be the ancestral nucleotide.
We assume there is no recurrent mutation and that the allele on the chromosome from species 3 is ancestral. Species 1 is polymorphic for the ancestral allele and a derived allele that arose by mutation since the three species had a common ancestor (node 5 in Figure 1). The theoretical problem is to predict the frequency spectrum of derived alleles in species 1 when the chromosome from species 2 has the ancestral or derived allele.
We assume a sample of n chromosomes is chosen randomly from species 1. The frequency spectrum is the density of loci at which i chromosomes carry the derived allele, fi (0 < i < n). If K loci are typed on each chromosome, Kfi is the expected number of loci for which the derived allele is on i chromosomes and  is the expected number of polymorphic loci, i.e., the expected number of segregating sites. If the chromosome from species 2 has the ancestral allele, we call the spectrum in species 1 the 2-ancestral spectrum and denote it by
is the expected number of polymorphic loci, i.e., the expected number of segregating sites. If the chromosome from species 2 has the ancestral allele, we call the spectrum in species 1 the 2-ancestral spectrum and denote it by 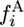 . If the chromosome from species 2 has the derived allele, we call the spectrum in species 1 the 2-derived spectrum and denote it by
. If the chromosome from species 2 has the derived allele, we call the spectrum in species 1 the 2-derived spectrum and denote it by 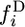 . The whole population is characterized by the continuous spectra,
. The whole population is characterized by the continuous spectra, 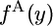 and
and 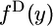 , where y is the frequency of the derived allele in species 1. If the population is sufficiently large that sampling with replacement can be assumed, then
, where y is the frequency of the derived allele in species 1. If the population is sufficiently large that sampling with replacement can be assumed, then
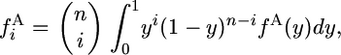 |
(1) |
with a similar expression relating 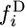 to
to 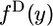 (Griffiths 2003). The unconditional spectra are
(Griffiths 2003). The unconditional spectra are 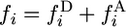 and
and 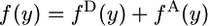 .
.
At t = 0, species 1 and 2 had a common ancestor (node 4 in Figure 1). We assume speciation was instantaneous: at t = 0, a single population containing N0 individuals split into two noninterbreeding populations each of which initially contained N0 individuals. We will see that, for neutral alleles, the population size of species 2 after t = 0 does not matter. The population size of species 1 is denoted by N(t), 0 ≤ t ≤ T, where time T is the present. The frequency spectrum of derived alleles at t = 0 is 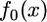 . We refer to derived alleles in the common ancestor as old alleles and derived alleles that arose by mutation in species 1 after t = 0 as new alleles.
. We refer to derived alleles in the common ancestor as old alleles and derived alleles that arose by mutation in species 1 after t = 0 as new alleles.
Let x be the frequency of the derived allele in the common ancestor (node 4), y be the frequency in species 1 at T, and z be the frequency in species 2 at T. Given x at t = 0, the distribution of y is 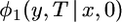 , where φ1 is the solution to the forward diffusion equation that describes the effects of genetic drift in the absence of mutation. The distribution in species 2,
, where φ1 is the solution to the forward diffusion equation that describes the effects of genetic drift in the absence of mutation. The distribution in species 2, 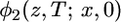 , may differ because of differences in the history of population size in the two species. Selection can be incorporated into the diffusion equation but we consider only neutral alleles here. The joint spectrum at T is obtained by averaging over x :
, may differ because of differences in the history of population size in the two species. Selection can be incorporated into the diffusion equation but we consider only neutral alleles here. The joint spectrum at T is obtained by averaging over x :
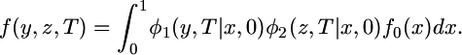 |
(2) |
The probability that a single chromosome sampled from species 2 carries the derived allele is z. Therefore,
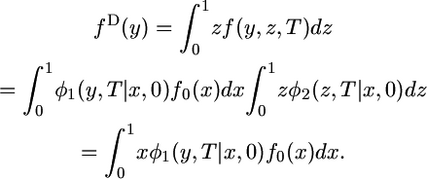 |
(3) |
The last step uses the fact that the expected frequency of the derived allele does not change under genetic drift alone, which implies that the expectation of z is x independently of the history of population growth in species 2.
If the ancestral population was at equilibrium under drift and mutation, 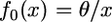 , where
, where 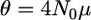 and μ is the mutation rate (Griffiths 2003). In that case, the last integral in Equation 3 reduces to
and μ is the mutation rate (Griffiths 2003). In that case, the last integral in Equation 3 reduces to
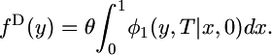 |
(4) |
Kimura (1955) provided the analytic solution for φ for a population of constant size. When the population varies in size, Kimura's solution can be written
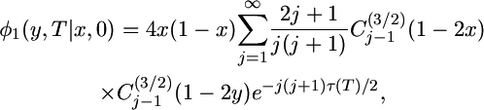 |
(5) |
where 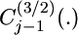 is the Gegenbauer polynomial of order j − 1 (Abramowitz and Stegun 1965), and
is the Gegenbauer polynomial of order j − 1 (Abramowitz and Stegun 1965), and
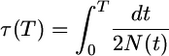 |
(6) |
The orthogonality of Gegenbauer polynomials implies
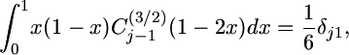 |
(7) |
where 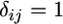 if i = j and 0 otherwise (Abramowitz and Stegun 1965). Therefore, when the right-hand side of Equation 5 is integrated term by term, only the j = 1 term is nonzero, implying
if i = j and 0 otherwise (Abramowitz and Stegun 1965). Therefore, when the right-hand side of Equation 5 is integrated term by term, only the j = 1 term is nonzero, implying
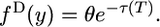 |
(8) |
Substituting this result into Equation 1 and integrating gives
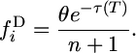 |
(9) |
Both the discrete and the continuous spectra of 2-derived alleles are uniform, independently of the history of population sizes in both species.
The intuitive reason for such a simple result is that, because the expected frequency of a neutral allele does not change with time, finding a derived allele on the chromosome from species 2 provides the same information that is provided if we know that a SNP has been ascertained by testing a single chromosome in species 1 for the presence of a derived allele: the probability that an allele in frequency y is ascertained is y and the equilibrium spectrum is 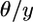 (Nielsen 2000). Multiplying results in cancellation of y and implies that the equilibrium spectrum with ascertainment based on a single chromosome is uniform (Nielsen 2000).
(Nielsen 2000). Multiplying results in cancellation of y and implies that the equilibrium spectrum with ascertainment based on a single chromosome is uniform (Nielsen 2000).
If species 2 has the ancestral allele, either the derived allele was present in the ancestral population at t = 0 (an old allele) and was not on the chromosome sampled from species 2 or it arose by mutation in species 1 after t = 0 (a new allele). The contribution of old alleles is
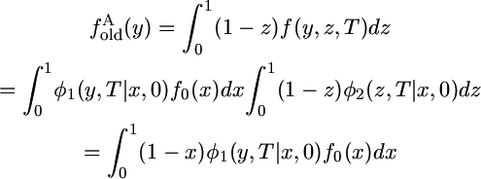 |
(10) |
because the expected value of 1 − z is 1 − x. In this case, the infinite sum does not reduce to a single term,
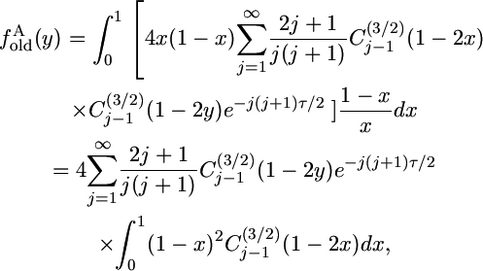 |
(11) |
which can be simplified by noting that
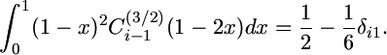 |
(12) |
The contribution to the finite spectrum of old alleles is
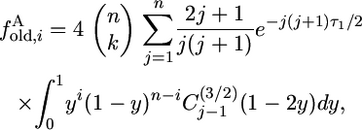 |
(13) |
which is a finite sum because the integral is 0 for j > n.
To compute the contribution of new alleles in species 1, we use the theory developed recently by Evans et al. (2007). The method is summarized in appendix a. The total 2-ancestral spectrum is
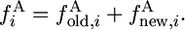 |
(14) |
The 2-ancestral spectrum can more easily be calculated by subtracting the 2-derived spectrum from the unconditional spectrum: 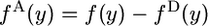 . We present the alternative derivation because it will be of interest to determine the probabilities that 2-ancestral alleles are old or new.
. We present the alternative derivation because it will be of interest to determine the probabilities that 2-ancestral alleles are old or new.
The expected numbers of 2-derived and 2-ancestral segregating sites in a sample of n chromosomes are obtained by summing 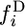 and
and 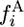 from i = 1 to n − 1.
from i = 1 to n − 1.
Alleles ages:
The age of an allele, meaning the time in the past it arose by mutation, depends on its current frequency and on the history of population sizes. In general, the probability that an allele found in frequency y arose at time t in the past is proportional to the forward transition function
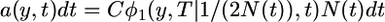 |
(15) |
(Kimura and Ohta 1973; Slatkin 2002), where C is a normalization constant that depends on the time interval being considered. Equation 15 simplifies considerably in the limit of large N0.
Knowing whether the allele found on the chromosome in species 2 is derived or ancestral provides additional information about allele age. An old allele necessarily arose at least T generations in the past. How much earlier it arose depends on x, the frequency at t = 0, and on whether it is 2-derived or 2-ancestral. The conditional distributions of x given y are derived in appendix b. The overall distribution of age is obtained by averaging Equation 15 over x and normalizing. The average age of old alleles depends on whether they are 2-derived or 2-ancestral. For example, if y = 0.1 the average age if it is 2-derived is 1.24 + T and the average age if it is 2-ancestral and arose before speciation is 0.77 + T, both in units of 2N0 generations.
Fixation within species 1:
The above theory also allows us to predict the densities of derived alleles that are fixed in species 1 when the chromosome from species 2 has the ancestral allele. There are four possibilities: a derived allele may be either old or new and may be either fixed (y = 1) or still segregating but found on all n chromosomes sampled (y < 1). The contribution of old alleles for which y < 1 is
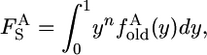 |
(16) |
where 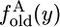 is given by Equation 11. The integral can be evaluated term by term but, unlike in Equation 13, the sum does not reduce to a finite sum. The probability that an old allele is fixed at T, given frequency x at 0 is given by Kimura's (1955) formula
is given by Equation 11. The integral can be evaluated term by term but, unlike in Equation 13, the sum does not reduce to a finite sum. The probability that an old allele is fixed at T, given frequency x at 0 is given by Kimura's (1955) formula
 |
(17) |
The net contribution is obtained after multiplying by  and integrating over 0 < x < 1. The density of new alleles that are fixed in a sample of n chromosomes is
and integrating over 0 < x < 1. The density of new alleles that are fixed in a sample of n chromosomes is
 |
(18) |
where the first term represents the new alleles still segregating and the second term represents alleles that are fixed. Both functions in Equation 18 are obtained from the theory summarized in appendix a.
IDEALIZED MODEL OF MODERN HUMANS AND NEANDERTHALS
The analysis in this article was motivated by the recently published nuclear sequences of Neanderthal DNA. To illustrate the use of our theory in a simple context, we assume an idealized model of the history of modern humans. The lineage leading to modern humans diverged from Neanderthals t4 years ago (the 4 indicates node 4 in Figure 1), was of constant effective size N0 until 100,000 years ago, and then grew exponentially to an effective size GN0 at the present time.
We assume N0 = 10,000 and that t4 = 500,000, 250,000, or 100,000 years ago (20,000, 10,000, or 4000 generations if a generation time of 25 years is used). It is convenient to measure time in units of 2N0 generations, so T = 1, 0.5, and 0.2. With this choice of N0, the time at which exponential growth began is 4000 generations (0.2 scaled time units) ago. The functional form of N(t) is as follows:  for 0 ≤ t ≤ T − 0.2 and,
for 0 ≤ t ≤ T − 0.2 and, 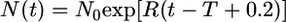 for T − 0.2 ≤ t ≤ T, where
for T − 0.2 ≤ t ≤ T, where 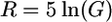 . Equation 6 gives the scaled time
. Equation 6 gives the scaled time 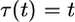 for t ≤ T − 0.2 and
for t ≤ T − 0.2 and 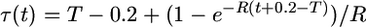 for t > T − 0.2. We refer to this as the model of delayed exponential growth.
for t > T − 0.2. We refer to this as the model of delayed exponential growth.
The frequency spectrum of 2-derived alleles is given by Equation 9. The finite spectrum is uniform on i and is proportional to 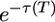 . This factor depends only weakly on G. If t4 = 500,000: it is e−1 ≈ 0.37 for G = 1 (no growth of modern humans) and increases only to
. This factor depends only weakly on G. If t4 = 500,000: it is e−1 ≈ 0.37 for G = 1 (no growth of modern humans) and increases only to 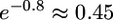 as G becomes very large. If t4 = 250,000 years (T = 0.5),
as G becomes very large. If t4 = 250,000 years (T = 0.5), 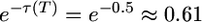 for G = 1 and increases to e−0.3 ≈ 0.74 as G becomes very large. In both cases, the spectrum of old alleles is relatively insensitive to the rate of recent growth because their loss occurs mostly before growth begins.
for G = 1 and increases to e−0.3 ≈ 0.74 as G becomes very large. In both cases, the spectrum of old alleles is relatively insensitive to the rate of recent growth because their loss occurs mostly before growth begins.
The frequency spectrum of 2-ancestral alleles has contributions from both old and new alleles. The probability that a 2-ancestral allele is new depends on G, as shown in Figure 2. Knowing that an allele is 2-ancestral changes somewhat the spectrum from the unconditional spectrum. The unconditional frequency spectrum is obtained by adding the 2-ancestral and 2-derived spectra. For t4 = 500,000, 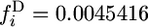 for G = 1 and
for G = 1 and 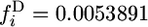 for G = 1000. Figure 3 shows that the difference between the 2-ancestral spectrum and the unconditional spectrum is small for small i but increases for larger i.
for G = 1000. Figure 3 shows that the difference between the 2-ancestral spectrum and the unconditional spectrum is small for small i but increases for larger i.
Figure 2.—

The probability that a 2-ancestral allele in frequency y in species 1 is new (meaning that it arose by mutation after species 1 and 2 separated). The results shown are for t4 = 500,000 years (T = 1 in generations scaled by 2N0), θ = 1, and n = 80. These results were obtained by computing the two components of the 2-ancestral spectra. The contribution from old alleles was computed by evaluating Equation 13 in the text. The contribution of new alleles was obtained by implementing the Evans et al. (2007) method described in appendix a. The probability that a 2-ancestral allele is new was found by taking the ratio of the new component to the sum. The delayed exponential growth model with N0 = 10,000 was used. G is the factor by which population size increased during the past 100,000 years of exponential growth.
Figure 3.—

Comparison of 2-ancestral spectra with the unconditional spectra for t4 = 500,000 years. The 2-ancestral spectra were computed as described for Figure 2 for the delayed exponential growth model. The unconditional spectra were obtained by adding the 2-derived spectra, 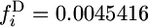 for G = 1 and
for G = 1 and 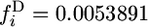 for G = 1000.
for G = 1000.
For t4 = 500,000, the average frequencies of 2-ancestral alleles are 0.176 for G = 1 and 0.079 for G = 1000. The results are qualitatively similar for t4 = 250,000 and t4 = 100,000.
It will be of interest to know whether 2-ancestral alleles arose before or after the onset of exponential growth. The unconditional probability that an allele in frequency y arose between the present and tlim generations in the past is
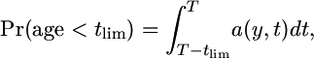 |
(19) |
where a(y, t) is given by Equation 15. The probability that a 2-ancestral allele arose in the same time period is the unconditional probability divided by the probability that a 2-ancestral allele arose after t = 0 (cf. Figure 2). Figure 4a shows that the unconditional age distribution of new alleles in frequency y = 0.1 depends strongly on G for T = 1. Even if G is only 100, the probability that it arose before t = 0.8 is almost 99%, in contrast to a probability of 68% for G = 1. Figure 4b shows that low-frequency alleles almost certainly arose before the onset of growth if G = 1000.
Figure 4.—

(a) Unconditional distributions of times of origin of alleles that arose by mutation in species 1 and that are found in frequency y = 0.1 at t = 1. The results were obtained by numerically evaluating Equation 15 in the text in the limit of large N0. The delayed growth model was assumed with t4 = 500,000 and three values of G. (b) The unconditional probability that a new allele in frequency y arose before t = 0.8 under the delayed growth model. The results were obtained from numerically evaluating Equation 19 in the text and subtracting from 1.
The densities of 2-ancestral and 2-derived segregating sites depend strongly on G and somewhat on t4, as shown in Table 1. The density of 2-ancestral alleles that are fixed in species 1 is low even if there is no growth (G = 1) and decreases as G increases. Some results are summarized in Table 1.
TABLE 1.
Predicted densities of segregating and fixed sites (multiplied by θ = 4N0μ) for the model of delayed exponential growth described in the text
|
t4 = 500,000
|
t4 = 250,000
|
t4 = 100,000
|
||||
|---|---|---|---|---|---|---|
| G = 1 | G = 1,000 | G = 1 | G = 1,000 | G = 1 | G = 1,000 | |
| 2-Ancestral segregating | 4.59 | 10.88 | 4.36 | 10.60 | 4.15 | 9.51 |
| 2-Derived segregating | 0.36 | 0.43 | 0.59 | 0.70 | 0.80 | 0.95 |
| 2-Ancestral, fixed | 0.18 | 0.13 | 0.06 | 0.03 | 0.01 | 0.00 |
NEANDERTHAL DATA SET
Our predictions can be compared with the recently published DNA sequences obtained from a Neanderthal bone (Green et al. 2006). The Neanderthal data set comprises short fragments aligned to both human and chimpanzee genomic sequences. The data were filtered as described by Green et al. (2006) to remove uncertain alignments and obvious sequencing errors. We determined whether each fragment contained a HapMap SNP found in build 36.1 of the HapMap data set. We retained only those SNPs polymorphic in at least one of the four HapMap populations (Europeans, Han Chinese, Japanese, and Yorubans) for which the Neanderthal sequence has one of the two SNP alleles, as illustrated in Figure 1. We assumed the nucleotide in the chimp reference sequence (build 2, version 1, also called pantro2) is ancestral and the other is derived. We divided the SNPs into two groups depending on whether the Neanderthal sequence had the ancestral (N-ancestral) or derived (N-derived) nucleotide. There were 461 N-ancestral SNPs and 177 N-derived SNPs. We then determined the frequency spectra of the two groups in each of the four HapMap populations.
The frequency spectra are shown in Figure 5 for the N-ancestral SNPs and in Figure 6 for the N-derived SNPs. Neither set of graphs agrees closely with the predictions made above. The histograms for the N-ancestral SNPs have too many high-frequency alleles. The histograms for N-derived SNPs have fewer very-low-frequency alleles than expected and also a deficiency in alleles of 0.2–0.5 frequency range, especially in the Han Chinese and Japanese samples. Our demographic model is highly simplified and does not account for a potential bottleneck in population size and other demographic complexities that were considered by Noonan et al. (2006).
Figure 5.—

Frequency spectra in the four HapMap populations of the 461 sites at which the Neanderthal sequences obtained by Green et al. (2006) had the ancestral nucleotide (N-ancestral SNPs).
Figure 6.—

Frequency spectra in the four HapMap populations of the 177 sites at which the Neanderthal sequences obtained by Green et al. (2006) had the derived nucleotide (N-derived SNPs).
Our prediction that the N-derived spectrum is uniform is based on the assumption that the spectrum at the time of speciation is the equilibrium spectrum for a population of constant size, 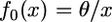 . Had there been population growth before speciation, f0 would be more heavily weighted to low-frequency alleles (Griffiths and Tavaré 1998; Griffiths 2003) and the 2-derived spectrum computed from Equation 3 would predict a higher proportion of low-frequency alleles. Therefore, the deficiency of low-frequency alleles in the N-derived spectrum cannot be attributed to population growth before speciation.
. Had there been population growth before speciation, f0 would be more heavily weighted to low-frequency alleles (Griffiths and Tavaré 1998; Griffiths 2003) and the 2-derived spectrum computed from Equation 3 would predict a higher proportion of low-frequency alleles. Therefore, the deficiency of low-frequency alleles in the N-derived spectrum cannot be attributed to population growth before speciation.
Table 2 shows the average frequencies of SNPs in the four populations. The average frequency of N-derived SNPs is larger in the European and Asian samples than in the Yoruban sample. The higher frequencies in the European and Asian samples than in the Yoruban sample can be attributed in part to the fact that HapMap SNPs were ascertained primarily in Europeans or people of European ancestry.
TABLE 2.
Average frequencies of the derived nucleotide in various subsets of SNPs in regions aligned to the Neanderthal and chimpanzee sequences
| HapMap sample
|
||||
|---|---|---|---|---|
| European | Han Chinese | Japanese | Yoruban | |
| N-derived | 0.654 | 0.660 | 0.659 | 0.582 |
| N-derived (CpG) | 0.692 | 0.772 | 0.766 | 0.606 |
| N-ancestral | 0.267 | 0.279 | 0.278 | 0.282 |
| N-ancestral (CpG) | 0.281 | 0.326 | 0.331 | 0.282 |
N-derived are sites at which the Neanderthal has a different nucleotide than in the chimpanzee sequence (n = 177); N-derived (CpG) are N-derived sites at which the human SNP is in a CpG pair (n = 24); N-ancestral are sites at which the Neanderthal and chimpanzee have the same nucleotide (n = 461); N-ancestral (CpG) are the N-ancestral sites at which the human SNP is in a CpG pair (n = 84).
In Table 2, we also present the results from analyzing separately N-ancestral and N-derived sites at which there is a CpG pair that might result in an elevated mutation rate. If two or more mutations have occurred at a site, then it is possible that some sites identified as N-ancestral are actually N-derived and some sites identified as N-derived are actually N-ancestral. If there are many such sites, the effect would be to reduce the average frequency of sites identified as N-derived because some of them are actually N-ancestral and to increase the average frequency of sites identified as N-ancestral because some of them are actually N-derived. This effect should be largest in the CpG subsets of sites because they are likely to have higher mutation rates. The average frequencies at the N-ancestral CpG sites are indeed elevated, but the average frequency in the N-derived CpG sites is also elevated, which is the opposite of what is expected if recurrent mutation is important.
Ascertainment bias may explain both the higher than expected fraction of N-derived SNPs and the higher than expected frequency of N-ancestral SNPs, ∼0.27 as compared to the expectation of 0.176 for G = 1 and 0.079 for G = 1000 (t4 = 500,000 years). The expected average frequencies of N-derived alleles under our model are not sensitive to changes in t4. If t4 = 100,000 years, they decrease only to 0.142 for G = 1 and 0.062 for G = 1000.
Green et al. (2006) estimated the average nuclear divergence time of Neanderthals and modern humans by comparing the Neanderthal fragments with the human and chimpanzee reference sequences. They noted that, because of errors in the Neanderthal sequences caused by the degradation of ancient DNA, the number of nucleotide changes assigned to the Neanderthal branch was much larger than the number assigned to the human branch. To avoid having degradation bias the estimated divergence time, Green et al. used only sequence changes assigned to the human branch, i.e., sites at which the Neanderthal and the chimp sequence were the same and the human reference sequence was different. We can use our theory to compute the probability that such human-specific substitutions are new, meaning that they arose since speciation. Results are shown in Table 3. Note that if humans and Neanderthals diverged relatively recently, then at most sites the human-specific nucleotide arose before the separation of those lineages, a result consistent with the simulation results of Noonan et al. (2006).
TABLE 3.
Probability that a neutral allele found only in the human reference sequence arose by mutation after the separation of the human and Neanderthal lineages (i.e., is a new allele) under the delayed growth model described in the text
| Probability of being a new allele
|
|||
|---|---|---|---|
| t4 = 500,000 | t4 = 250,000 | t4 = 100,000 | |
| G = 1 | 0.506 | 0.333 | 0.167 |
| G = 1,000 | 0.474 | 0.283 | 0.086 |
The Neanderthal sequences provide us with some information about the age of SNPs. Assuming no recurrent mutation, N-derived SNPs are necessarily old; they arose before the divergence of Neanderthals and modern humans. N-ancestral SNPs may be old or new. The results in Figure 3b suggest that low-frequency N-ancestral SNPs are probably new. The question is whether they arose before or after the divergence of modern human populations. Nucleotides that arose after the divergence of modern human populations will likely show more differentiation among populations than will nucleotides polymorphic in the population ancestral to all modern humans.
A rough test of whether many of the N-ancestral SNPs arose before the divergence of modern human populations is obtained by determining whether low-frequency N-derived SNPs show less differentiation among HapMap populations than do low-frequency N-ancestral SNPs. We quantified the extent of differentiation by computing 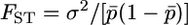 , with
, with 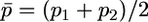 and
and 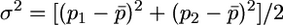 . Figure 7 shows FST values for the N-derived and N-ancestral SNPs computed for the Yoruban and European samples for derived nucleotides in frequency <0.2 in the European sample. Values of FST for N-derived SNPs are not consistently lower: in fact the average for the 10 N-derived alleles shown in Figure 7b, 0.072, is slightly higher than the average for the 253 N-ancestral alleles shown in Figure 7a, 0.053. The very few low-frequency N-ancestral SNPs with relatively high FST values might indicate recent origin or recent selection, but the vast majority were probably present in the population ancestral to all modern humans, as would be expected from the above theory if the ancestral human population began to grow rapidly before populations ancestral to the HapMap populations diverged from one another.
. Figure 7 shows FST values for the N-derived and N-ancestral SNPs computed for the Yoruban and European samples for derived nucleotides in frequency <0.2 in the European sample. Values of FST for N-derived SNPs are not consistently lower: in fact the average for the 10 N-derived alleles shown in Figure 7b, 0.072, is slightly higher than the average for the 253 N-ancestral alleles shown in Figure 7a, 0.053. The very few low-frequency N-ancestral SNPs with relatively high FST values might indicate recent origin or recent selection, but the vast majority were probably present in the population ancestral to all modern humans, as would be expected from the above theory if the ancestral human population began to grow rapidly before populations ancestral to the HapMap populations diverged from one another.
Figure 7.—

FST values for 253 N-ancestral and 10 N-derived SNPs in European and Yoruban HapMap samples for which the frequency of the derived allele was <0.2 in Europeans. The values of FST were computed using the formulas in the text.
The six SNPs found in exons identified in the human reference sequence are listed in Table 4. The one N-derived SNP results in a synonymous change. The relatively high frequencies of the derived nucleotide in the four HapMaps is consistent with neutrality. Of the four N-ancestral SNPs that result in nonsynonymous changes, the derived nucleotide is in low frequency for two of them (rs8192506 and rs449643) as would be expected if they are neutral or slightly deleterious. The derived nucleotide is in relatively high frequency at the other two (rs11208299 and rs2304824), which is suggestive of selection. For these two SNPs, we computed the iHS statistic defined by Voight et al. (2006). Under the null hypothesis of no selection, iHS has a normal distribution with mean 0 and variance 1. For rs11208299, iHS = −1.765 in Europeans, −1.224 in Yorubans, and 1.156 in the combined Chinese and Japanese samples. For rs2304824, iHS = 0.827 in Europeans, 0.183 in Yorubans, and −1.119 in the combined Chinese and Japanese samples. On the basis of the iHS value alone, neither SNP shows significant evidence of selection in any population, but the consistently high values of iHS for rs11208299, combined with the substantial variation in allele frequency among populations and the fact that the Neanderthal sequence has the ancestral allele, indicate that the genomic region containing this SNP may well have been subject to recent selection in modern humans.
TABLE 4.
HapMap SNPs in exons for which the Neanderthal nucleotide could be determined
| Name | Chr | Syn/Non | Nean | Chim | Hum | Ceu | Hcb | Jpt | Yri |
|---|---|---|---|---|---|---|---|---|---|
| N-derived | |||||||||
| rs157397 | 17 | Syn | C | T | C/T | 0.95 | 0.56 | 0.7 | 0.77 |
| N-ancestral | |||||||||
| rs11208299 | 1 | Non | T | T | G/T | 0.708 | 0.567 | 0.409 | 0.30 |
| rs8192506 | 2 | Non | A | A | G/A | 0.05 | 0 | 0 | 0 |
| rs449643 | 6 | Non | G | G | A/G | 0.092 | 0.024 | 0.058 | 0.24 |
| rs2304824 | 15 | Non | G | G | A/G | 0.525 | 0.722 | 0.716 | 0.18 |
| rs17009819 | 4 | Syn | C | C | T/C | 0 | 0 | 0 | 0.12 |
Name, SNP identifier used by build 36.1 of the HapMap database; Chr, number of human chromosome; Syn, synonymous change; Non, nonsynonymous change; Nean, nucleotide of the Neanderthal; Chim, nucleotide of build 2, version 1 of the chimpanzee reference sequence (pantro2); Hum, pair of nucleotides polymorphic in the HapMap database; Ceu, frequency of the derived nucleotide in the European HapMap database; Hcb, frequency of the derived nucleotide in the Han Chinese HapMap database; Jpt, frequency of the derived nucleotide in the Japanese HapMap database; Yri, frequency of the derived nucleotide in the Yoruban HapMap database. The nucleotide in the chimpanzee sequence is assumed to be ancestral.
DISCUSSION AND CONCLUSIONS
In the preceding text we presented a model of the joint frequency spectra of alleles in closely related species. The theory is developed in terms of classical diffusion theory that allows us to obtain analytic and numerical results. Our approach is quite general and can be modified to allow for selection and different histories of population sizes in the two species.
One question we addressed is how much is gained by having even a single chromosome from a second species. Although the unconditional frequency spectrum already provides a way to estimate past population growth rates and selection intensities (Sawyer and Hartl 1992; Nielsen 2000; Williamson et al. 2005), being able to partition the unconditional spectrum on the basis of whether the closely related species has the ancestral or derived allele permits a clearer understanding of when changes in allele frequency occurred. If mutation is irreversible, alleles found in both species necessarily arose before speciation. Alleles found in only one species probably arose after speciation or were probably in low frequency at the time of speciation. A high frequency of such alleles indicates positive selection.
Selection can be incorporated into our model. No analytic solutions can be found but numerical analysis is straightforward. The Matlab programs available at the Slatkin laboratory web site allow for alleles with an additive effect on fitness. The only modification needed to the theory developed by Evans et al. (2007) is that the initial frequency spectrum must be adjusted to reflect whether the chromosome from species 2 carries the derived or the ancestral allele.
We can anticipate the effects of directional selection. Selected alleles tend to be younger (Slatkin 2002), implying that selected SNPs in modern humans have arisen more recently and exhibit more divergence among populations. In addition, recent positive selection could carry advantageous derived alleles to fixation. As a consequence, selection could result in N-ancestral sites that are fixed for the derived allele in modern humans, something that is very unlikely for neutral alleles.
SNPs subject to balancing selection would tend to be old alleles that arose before the separation of Neanderthals and modern humans. If the frequency of a SNP had been maintained by balancing selection for a long time, the probability that the Neanderthal chromosome would carry the derived nucleotide would be approximately its frequency in modern humans.
The theory developed in this article was motivated by the recently published data set containing ∼1 Mb of autosomal Neanderthal sequence (Green et al. 2006), but the application of our theory to HapMap SNPs for which the state of the Neanderthal chromosome can be assessed is largely illustrative. There is extensive ascertainment bias in HapMap SNPs, as well as an elevated probability of sequencing error in the Neanderthal data because of the degradation of ancient DNA, that we have not accounted for. Nevertheless, comparison of the model's predictions with observations is instructive. The prediction of a uniform distribution of an N-derived spectrum of neutral SNPs depends only on neutrality and is robust to changes in the details of the demographic history of modern humans and Neanderthals. The evident deviations from uniformity in the N-derived spectra of all four HapMap populations (Figure 6) are difficult to account for only with ascertainment bias. Selection, admixture, and factors associated with the genomic analysis of ancient DNA will also have to be considered.
ELECTRONIC RESOURCES
The HapMap data analyzed were obtained from build 36.1 of the HapMap project, available at http://www.hapmap.org/.
The alignment of the Neanderthal fragments to the human and chimpanzee reference sequences, the N-derived and N-ancestral HapMap SNPs for each HapMap population, and the Matlab program used to obtain the spectra of new alleles are available from the Slatkin laboratory site, http://ib.berkeley.edu/labs/slatkin/.
Acknowledgments
We thank G. Coop, S. Kudaravalli, M. Lachmann, S. Ptak, J. Wakeley, and the reviewers for helpful discussions of this topic and comments on earlier versions of our results. This research was supported in part by National Institutes of Health grant R01-GM40282 to M.S.
APPENDIX A
We show here how to apply the theory of Evans et al. (2007). The continuous spectrum of new neutral alleles satisfies the differential equation
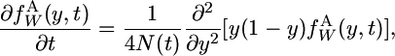 |
(A1) |
where the subscript W indicates new alleles. The initial condition is 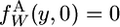 and the two boundary conditions are
and the two boundary conditions are
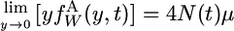 |
(A2a) |
and
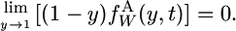 |
(A2b) |
Equation A1 is the standard forward diffusion equation. Evans et al. (2007) show that with (A2a) and (A2b) as boundary conditions it also describes the forward evolution of the frequency spectrum when there is a steady flux of mutations and variable population size.
Equation A1 can be solved numerically by first transforming the timescale from t to τ, as in Equation 6 in the main text, and then defining
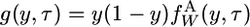 |
(A3) |
to obtain
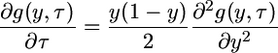 |
(A4) |
with initial condition g(y, 0) = 0 and boundary conditions
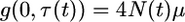 |
(A5) |
and
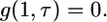 |
(A6) |
The contribution of new alleles to the finite spectrum is
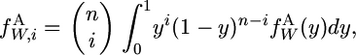 |
(A7) |
which we evaluated using the Matlab program described by Evans et al. (2007).
APPENDIX B
We derive here the conditional distribution of x, the frequency at t = 0, given y the frequency in species 1 and whether the chromosome from species 2 has the ancestral or derived allele. If species 2 has the derived allele, then the derived allele in species 1 has to be old. We can infer its frequency at t = 0 in terms f0 by applying Bayes' theorem:
 |
(B1) |
As was established in Equation 3, 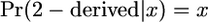 and hence, if
and hence, if 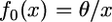 ,
,
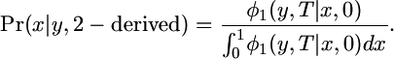 |
(B2) |
Therefore, the frequency of the derived allele at t = 0 given that species 2 has the derived allele is the solution to the forward diffusion equation, normalized to be a probability distribution on x.
The results are slightly different if species 2 has the ancestral allele. The derived allele in species 1 may be new or old. In a sample of n chromosomes, it is new with probability
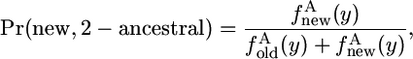 |
(B3) |
where 2-ancestral means that the chromosome from species 2 carries the ancestral allele, and in that case, x = 0.
If the derived allele is old, then
 |
(B4) |
Because 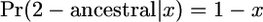 ,
,
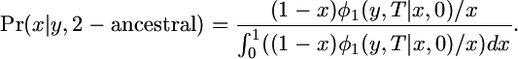 |
(B5) |
The distribution of x given y for 2-ancestral alleles has two parts, a spike at x = 0 and a continuous part given by Equation B5. The expectation of x is substantially larger than y when y is small because, if an allele is still polymorphic at t = T, it had to have been relatively frequent at t = 0. If y = 0.1 and t = 1, E(x) = 0.45 for 2-derived alleles and E(x) = 0.22 for an old 2-ancestral allele.
References
- Abramowitz, M., and I. A. Stegun (Editors), 1965. Handbook of Mathematical Functions. Dover, New York.
- Bustamante, C. D., J. Wakeley, S. Sawyer and D. L. Hartl, 2001. Directional selection and the site-frequency spectrum. Genetics 159: 1779–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, A. G., M. J. Hubisz, C. D. Bustamante, S. H. Williamson and R. Nielsen, 2005. Ascertainment bias in studies of human genome-wide polymorphism. Genome Res. 15: 1496–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, S. N., Y. Shvets and M. Slatkin, 2007. Non-equilibrium theory of the allele frequency spectrum. Theor. Popul. Biol. 71: 109–119. [DOI] [PubMed] [Google Scholar]
- Fisher, R. A., 1930. The distribution of gene ratios for rare mutations. Proc. R. Soc. Edinb. 50: 205–220. [Google Scholar]
- Green, R. E., J. Krause, S. E. Ptak, A. W. Briggs, M. T. Ronan et al., 2006. Analysis of one million base pairs of Neanderthal DNA. Nature 444: 330–336. [DOI] [PubMed] [Google Scholar]
- Griffiths, R. C., 2003. The frequency spectrum of a mutation, and its age, in a general diffusion model. Theor. Popul. Biol. 64: 241–251. [DOI] [PubMed] [Google Scholar]
- Griffiths, R. C., and S. Tavaré, 1998. The age of a mutation in a general coalescent tree. Stoch. Models 14: 273–295. [Google Scholar]
- Hudson, R. R., 2002. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18: 337–338. [DOI] [PubMed] [Google Scholar]
- Johnson, P. L. F., and M. Slatkin, 2006. Inference of population genetic parameters in metagenomics: a clean look at messy data. Genome Res. 16: 1320–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, M., 1955. Solution of a process of random genetic drift with a continuous model. Proc. Natl. Acad. Sci. USA 41: 144–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, M., 1964. Diffusion models in population genetics. J. Appl. Probab. 1: 177–232. [Google Scholar]
- Kimura, M., 1969. The number of heterozygous nucleotide sites maintained in a finite population due to steady flux of mutations. Genetics 61: 893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, M., and T. Ohta, 1973. The age of a neutral mutant persisting in a finite population. Genetics 75: 199–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, R., 2000. Estimation of population parameters and recombination rates from single nucleotide polymorphisms. Genetics 154: 931–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noonan, J. P., G. Coop, S. Kudaravalli, D. Smith, J. Krause et al., 2006. Sequencing and analysis of Neanderthal genomic DNA. Science 314: 1113–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer, S. A., and D. L. Hartl, 1992. Population genetics of polymorphism and divergence. Genetics 132: 1161–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatkin, M., 2002. The age of alleles, pp. 233–259 in Modern Developments in Theoretical Population Genetics. Oxford University Press, Oxford.
- Voight, B. F., S. Kudaravalli, X. Wen and J. K. Pritchard, 2006. A map of recent positive selection in the human genome. PLoS Biol. 4: e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeley, J., and J. Hey, 1997. Estimating ancestral population parameters. Genetics 145: 847–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeley, J., R. Nielsen, S. N. Liu-Cordero and K. Ardlie, 2001. The discovery of single-nucleotide polymorphisms: and inferences about human demographic history. Am. J. Hum. Genet. 69: 1332–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson, S. H., R. Hernandez, A. Fledel-Alon, L. Zhu, R. Nielsen et al., 2005. Simultaneous inference of selection and population growth from patterns of variation in the human genome. Proc. Natl. Acad. Sci. USA 102: 7882–7887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S., 1938. The distribution of gene frequencies under irreversible mutation. Proc. Natl. Acad. Sci. USA 24: 253–259. [DOI] [PMC free article] [PubMed] [Google Scholar]