

Abstract
Homolog pairing refers to the alignment and physical apposition of homologous chromosomal segments. Although commonly observed during meiosis, homolog pairing also occurs in nonmeiotic cells of several organisms, including humans and Drosophila. The mechanism underlying nonmeiotic pairing, however, remains largely unknown. Here, we explore the use of established Drosophila cell lines for the analysis of pairing in somatic cells. Using fluorescent in situ hybridization (FISH), we assayed pairing at nine regions scattered throughout the genome of Kc167 cells, observing high levels of homolog pairing at all six euchromatic regions assayed and variably lower levels in regions in or near centromeric heterochromatin. We have also observed extensive pairing in six additional cell lines representing different tissues of origin, different ploidies, and two different species, demonstrating homolog pairing in cell culture to be impervious to cell type or culture history. Furthermore, by sorting Kc167 cells into G1, S, and G2 subpopulations, we show that even progression through these stages of the cell cycle does not significantly change pairing levels. Finally, our data indicate that disrupting Drosophila topoisomerase II (Top2) gene function with RNAi and chemical inhibitors perturbs homolog pairing, suggesting Top2 to be a gene important for pairing.
ONE way in which the genome can be organized is through the physical pairing of homologous chromosomes. Such pairing occurs during meiosis, when it aligns chromosomes in preparation for recombination and segregation, as well as in nonmeiotic nuclei. Although most extensively studied in the Dipteran insect Drosophila melanogaster, nonmeiotic association of homologous chromosomal regions has also been observed elsewhere, where it participates in gene expression and other vital processes (reviewed by Wu and Morris 1999; Duncan 2002; Grant-Downton and Dickinson 2004; Mckee 2004; Zickler 2006). Importantly, nonmeiotic pairing can influence gene regulation and DNA repair through the processes of transvection (reviewed by Pirrotta 1999; Wu and Morris 1999; Duncan 2002; Kassis 2002; Kennison and Southworth 2002) and recombination (reviewed by Gloor 2002; Wyman et al. 2004; also see Rong and Golic 2003), respectively. Pairing of homologous chromosomal regions has also been implicated in mammalian X-inactivation (Marahrens 1999; Bacher et al. 2006; Diaz-Perez et al. 2006; Xu et al. 2006) and imprinting (Lasalle and Lalande 1996; Riesselmann and Haaf 1999). Intriguingly, a reduced level of pairing at imprinted regions may be associated with syndromes such as autism and the Prader–Willi and Angelman syndromes (Lasalle and Lalande 1996; Thatcher et al. 2005). Despite its role in multiple phenomena, however, the mechanism(s) of nonmeiotic homolog pairing remains largely unknown.
To better understand nonmeiotic pairing, we have focused on Drosophila, where homologous chromosomes are essentially paired in all somatic cells throughout development (Stevens 1907, 1908; Metz 1916; reviewed by Duncan 2002; Mckee 2004). FISH analyses of embryos and tissues such as from the larval brain or imaginal discs indicate that, typically, a given chromosome region is paired in 60–100% of nuclei, but that this level of pairing can vary depending on the time of development, region being assayed, and phase of the cell cycle (Kopczynski and Muskavitch 1992; Csink and Henikoff 1998; Fung et al. 1998; Gemkow et al. 1998; Sass and Henikoff 1999; Vazquez et al. 2002; Ronshaugen and Levine 2004; Fritsch et al. 2006). For example, analysis of multiple chromosomal locations during early embryogenesis indicates that some regions initiate pairing earlier than do others, but that all regions eventually achieve high levels of pairing (Fung et al. 1998). Other studies have examined the impact of the cell cycle on pairing. In one case, BrdU labeling of the larval central nervous system (CNS) revealed that the percentage of nuclei in which a euchromatic region, 59E, and a pericentromeric repeat, AACAC, were paired decreased during S phase from >75% to ∼60% and from ∼85% to <70%, respectively, and remained relatively low until after mitosis (Csink and Henikoff 1998). In contrast, pairing at the internally repeated histone locus during the 13th embryonic mitotic cycle was found to be unperturbed by progression through S phase and well into metaphase, decreasing only during anaphase, when the percentage of nuclei in which the locus was paired dropped from 63% to 19% (Fung et al. 1998).
To date, efforts to identify genes that participate in somatic pairing have stemmed largely from observations of in vivo phenotypes that had been previously hypothesized to be sensitive to homolog pairing. For example, zeste, which encodes a transcription factor, was identified as a gene that modifies transvection-associated phenotypes of several loci (reviewed by Pirrotta 1999; Wu and Morris 1999; Duncan 2002; Kennison and Southworth 2002). Interestingly, zeste protein shows a propensity to self-aggregate, suggesting that it may promote pairing by binding to chromosomes and pulling or holding them together (Chen and Pirrotta 1993). However, it has yet to be determined how directly zeste affects pairing.
A more immediate connection has been made with Polycomb group (PcG) genes, which can mediate the pairing of PcG response elements (PREs) to effect long-range interactions and pairing-sensitive silencing (reviewed by Pirrotta 1999; Kassis 2002; Kavi et al. 2006; also see Vazquez et al. 2006). For example, pairing of the PRE-containing element Fab-7 requires several PcG proteins, such as Polycomb, Polyhomeotic, Posterior sex combs, and Polycomb-like (Bantignies et al. 2003). A more recent study has shown that the pairing-sensitive silencing associated with Fab-7 may also require members of the RNAi machinery, such as Dicer-2, PIWI, and Argonaute1 (Grimaud et al. 2006).
Another genetic element implicated in long-range interactions between homologous chromosomal regions is an insulator carried by the gypsy retrotransposon (reviewed by Kuhn and Geyer 2003; Capelson and Corces 2004; Brasset and Vaury 2005; Gaszner and Felsenfeld 2006; Valenzuela and Kamakaka 2006; also see Kravchenko et al. 2005). Of note, two factors that work in conjunction with the gypsy insulator, Suppressor of Hairy Wing [Su(Hw)] and Modifier of mdg4 [Mod(mdg4)] (Kravchenko et al. 2005), colocalize at hundreds of chromosomal sites that do not correspond to gypsy elements and, along with another protein, Centrosomal Protein 190 (CP190), mediate the clustering of these sites into “insulator bodies” (reviewed by Kuhn and Geyer 2003; Capelson and Corces 2004; Brasset and Vaury 2005; Gaszner and Felsenfeld 2006; Valenzuela and Kamakaka 2006). Consistent with this finding, loss of Su(Hw) function has been observed to compromise homolog pairing (Fritsch et al. 2006). Intriguingly, a protein that is associated with insulator function in mammals, the CCCTC binding factor (CTCF), has also been implicated in long-range interactions (Ling et al. 2006; Donohoe et al. 2007).
Despite these advances, we are still far from a full understanding of how homologs sense, find, and align with each other. To further our knowledge of somatic pairing in Drosophila, we have extended our studies to cell culture in anticipation that such an experimental platform will facilitate analyses requiring the manipulation of homogenous populations of cells. A cell line-based approach could also allow researchers to identify genes involved in pairing solely on the basis of their capacity to support pairing, as assayed directly by visual examination. Importantly, because cell culture is unencumbered by developmental programs, it would permit the identification of genes that may also be essential for organismal viability.
Our study was encouraged by two reports. One, by Cherbas and Cherbas (1997), documented the capacity of a Drosophila immortal cell line to support parahomologous recombination, a form of transformation attributed to somatic homolog pairing in which exogenous sequences integrate in the vicinity of homologous chromosomal regions. The second report, by Halfer and Barigozzi (1973), described two newly established Drosophila cell lines in which condensed mitotic chromosomes appeared entangled or aligned with their homologs in orcein- or quinacrine-stained spreads. These images were obtained with diploid as well as polyploid cell lines and recall earlier studies of Dipteran insects demonstrating the capacity of somatic pairing to accommodate polyploidy in vivo (Metz 1916, 1922, 1925; Holt 1917). Interestingly, aligned condensed chromosomes have also been observed in cell lines of Aedes (mosquito), another Dipteran insect (Nichols et al. 1971). In our analysis, we used fluorescent in situ hybridization (FISH) to document somatic pairing of decondensed nonmitotic chromosomes in cell lines established decades ago and then characterized the consistency and breadth of that pairing. In addition, we obtained evidence indicating that topoisomerase II is a gene important for homolog pairing.
MATERIALS AND METHODS
Cell culture:
Kc167 (Echalier and Ohanessian 1969; Perrimon 2007), S2 (Schneider 1972; Perrimon 2007), D (Debec 1978, 1984; Cherbas 2007), DH-33 (Sondermeijer et al. 1980; Cherbas 2007), mbn2 (Haars et al. 1980; Cherbas 2007), clone 8 (Peel et al. 1990; Perrimon 2007), and ML-DmBG2-c6 (Ui et al. 1994; Cherbas 2007) cells were grown at 25° following standard protocols. Kc167 and S2 cultures were grown in sterile filtered Schneider's medium [GIBCO (Grand Island, NY) no. 11720-034] supplemented with heat-inactivated fetal bovine serum (FBS, to a final concentration of 10% v/v; JRH 12103-78P) and penicillin–streptomycin (50 units/ml penicillin, 50 μg/ml streptomycin; GIBCO no. 15070-63). D, mbn2, DH-33, and ML-DmBG2-c6 cultures were grown in sterile filtered Shields and Sang M3 insect medium (S8523; Sigma, St. Louis) supplemented with FBS (10% v/v), BPYE [2.5 g/liter bactopeptone (211677; Difco, Detroit) and 1.0 g/liter yeast extract (Y-1000; Sigma)], and penicillin–streptomycin (50 units/ml penicillin, 50 μg/ml streptomycin), with ML-DmBG2-c6 cultures being further supplemented with insulin (10 μg/ml; I6634, Sigma). Clone 8 cultures were grown in sterile filtered Shields and Sang M3 insect medium supplemented with FBS (2% v/v), insulin (0.0125 IU/ml), fly extract (2.5% v/v; Perrimon 2007), and penicillin–streptomycin (50 units/ml penicillin, 50 μg/ml streptomycin). To ensure that experiments were done with log-phase cells, active cultures were split at a 1:≥3 ratio, cultured for 1–2 days, and then passaged at 2–4 × 106 cells/ml prior to the analyses.
The Kc167 and S2 cell lines were obtained from Norbert Perrimon; the D, DH-33, mbn2, and ML-DmBG2-c6 cell lines were obtained from the Drosophila Genome Resource Center; and the clone 8 cell line was obtained from Mitzi Kuroda.
FISH:
Our FISH protocol was adapted from previously published protocols (Marshall et al. 1996; Dernburg and Sedat 1998; Dernburg 2000; Bantignies et al. 2003, 2005) and involved the following steps: Cells from log-phase cultures were adhered to either gelatin-coated (0.2%; G1393, Sigma) or lysine-treated (P8920; Sigma) 10-well glass slides (ER208W; Erie Scientific) for 1–3 hr. Slides were then gently washed with PBS (pH 7.2), fixed for 5 min with 4% formaldehyde in PBS (15700; Electron Sciences) at room temperature (RT), covered with a coverslip, frozen on an aluminum block (which had been precooled on dry ice), freed of their coverslips, and stored in 95% ethanol at −20°. After at least 20 min, slides were washed in 2× SSCT (0.3 m NaCl, 0.03 m sodium citrate, 0.1% Tween-20)/formamide (5 min each in 0, 20, 40, and 50% formamide at RT and 30 min in 50% formamide/2× SSCT at 37°). DNA probe in hybridization buffer was then added to the slides, covered with a coverslip, and denatured in an MJ Research (Watertown, MA) PTC-200 thermocycler with an Alpha Unit lock assembly block for 2 min at 91°, after which slides were transferred to a humidifying chamber, incubated overnight at 37°–40°, and freed of their coverslips while being washed (30 min in 50% formamide/2× SSCT at 37°, 5 min in 25% 2× SSCT/formamide at RT, and three times in 5 min in 2× SSCT at RT).
To visualize probes, slides were blocked in blocking buffer (0.1% BSA in 2× SSCT) at RT for 30 min, incubated with either rhodamine-conjugated anti-DIG antibody (1207750; Roche, Indianapolis) or fluorescein anti-biotin (SP-3040; Vector, Burlingame, CA) in blocking buffer for 1.5 hr, and washed for 1 hr in 2× SSCT, after which Vectashield with DAPI (H-1200; Vector) was added. Coverslips were applied and sealed to the slides with nail polish.
DNA probes were synthesized according to standard protocols. P1 plasmids (Berkeley Drosophila Genome Project) containing cloned Drosophila genomic DNA corresponding to chromosomal regions 21E3–4 (abbreviated as 21E3; DS03071; Fung et al. 1998), 28B1–28B2 (abbreviated as 28B1; DS01529; Fung et al. 1998), 40A2–40A3 (abbreviated as 40A2; DS09165; Fung et al. 1998), and 69C2–69C8 (abbreviated as 69C2; DS02752; Dej and Spradling 1999) were digested with restriction enzymes (Dernburg 2000) and then labeled with either Digoxigenin (DIG-Nick translation mix, 1 745 816; Roche Diagnostics) or, for dual label experiments, Biotin (BioNick Labeling System, LT18247-015; Invitrogen) following the manufacturers' protocols. Probe for 16E1–16E2 (abbreviated as 16E1) was synthesized from the bacterial artificial chromosome BACR17D02 RP98-17D2 (AC012163; AE003507) by Nick translation/direct labeling (32-801300; Vysis) following the manufacturer's protocol. The 359-bp repeat probe was synthesized by PCR (Dernburg 2000). Probes for 8C8 and 44F1 were synthesized from PCR products according to F. Bantignies (personal communication): 8–10 1- to 1.4-kbp PCR products corresponding to genomic regions separated by ∼1 kbp and spanning ∼30 kbp were combined, purified, and labeled by Nick translation (FISH-Tag DNA kit; Invitrogen). Probes were diluted into hybridization buffer (50% formamide/2× SSCT, 10% dextransulfate) to a final concentration of ∼150–500 ng/30 μl.
Oligo probes for the AACAC and dodeca heterochromatic repeats (Dernburg and Sedat 1998; Dernburg 2000) were synthesized with either a 5′ cy3 or cy5 fluorescent dye (Phoenix BioTechnologies) and contained locked nucleic acid (LNA) (Silahtaroglu et al. 2003, 2004) bases to increase melting temperature (AaCaCaAcAcAaCaCaAcAc and AcGgGaCcAgTaCgG, for “AACAC” and “dodeca” probes, respectively, where uppercase letters denote LNA-modified nucleotides). An abbreviated FISH protocol was developed for LNA containing oligos: After fixation, cells were incubated for 30 min in 2× SSCT at 37°, after which probe (1–100 nm in hybridization buffer) was added and slides were denatured at 91° for 2 min, washed immediately in 2× SSCT for 30 min at 37°, and then mounted with Vectashield.
Microscopy:
Through the generosity of Monica Colaiácovo, we were able to obtain images using a DeltaVision system with an Olympus IX-70 microscope and record data with a cooled CCD camera (model CH350; Roper Scientific). A U-APO 40× lens (NA 0.65–1.35) was used for all images except for those in Figure 1, where a PlanApo 60× lens (NA 1.4) was also used. Optical sections were collected at 0.50-μm increments and deconvolved using a conservative algorithm with 15 iterations. Data were analyzed with SoftWoRx Explorer software (Versions 1.0.1 and 1.1; Applied Precision). Three-dimensional images of nuclei were flattened for the figures.
Figure 1.—

Karyotypes and chromosomal pairing (40–96× magnification). (A) Kc167 mitotic figure stained with DAPI. Chromosome IV is not noted. (B) Kc167 mitotic figures labeled with DAPI (blue) and probes targeting the AACAC (green, chromosome II) and dodeca (red, chromosome III) pericentric heterochromatin repeats confirm four copies of X and chromosome II and three to five copies of chromosome III. The X chromosome was identified by its unique morphology. (C–F) Indicative of pairing, chromosomes (red lines) of similar morphology are occasionally intertwined or grouped in Kc167 (C and D) and clone 8 (E and F) mitotic figures. Clone 8 cells have a single X, a single Y, and two copies each of chromosomes II and III; chromosome IV is not noted. (G and H) ML-DmBG2-c6 mitotic figures stained with DAPI (G) or labeled with AACAC (yellow) and dodeca (red, H) include chromosomes with aberrant morphology.
Mitotic spreads:
Chromosomes were spread using standard protocols (Veile 1990; Echalier 1997), with or without incubating cells with colchicine (C3915; Sigma) for 2–24 hr.
Determining mitotic index:
Anti-α-tubulin (T5168; Sigma) and secondary antibodies (SC-2010; Santa Cruz Biotechnology) were used following the manufacturers' instructions for labeling tissue culture cells. Mitotic cells were then identified by morphology. We confirmed the mitotic indexes corresponding to Figure 6, F and G (inset, Figure 6F) through assays using anti-phosphorylated-histone H3 (anti-PH3; 06-570; Upstate Biotechnology, Lake Placid, NY): control, 3.5% (N = 2152); camptothecin, 0.3% (N = 1000); m-amsacrine, 0.3% (N = 1280); and ICRF-193, 3.5% (N = 976).
Figure 6.—

Disruption of Top2 perturbs homolog pairing. (A–C) dsRNA directed against Top2 reduces pairing at 28B1 in Kc167 cells as illustrated by nuclei imaged with DAPI and FISH (A), a pairing profile (B), and a percentile plot (C). dsRNA directed against Top1 has little effect. B shows the results (±SD) of experiments using three distinct dsRNAs targeting Top2 and two distinct dsRNAs targeting Top1, as well as two control experiments run without dsRNA. The percentile plot combines data described in B. (D and E) ICRF-193 (10 μm) reduces pairing at 28B1 (D) and 16E1 (E) in Kc167 cells. Camptothecin (4 μm) and m-amsacrine (5 μm) are less perturbing. Control nuclei were from cells treated with DMSO. Data represent two trials. (F and G) Pairing is more disrupted at 28B1 (F; P < 0.0001) than it is at 16E1 (G; P = 0.003) by ICRF-193 in clone 8 cells. Little disruption was observed when cells were treated with camptothecin (4 μm), m-amsacrine (5 μm), or DMSO (control). The data are from two independent experiments, one in which cells were treated with drugs for 24 hr and the other in which cells were treated for 30 hr. For each time point, nuclei imaged with FISH targeting 28B1 or 16E1 were from the same culture. N, number of nuclei from both time points combined. (Insets in D and F) Mitotic indexes correspond to one of the two independent trials shown.
Cell sorting:
Live cells incubated with Hoechst 33342 (H3570; Molecular Probes, Eugene, OR) were sorted on a Vantage SE high-speed sorter (FACS Sorting Core Facilities; Children's Hospital, Boston), adhered to slides for 30–60 min, and then subjected to FISH. We confirmed that cells had been successfully sorted into G1, S, and G2 subpopulations by assessing nuclear DNA content as determined by DAPI staining (data not shown).
RNAi:
Synthesis of dsRNA and application of RNAi to cells was carried out according to published protocols (Clemens et al. 2000; Perrimon 2007). Control cells were treated with a blank of deionized water. Cells were fixed 3–4 days after treatment. Three distinct nonoverlapping dsRNAs were tested independently in studies of Top1 as well as of Top2; outcomes did not vary among dsRNAs targeting the same gene.
Application of topoisomerase inhibitors:
Our studies using camptothecin (C9911; Sigma; Solier et al. 2004), m-amsacrine (A9809; Sigma; Solier et al. 2004), and ICRF-193 (GR-332; Biomol; Hossain et al. 2004) were guided by a sampling of studies carried out in mammals and Drosophila. Camptothecin and ICRF-193 were dissolved in DMSO (10 mm each), and m-amsacrine was dissolved in 30% ethanol (10 mm) before being added to log-phase cells to a final concentration of 10, 4, and 5 μm, respectively. Under the culture conditions used for this study, the doubling times for Kc167 and clone 8 cells were estimated to be ∼18 and ∼15 hr, respectively. Accordingly, Kc167 and clone 8 cells were incubated for 30 and 24 hr, respectively, except in the case of the clone 8 cells described in Figure 6, F and G, where, in one of the two trials, cells were incubated for an extended time of 30 hr. Control cells were treated with DMSO only.
Data analysis, percentile plots:
All analyses were carried out two or more times, except for that of G1 cells (Figure 5), which was conducted only once. To simplify the distance analyses for percentile plots, we assumed that cell lines with complex ploidies had the ploidy that was the most observed; for example, we assumed tetraploidy for Kc167 cells, 94% of which had four copies of the X chromosome, four copies of chromosome II, and three to five copies of chromosome III. In support of this strategy, the conclusions of our analyses were not altered when the data were reanalyzed assuming other possible combinations of the observed ploidies. For nuclei with more than one signal, we identified signals likely to represent more than one copy of the targeted region by considering the expected ploidy for the nucleus, the number of signals obtained, and relative signal intensities. Often signal intensities did not permit us to assign copy number and, in these cases, we assigned copy number at random. We rarely observed Kc167 nuclei (<1 of 2 × 103) with more than four signals; instances when this did occur may have been due to background or represent G2 nuclei with separated sister chromatids.
Figure 5.—

Pairing is maintained through G1, S, and G2 phases of the cell cycle. (A) Pairing profiles of G1, S, G2, and unsorted Kc167 cells imaged with FISH targeting 28B1. Analysis of G2, S, and unsorted cells was repeated with similar results (not shown). (B) Percentile plot of data in A showing that distance distributions of G1, S, G2, and unsorted cells do not differ significantly from each other (Ps ≥ 0.95). Inset, cells were stained with Hoechst 33342 and sorted by DNA content.
Nuclei that could not be scored due to lack of FISH signal, a weak signal-to-noise ratio, or high background were not included in the N values. With respect to assays that did not involve RNAi or chemical agents, the percentage of such nuclei varied from experiment to experiment. For example, Kc167 and clone 8 cells yielded, respectively, 15 ± 8% (four trials) and 35 ± 15% (three trials) of nuclei that were not labeled in assays targeting 8C8, 15 ± 7% (two trials) and 9 ± 1% (two trials) in assays targeting 16E1, 7 ± 5% (seven trials) and 7 ± 10% (eight trials) in assays targeting 28B1, and 15 ± 11% (two trials) and 25 ± 2% (two trials) in assays targeting 44F1. Note that there was no obvious correlation between the degree of pairing and percentage of nuclei lacking signal in these cells (our unpublished observation).
The total percentage of nuclei without signal in Kc167 cells treated with ICRF-193 was increased by an additional 2–27% (for example, a 27% increase would be one from 10 to 37%; three trials) in assays targeting 8C8, 12–14% (two trials) in assays targeting 16E1, 12–34% (five trials) in assays targeting 28B1, and 10–15% (two trials) in assays targeting 44F1. With regard to clone 8 cells treated with ICRF-193, we observed increases of 4–9% (three trials) in assays targeting 8C8, 12–16% (two trials) in assays targeting 16E1, 7–24% (six trials) in assays targeting 28B1, and 0–3% (two trials) in assays targeting 44F1. These observations also confirm the capacity of Drosophila cells to progress through mitosis in the presence of ICRF-193, consistent with other studies in which Top2 activity has been disrupted (Chang et al. 2003; Coelho et al. 2003). Note that nuclei that are abnormally monosomic for a region targeted by FISH would artificially inflate the percentage of nuclei with single signals representing paired regions, suggesting that our assessment of the impact of ICRF-193 on pairing may be an underestimate.
To simulate a random distribution of FISH signals in the nucleus, we implemented a simple algorithm in Perl in which the nuclear shape of a typical Kc167 nucleus was approximated as an ellipsoid defined by three radii (2.0, 2.0, and 2.5 μm; our unpublished observations), and the x, y, and z coordinates of any point within or on the surface of that ellipsoid were described by the following equation:
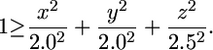 |
In our studies, we generated a random set of points (using the pseudorandom rand Perl function) with x, y, and z coordinates that fell in the ranges of [−2.0, 2.0] μm, [−2.0, 2.0] μm, and [−2.5, 2.5] μm, respectively. To model a tetraploid nucleus with randomly placed FISH signals, we generated four points per ellipsoid and calculated the distances between them.
Matlab 7.0.1SP1 and Microsoft Excel 11.2 were used to analyze and plot the data. A two-tailed chi-square test was used to compare matched populations of nuclei with respect to the frequency of nuclei with a single signal. When experiments with more than one trial were compared, trials were combined. A two-tailed two-sample Kolmogorov–Smirnov test was used to compare the distances between populations displayed in percentile plots.
RESULTS
Drosophila immortal cell lines support pairing:
We first asked whether Drosophila immortal cell lines support homolog pairing by examining the commonly used D. melanogaster Kc167 cell line, our subline of which is primarily tetraploid; among >50 nuclei (more than four trials), 94% had four copies of the X chromosome, four copies of chromosome II, and three to five copies of chromosome III (Table 1; Figure 1, A and B). To determine whether Kc167 cells pair homologous chromosomes, we fixed log-phase cells with formaldehyde and then labeled them with fluorescent probes and used wide-field deconvolution microscopy to determine the positions of the signals in three-dimensional space. Regions targeted by the probes were considered paired when the center-to-center distance between FISH signals was ≤0.50 μm, the approximate distance at which adjacent FISH signals no longer overlap.
TABLE 1.
Pairing is observed in seven different cell lines
| Year established | Karotypeb
|
||||
|---|---|---|---|---|---|
| Line | Origina | II | III | % pairedc | |
| Kc167 | Embryos | 1969 | 4 | 3–5 | 78 |
| S2 | Embryos | 1972 | 4, 6 | 4, 6 | 76 |
| D | Embryonic haploid cells | 1978 | 2, 4 | 2, 4 | 83 |
| DH-33 | Embryos, D. hydei | 1980 | 4 | 4–5 | 82 |
| mbn2 | Hemocytes | 1980 | NDd | NDd | 69 |
| Clone 8 | Imaginal wing discs | 1990 | 2 | 2 | 87 |
| ML-DmBG2-c6 | Larval central nervous system | 1994 | 3e | 2e | 86 |
See Figure 1 for examples of mitotic figures and materials and methods for references for cell lines.
All cells lines were from D. melanogaster, except as noted for DH-33.
N > 50 for Kc167, N = 20 for clone 8, and N > 7 for all other lines except mbn2 (ND, not determined).
Data represent the percentage of single-signal nuclei as assayed at the 28B1 region (Figure 4A except in the case Kc167, where the data correspond to Figure 2C); N > 55 for each line.
Assumed tetraploid.
Aberrant chromosome morphology (Figure 1, G and H).
The first probe we used targeted region 28B1, located in a euchromatic portion of chromosome II (Figure 2A). Strikingly, 78.4 ± 8.4% of labeled nuclei produced a single FISH signal (N = 1507, nine trials; Figure 2, B and C). This high percentage of single-signal nuclei suggests that the majority of Kc167 nuclei pair all four copies of the 28B1 region. To quantitate the range of distances between any two copies of 28B1, we assumed all nuclei to be tetraploid, calculated the distances between each pairwise combination of 28B1, as represented by FISH signals (Figure 2D, inset), and then arranged the distance data in a percentile plot (Figure 2D). Although distances between copies of 28B1 ranged from 0 μm to the full diameter of a nucleus (typically ∼4–5 μm), 88% of all possible pairwise combinations were paired. Consistent with our observations of 28B1, significant pairing was also found for five other euchromatic regions (Figure 2, A, E, and F). We found that 72, 76, 82, 86, and 75% of nuclei gave a single signal when FISH was targeted, respectively, to 8C8 and 16E1 in the middle of the X, 21E3 and 44F1 at the end of the left arm and in the middle of the right arm of chromosome II, and 69C2 in the middle of the left arm of chromosome III. This consistent and high level of pairing across all six regions assayed is remarkable; regardless of probe used, between 72 and 86% of nuclei contained only a single signal even though there is variability in the efficacy of FISH, as determined by signal strength, background staining, and the percentage of nuclei lacking signal (materials and methods).
Figure 2.—

Drosophila cell culture supports pairing. (A) Regions targeted by probes. Chromosome IV is not shown. (B) Flattened images of Kc167 nuclei stained with DAPI and FISH targeting 28B1. (C) Percentage of Kc167 nuclei ±SD (nine trials) with the indicated number of signals per nucleus identifying 28B1. (D) Percentile plot of data shown in C, assuming tetraploidy. All intersignal distances within each nucleus are ranked, shortest to longest, left to right, where 100% corresponds to the greatest distance observed. Signals separated by ≤0.50 μm (- - -) are considered one signal. Open circles, data from inset illustrating distance calculations of nucleus with three signals. (E and F) Pairing profile and percentile plot when FISH was targeted to euchromatic regions. (G and H) Pairing profile and percentile plot when FISH was targeted to regions in or near heterochromatin. In this and all other figures, bars, 5 μm; N, number of nuclei.
Using the percentile plot, we then assessed the statistical significance of the pairing we observed. First, we determined the frequency at which two or more of the four copies of 28B1 would be expected to colocalize solely by chance by computationally simulating three-dimensional nuclei containing four randomly placed FISH signals. Virtually no computer-simulated nuclei colocalized all four signals. In fact, 90% of these nuclei showed four completely separated signals. In addition, percentile plots revealed that only 2% of all pairwise combinations of signals were colocalized (black line, Figure 3A). In our second approach, we applied dual-labeled FISH to experimentally establish the frequency of colocalization for two regions that do not share homology. Here, we measured the distances between two sets of two loci each: 21E3 and 28B1, both on chromosome II, and 28B1 and 69C2, on chromosomes II and III, respectively (Figure 3B). Colocalization was observed for only 5 and 0.8% of all pairwise combinations of the 21E3 and 28B1 regions and the 28B1 and 69C2 regions, respectively (Figure 3B). These data demonstrate that euchromatic regions rarely overlap by chance, strongly reinforcing our observation that somatic homolog pairing can occur genomewide and be maintained in transformed cell lines. Importantly, the amount of pairing we obtained approximates that observed in vivo (Csink and Henikoff 1998; Fung et al. 1998; Gemkow et al. 1998; Sass and Henikoff 1999). As Kc167 cells represent a departure from diploidy, these data further indicate that somatic pairing can tolerate levels of aneuploidy that are typically found in established cell lines.
Figure 3.—

(A) Percentile plot for randomly placed FISH signals in theoretical nuclei with radii of 2, 2, and 2.5 μm compared to the percentile plot of an experiment in which Kc167 nuclei were labeled with probe targeting 28B1. Distances between randomly placed FISH signals are significantly greater than those between signals representing 28B1 (P < 1 × 10−10). (B) Percentile plot of distances between signals representing 21E3 and 28B1 and between signals representing 28B1 and 69C2. Data from A are added for comparison. Distances between signals representing 21E and 28B1 or 28B1 and 69C2 are significantly greater than those between signals representing 28B1 in A (Ps < 1 × 10−10).
In contrast to our findings at euchromatic regions, we found significantly less pairing at regions located in or near the centromeric heterochromatin of all three major chromosomes (P ≤ 0.05; Figure 2, G and H). Using probes targeting a 359-bp repeated element of the centromeric heterochromatin of X, region 40A2 located just outside the centromeric heterochromatin of chromosome II, and the dodeca-repeated element located in the centromeric heterochromatin of chromosome III (Figure 2A), we observed only 24, 51, and 13%, respectively, of nuclei giving a single signal (Figure 2G) and correspondingly modest pairing levels of 45, 67, and 49% (Figure 2H). While it is possible that this lower level of pairing resulted not from the unpaired state of homologs but rather from the potential of repeated elements to form multiple distinct clusters, the apparent reduction in pairing we observed when visualizing unique sequences in the pericentromeric region of 40A2 argues against this as the sole interpretation. Interestingly, Halfer and Barigozzi (1973) also observed that the heterochromatic regions of condensed mitotic chromosomes were less paired than their euchromatic counterparts. Note that as mitotic cells represented only ∼2.1% (see below) of our cultures, our findings cannot be explained solely by the behavior of mitotic chromosomes.
Homolog pairing occurs in a variety of cell lines:
The widespread and persistent nature of homolog pairing in Kc167 cells encouraged us to ask whether the mechanism underlying pairing has also been maintained in other cell lines and, if so, whether the degree of pairing would vary with cell type or culture history. Accordingly, we assayed pairing at 28B1 in six additional established cell lines (Table 1). These lines differ in terms of their species of origin (D. melanogaster and D. hydei), morphology, tissue of origin (embryonic, epithelial, hemocyte, imaginal disc, and CNS), year of establishment (1969–1994), and karyotype/ploidy (diploid, polyploid/aneuploid). They include the popular S2 cell line (Schneider 1972), which was derived from late-stage embryos and, like its derivative sublines such as S2R+ (Yanagawa et al. 1998), is used in a wide variety of studies. The S2 cells we used proved to be generally tetraploid. In contrast, the male (X/Y) clone 8 cell line, which originated from imaginal wing discs, is diploid (Table 1) (Peel et al. 1990; Peel and Milner 1991). The D cell line is also noteworthy, as it was established from haploid embryos (Debec 1978, 1984), even though it is currently a mixed population of diploid and tetraploid cells (Table 1); as such, there is no history of in vivo homolog pairing for chromosomes of the D cell line. Following standard protocols (materials and methods), aliquots of each of the cell lines were cultured to generate log-phase cells. Using probes targeting 28B1, we found a nearly uniform high level of pairing that was reminiscent of that observed in Kc167 cells (Figure 4).
Figure 4.—

Seven different cell lines support pairing. (A) Pairing profiles and (B) percentile plots of nuclei imaged with FISH directed against 28B1.
Homolog pairing remains equally strong in G1, S, and G2:
Taking advantage of the amenability of cell culture to cell sorting, we addressed the impact of cell cycle on pairing. Log-phase Kc167 cells incubated in Hoechst dye were sorted into G1, S, and G2 subpopulations on the basis of fluorescence intensity. Sorted cells were then allowed to adhere to glass slides for ∼30 min in medium prior to being fixed in formaldehyde and then subjected to FISH analysis. Our data indicate that the level of pairing, as assayed at 28B1, is uniform across all three subpopulations (Figure 5) and does not differ significantly from that of unsorted cells (Ps ≥ 0.95); the slightly lower level of pairing observed for unsorted cells may reflect its inclusion of mitotic cells.
Although we cannot rule out that the observed pairing derives from a potential technical bias that enriches for cells in which homologs are paired, it is noteworthy that our observation is in line with the report by Fung et al. (1998) that pairing can be maintained through S phase and into metaphase. A constant level of pairing in the G1, S, and G2 portions of the cell cycle differs, however, from the disruption of pairing described by Csink and Henikoff (1998) in S-phase larval CNS cells. These diverse patterns of pairing may reflect different sensitivities of pairing to cell type, ploidy, and/or length of the cell cycle (Golic and Golic 1996; Gubb et al. 1997).
To examine mitotic chromosomes, we treated cells with a hypotonic solution and then fixed them in a mix of acetic acid and methanol (Echalier 1997). In this way, we were occasionally able to observe an intertwining of chromosomes in Kc167 cells (Figure 1, C and D) as well as unambiguous alignments of homologs as fully condensed mitotic chromosomes, in the form of paired dyads, in clone 8 cells (Figure 1, E and F). These findings are reminiscent of the homolog entanglements and alignments characterized in vivo by Stevens (1907, 1908), Metz (1916, 1922, 1925), and Holt (1917) and in Drosophila cell lines by Halfer and Barigozzi (1973), as well as in Aedes cell lines by Nichols et al. (1971). They demonstrate that homologous chromosomes have a capacity to remain paired in at least some stages of mitosis and are consistent with FISH analyses showing that pairing is not perturbed at the histone locus during the 13th embryonic mitosis until after metaphase (Fung et al. 1998). That M-phase pairing is tenacious is furthered by its ability to withstand hypotonic treatment.
Disruption of Top2 perturbs homolog pairing:
Taken together, our data establish that pairing in cell culture approximates pairing in vivo in terms of level and tenacity. This finding suggests that Drosophila cell culture can serve as a powerful experimental system for identifying genes responsible for somatic homolog pairing. In particular, FISH-based screens in cell culture would enable direct visual assessments of pairing in different RNAi-driven mutant backgrounds. In fact, focusing our analysis on euchromatic regions, where homolog pairing is maximal, we have found that dsRNA targeting Drosophila Top2 expression can reduce the level of pairing in Kc167 cells.
Top2 encodes the single Drosophila type II DNA topoisomerase, which represents a class of enzymes that generate double-strand breaks (DSBs), can catenate and decatenate DNA, and are well known for their role in chromosome structure and condensation and sister chromatid segregation (reviewed by Porter and Farr 2004; also see Chang et al. 2003; Coelho et al. 2003; Savvidou et al. 2005; Smiley et al. 2007). We have found that treatment of log-phase Kc167 cells with any one of three dsRNAs directed against Top2 function followed by FISH targeting 28B1 reduced the percentage of nuclei with a single signal from 87 ± 1% to 55 ± 5% (P < 0.0001; Figure 6, A and B) and lowered pairing levels from 91 to 71% (P < 0.0001; Figure 6C).
To extend these findings, we determined the impact of two chemical agents that block topoisomerase II: ICRF-193, which stabilizes a noncovalent complex between the enzyme and DNA without generating DSBs, and m-amsacrine, which leads to the accumulation of DSBs (reviewed by Andoh and Ishida 1998; Larsen et al. 2003). Treatment with ICRF-193 (10 μm) reduced the percentage of nuclei with a single signal from 84 ± 4% to 54 ± 4% (P < 0.0001; Figures 6D and 7; also see Table 2, which includes data not shown in Figure 6D). This finding is unlikely to reflect an increase in the percentage of cells in M phase as the mitotic index of ICRF-193-treated cells is, if anything, lower than that of untreated controls (2.1 and 1.0%, respectively; Figure 6D, inset; Chang et al. 2003; Coelho et al. 2003). In contrast to treatment with ICRF-193, treatment with m-amsacrine (5 μm) had much less of an effect (Figures 6D and 7). This result is consistent with the fact that ICRF-193 and m-amsacrine have different mechanisms of action (reviewed by Andoh and Ishida 1998; Larsen et al. 2003), although we cannot rule out a technical basis for the ineffectiveness of m-amsacrine. Treatment with dsRNA (Figure 6, A–C) or an enzymatic inhibitor, camptothecin (4 μm), directed against a different topoisomerase, topoisomerase I (Top1; reviewed by Pommier 2006), had similarly minimal effects in our assays (Figures 6D and 7). Finally, we demonstrated that the impact of ICRF-193 was not specific for 28B1 by targeting FISH to 8C8, 16E1, and 44F1. We observed a decrease in the percentage of single-signal nuclei from 73 ± 2% to 55 ± 2%, 65 ± 7% to 32 ± 4%, and 79 ± 10% to 59 ± 9%, respectively, suggesting that the effect of ICRF-193 on pairing may be general, although variable, across euchromatic regions (Table 2, Ps < 0.0001; Figure 6E).
Figure 7.—

ICRF-193 disrupts pairing in Kc167 cells. Nuclei are from Kc167 cells treated with inhibitors of topoisomerase I (4 μm camptothecin) or topoisomerase II (5 μm m-amsacrine, 10 μm ICRF-193) and imaged with FISH targeting 28B1. Control nuclei are from cells treated with DMSO. Note the nuclei of cells treated with ICRF-193 can be misshapen. Nuclei are from cultures represented in Figure 6, D and E.
TABLE 2.
ICRF-193 disrupts pairing more effectively in the presence of a homolog
| 8C8 (%)
|
16E1 (%)
|
28B1 (%)
|
44F1 (%)
|
|||||
|---|---|---|---|---|---|---|---|---|
| Clone 8 | Kc167 | Clone 8 | Kc167 | Clone 8 | Kc167 | Clone 8 | Kc167 | |
| Control | 93 ± 2 | 73 ± 2 | 89 ± 1 | 65 ± 7 | 84 ± 4 | 84 ± 5 | 90 ± 6 | 79 ± 10 |
| ICRF-193 | 86 ± 2 | 55 ± 2 | 83 ± 1 | 32 ± 0.4 | 51 ± 5 | 52 ± 5 | 75 ± 3 | 59 ± 9 |
| Difference | 7 ± 3 | 18 ± 2 | 6 ± 1 | 33 ± 7 | 31 ± 6 | 32 ± 6 | 15 ± 7 | 20 ± 13 |
Data represent the percentage of single-signal nuclei (±SD). Ns = 149–583 (two to three trials), except in the case of Kc167 cells treated with ICRF-193 and labeled with probe targeting 16E1, where N = 93 (two trials). Underlining indicates data from FISH analyses targeting regions on the X chromosome in clone 8 cells, which carry only a single X.
These observations must be considered in light of the cell cycle and the two forms of homologous pairing that come into play: pairing between homologs (including the pairing of subchromosomal regions or fragments thereof) and pairing, or cohesion, between sister chromatids (Figure 8A). A priori, reduced pairing in G1 prior to the synthesis of sister chromatids would necessarily imply the unpaired state of homologs. In contrast, reduced pairing in G2 could result from the unpaired state of either homologs or sister chromatids or some combination thereof (see Figure 8 legend for further discussion). The data presented thus far do not distinguish between these possibilities.
Figure 8.—

Use of clone 8 cells to distinguish unpaired homologs from separated sister chromatids. (A) In tetraploid Kc167 cells, which carry four X chromosomes, multiple signals in a nucleus may represent unpaired homologs in either G1 or G2 or separated sister chromatids in G2, regardless of whether they identify X-linked or autosomal regions. (B) In clone 8 cells, which carry only one X chromosome, multiple signals identifying X-linked regions can represent only separated sister chromatids in G2. In contrast, multiple signals identifying autosomal regions can represent unpaired homologs in either G1 or G2 or separated sister chromatids in G2. Note that the number of homologs or sister chromatids that are unpaired can differ from that shown. Variation of signal intensity did not allow accurate experimental determination of copy number per signal (data not shown). Also note that homolog pairing could occur between the sister chromatid of a chromosome and the homolog of that same chromosome. As such, the forms of sister chromatid separation illustrated may also entail an unpairing of homologs. If so, any separation of sister chromatids will necessarily imply homolog unpairing. This figure does not address situations involving aneuploidy. N/A, sister chromatids do not exist prior to replication.
Equally important for consideration is that mitosis in the presence of Top2 inhibitors can cause sister chromatids to cosegregate to the same daughter nucleus (not shown in Figure 8; reviewed by Porter and Farr 2004) and produce cells that are monosomic, nullisomic, or segmentally aneuploid. Indeed, treatment with ICRF-193 generated a significant level of aneuploidy; we observed that the total percentage of nuclei lacking FISH signal can be increased by an additional 34% (for example, from 3 to 37%), indicating that the mitotic divisions producing these nuclei had cosegregated all copies of the targeted chromosomal region to only one daughter nucleus (see materials and methods for additional information). As such, an increase in the percentage of nuclei with multiple FISH signals from cultures treated with ICRF-193 could also reflect the separation in G1 aneuploid cells of what were once sister chromatids.
To decipher what form of unpairing underlies the effect of ICRF-193, we turned to male (X/Y) clone 8 cells where, a priori, lack of a homolog for the single X (N = 20 cells; Table 1) should allow the unpaired state of homologs to be more easily distinguished from sister chromatid separation (Figure 8B). Specifically, conditions that disrupt only homolog pairing should decrease the number of single-signal nuclei when FISH is targeted to autosomal regions but not when FISH is targeted to X-linked regions. In contrast, conditions that separate sister chromatids in G2 and/or in aneuploid G1 cells should decrease the number of single-signal nuclei regardless of whether FISH targets autosomal or X-linked regions.
We found that application of ICRF-193 to clone 8 cells reduced the percentage of single-signal nuclei by only 6.9% (of total nuclei scored) when FISH was targeted to 16E1 on the X even as it reduced the percentage by 37.8% when FISH was targeted to autosomal 28B1 (P = 0.02 and P < 0.0001, respectively; data included in Figure 6, F and G, and Table 2; Figure 9). Aiming to augment the impact of ICRF-193 at 16E1, we repeated our analysis while increasing the exposure time of the cells to ICRF-193. Again, we observed a much smaller effect when FISH was targeted to 16E1 as compared to 28B1, with the reduction of single-signal nuclei being 4.5 and 39.7%, respectively (P = 0.08 and P < 0.0001, respectively; data included in Figure 6, F and G, and Table 2). Similar results were obtained when we targeted FISH to another X-linked region, 8C8, and another autosomal locus, 44F1; here we observed that single-signal nuclei were reduced by 7 ± 3% and 15 ± 7%, respectively (Ps < 0.0001; Table 2). In short, although we observed small reductions in the percentage of single-signal nuclei when FISH was targeted to regions on the X, we observed greater reductions when FISH was targeted to autosomal regions. Importantly, this lesser effect at 16E1 and 8C8 is not due to an insensitivity of these regions to ICRF-193, as pairing at 16E1 and 8C8 in Kc167 cells is as susceptible to ICRF-193 as is pairing at autosomal regions (Figure 6, D and E; Table 2). These findings indicate that while sister chromatid separation may be occurring, it is unlikely to fully explain the effect of ICRF-193, arguing that loss of Top2 activity can disrupt homolog pairing.
Figure 9.—

ICRF-193 disrupts pairing in clone 8 cells. Nuclei are from clone 8 cells treated with inhibitors of topoisomerase I (4 μm camptothecin) or topoisomerase II (5 μm m-amsacrine, 10 μm ICRF-193) and imaged with FISH targeting 28B1 (A) or 16E1 (B). Control nuclei are from cells treated with DMSO. Nuclei are from cultures represented in Figure 6, F and G.
As mentioned just above, our data suggest that loss of Top2 function may also disturb sister chromatid cohesion to a limited extent. Consistent with this, we have found that treatment with ICRF-193 augments the number of nuclei whose single signals actually consist of two fluorescent spots that are, however, too closely spaced (≤0.50 μm) to be considered separate (materials and methods). The total percentage of nuclei with such closely spaced spots was increased by an additional 1–11% (three trials; for example, an 11% increase would be one from 1 to 12%) and 6–8% (two trials) in clone 8 cells when FISH was targeted to 8C8 and 16E1, respectively, and 0–11% (two trials) and 2–10% (five trials) when FISH was targeted to 28B1 and 44F1, respectively. This observation suggests that ICRF-193 may antagonize sister chromatid cohesion in G2 nuclei in addition to disrupting homolog pairing.
DISCUSSION
In this study, we demonstrated that Drosophila immortal cell lines can support homolog pairing. Using FISH, we found remarkably consistent and high levels of pairing in tetraploid Kc167 cells and observed that such levels equally characterize G1, S, and G2 cells. Strong pairing was also seen in six additional Drosophila cell lines representing two species and different tissue types and ploidies. Of particular note is the pairing that occurs in the D cell line. As this line originated from haploid cells, becoming a mix of diploid and tetraploid cells only after having become established (Table 1) (Cherbas 2007), the pairing observed in D cells indicates that homolog pairing can initiate ex vivo and does not require homologs to be of different parental origin.
We also observed homolog pairing during M phase. Such homolog alignment is reminiscent of meiotic pairing and may reflect a commonality between the two forms of pairing. It further suggests that homolog interactions may be able to endure cell division, perhaps via the persistence of chromatin marks, physical associations, nuclear territories (Fung et al. 1998; Bantignies et al. 2003), or perichromosomal layers (Van Hooser et al. 2005).
Theoretically, enduring associations are compatible with mitosis. For instance, if each sister chromatid of a G2 dyad is paired with one sister of the homologous dyad to produce two sets of paired homologs, pairing could be maintained through mitosis if paired homologs cosegregate. In fact, assuming sister chromatid separation of one dyad to be random with respect to that of its homologous dyad, paired homologs will as often cosegregate as they will be pulled apart (Fung et al. 1998). This scenario predicts a loss of pairing during mitosis, but not a complete loss, and is consistent with observations of embryonic mitoses (Fung et al. 1998). Note, however, that mitosis could also progress without fully unpairing any set of homologs while still accommodating the unpairing observed during mitosis. For example, if homologs were permanently attached but at only a few or just one locus, regions elsewhere on the chromosomes would be free to unpair. Alternatively, if paired homologs were permanently attached via one strand of DNA from each homolog such that replication produced two dyads that are paired via only one sister per dyad, mitoses would generate a mix of paired and unpaired homologs, the latter being expected to become paired in the subsequent G1. Mechanisms such as these raise the possibility of immortal chromosome attachments and recall observations of immortal DNA strands in asymmetrically dividing cells (reviewed by Cairns 2006; see also Conboy et al. 2007). Finally, we note that mitosis may be a means by which pairing and pairing-sensitive phenomena are regulated; if there are instances when pairing must be maintained, mitotic mechanisms could orchestrate paired homologs at the metaphase plate such that they cosegregate while, if ever pairing must be destroyed, the cell could trigger a mitosis in which paired homologs are forced apart.
Our studies also revealed that pairing of regions in or near centric heterochromatin is significantly less than that observed at euchromatic regions. This finding is consistent with observations of condensed mitotic chromosomes by Halfer and Barigozzi (1973) and could be explained if heterochromatic regions pair less, unpair more, or pair more slowly than do euchromatic regions. The prevalence of repeated sequences in heterochromatin may contribute to the unpaired state of homologs by, for example, promoting intrachromosomal pairing and/or homolog misalignment. Our data are also consistent with a loss of sister chromatid cohesion in centromeric regions. This explanation, however, would not be in line with studies showing that cohesin proteins, which hold sister chromatids together, persist at the centromeres longer than they do along chromosome arms (reviewed by Wang and Dai 2005; Watanabe 2005). As such, our data may indicate that the lower degree of homolog pairing observed in or near pericentric heterochromatin in Kc167 cells results from a preoccupation of centromeric regions with sister chromatid, vs. homolog, pairing.
This report also summarizes our identification of Top2 as a gene important for homolog pairing. Here, we found that loss of Top2 function reduced pairing to a greater degree at autosomal vs. X-linked regions in diploid male clone 8 cells. This finding indicates that the reduction in pairing cannot be explained solely by a loss of cohesion between sister chromatids and argues that Top2 plays a role in homolog pairing. How might Top2 influence homolog pairing? It might act indirectly through its role in cell cycle events, such as chromosome condensation, congression of chromosomes at the metaphase plate, and separation of sister chromatids during mitosis (reviewed by Porter and Farr 2004; also see Chang et al. 2003; Coelho et al. 2003; Hossain et al. 2004; Savvidou et al. 2005). For example, cosegregation of sister chromatids due to a lack of Top2 activity may generate a degree of disorganization that is incompatible with homolog pairing. Alternatively, abnormal presence of a sister chromatid may prohibit a chromosome from pairing with its homolog. That sister chromatid and homolog pairing may be mutually exclusive raises the possibility that the two are mutually regulated and may even explain how the genome can orchestrate sister chromatid separation during mitosis even as it promotes homolog pairing in the ensuing interphase. Interestingly, as sister chromatid cohesion has been implicated in gene regulation (Azuara et al. 2003; Mlynarczyk-Evans et al. 2006; also see Dorsett 2007), an interplay between homolog pairing and sister chromatid cohesion could also provide a mechanism by which homolog pairing can modulate gene regulation.
Top2 may also have a direct role in the paired state of homologs. For example, it may bring and/or hold homologs together by altering DNA topology, modifying chromatin or chromosome structure, or catenating DNA molecules (Bachant et al. 2002; Vazquez et al. 2002; Iwabata et al. 2005). Alternatively, it may dimerize while bound to DNA or via its role as a chromosome structural/scaffold protein (Earnshaw and Heck 1985; Earnshaw et al. 1985; Gasser et al. 1986; more recently Maeshima and Laemmli 2003; Maeshima et al. 2005); for instance, it may act in conjunction with Su(Hw) (Fritsch et al. 2006), whose DNA binding site contains the in vitro recognition sequence for topoisomerase II (Nabirochkin et al. 1998). Interestingly, if homolog pairing is required for the proper arrangement and segregation of chromosomes during mitosis, such unpairing could have contributed to the aneuploidy we observed in the presence of ICRF-193.
Top2 may also influence pairing via its capacity to decatenate chromosomes; just as the decatenation activity of Top2 is required for the separation of sister chromatids, so may it be required prior to or during mitosis for the separation of paired homologs, should such pairing involve catenation. In this case, loss of Top2 would lead to the cosegregation of catenated homologs, whose entanglement may then inhibit homolog pairing, and perhaps even sister chromatid cohesion, in the daughter nuclei.
Finally, it is worth considering the role of topoisomerase II activity in transcription (Ju et al. 2006), where it appears to generate transient DSBs during gene activation. Top2 may therefore affect pairing via its impact on chromatin structure during the transcriptional process. This interpretation is reminiscent of the implication of transcription, and the regulation thereof, in pairing (reviewed by Mckee 2004) and the association of Top2 with PcG target sequences and Polyhomeotic (Lupo et al. 2001), both of which participate in pairing-sensitive gene regulatory phenomena (reviewed by Pirrotta 1999; Kassis 2002; Kavi et al. 2006).
Our findings are consistent with proposals suggesting that topoisomerase II may contribute to pairing during meiosis (Cobb et al. 1997; Vazquez et al. 2002; Iwabata et al. 2005). In this regard, it is of interest that Spo11, which is homologous to a type II DNA topoisomerase, promotes meiotic pairing through the generation of DSBs in yeast, mice, and plants (reviewed by Mckee 2004; Gerton and Hawley 2005; Pawlowski and Cande 2005; Colaiacovo 2006; Zickler 2006). As such, the role of Top2 in somatic pairing may be analogous to that of type II topoisomerases in meiotic pairing. In fact, a contribution of Top2 to pairing in nonmeiotic cells may explain why Drosophila meiosis, which is believed to arise from the pairing that occurs in premeiotic cells (reviewed by Mckee 2004; Gerton and Hawley 2005; Zickler 2006), does not require the generation of meiotic DSBs.
Curiously, absence of Spo11, as well as other genetic backgrounds, can lead to the meiotic pairing of nonhomologous chromosomes (Mcclintock 1933; reviewed by Mckee 2004; Gerton and Hawley 2005; Pawlowski and Cande 2005; Colaiacovo 2006; Osman et al. 2006; Zickler 2006; also see Tsubouchi and Roeder 2005), which was proposed by Mcclintock (1933) to be competitive with homologous pairing. Hence, loss of somatic pairing brought about by disrupting Top2 activity may be accompanied, maintained, or even driven by nonhomologous pairing. The concept that homologous and nonhomologous pairing are mutually exclusive suggests that interplay between them may provide a general mechanism for modulating nuclear organization and gene expression. Indeed, interactions between nonhomologous chromosomal regions may be as potent as those between homologous regions and, as may be the case with homologous pairing, have the capacity to persist through and be regulated by mitosis. Such nonhomologous interactions range from those ensuing from the clustering of PREs (Grimaud et al. 2006) and insulators scattered throughout the genome (reviewed by Kuhn and Geyer 2003; Capelson and Corces 2004; Brasset and Vaury 2005; Gaszner and Felsenfeld 2006; Valenzuela and Kamakaka 2006) to those occurring in transcription factories (reviewed by Dillon 2006; Faro-Trindade and Cook 2006; Fraser 2006). Excitingly, they also include gene-specific interactions, such as those that occur in mammals between enhancers and distant promoters during olfactory receptor choice (Lomvardas et al. 2006), between the interferon-γ and TH2 cytokine loci during T-cell development (Spilianakis et al. 2005), and between the insulin-like growth factor 2/H19 imprinting control region and the chromosomal segment containing the Wsb1 and Nf1 loci in a manner that appears to require the CCCTC binding factor CTCF (Ling et al. 2006).
In closing, we note that while our studies have sought conditions that reduce somatic pairing in Drosophila cell culture, it will also be informative to seek situations in which pairing can be enhanced or induced. In particular, might the capacity to pair be an inherent property of chromosomes that is, however, precluded or suppressed in most organisms? Extending a suggestion by McClintock in 1933 of a “powerful force” that promotes pairing in meiosis (Mcclintock 1933), it may be that the paired state of chromosomes or chromosomal regions, be it homologous or not, is energetically, kinetically, or structurally favored (also see Lee et al. 2004) and must, therefore, be actively controlled. That somatic pairing may be a widespread potential of chromosomes is consistent with the abundance of homology- and pairing-dependent regulatory mechanisms as well as reports of nonhomologous interactions, while a necessity to suppress pairing is conceivable in situations where continual interallelic communication would be detrimental, such as may be the case when alleles are differentially regulated.
Acknowledgments
We thank Kami Ahmad for alerting us to the article by Halfer and Barigozzi (1973); Monica Colaiácovo for critical input and generous access to her Deltavision Imaging System; the Drosophila Genomics Research Center for cell lines; Kami Ahmad, Frédéric Bantignies, Giacomo Cavalli, and George Church for technical advice and discussion; John Aach, Mar Carmena, Lucy Cherbas, Charleston Chiang, Stephen Elledge, Richard Emmons, Allan Gurtan, Scott Hawley, Amber Hohl, Tao Hsieh, Matt Jakubik, Mitzi Kuroda, Erica Larschan, Anne Lee, Jeannie Lee, Lillian Merriam, Norbert Perrimon, and Johannes Walter for thoughtful input; and Anna Moran and Laura Stadelmann for technical assistance. We apologize to authors we could not cite due to space limitations. This study was supported by a National Science Foundation Graduate Research Fellowship to B.R.W. and by a Ruth L. Kirschstein National Research Service Award (GM-67460) and a grant (GM-61936) from the National Institutes of Health to J.R.B. and C.-t.W., respectively.
References
- Andoh, T., and R. Ishida, 1998. Catalytic inhibitors of DNA topoisomerase II. Biochim. Biophys. Acta 1400: 155–171. [DOI] [PubMed] [Google Scholar]
- Azuara, V., K. E. Brown, R. R. Williams, N. Webb, N. Dillon et al., 2003. Heritable gene silencing in lymphocytes delays chromatid resolution without affecting the timing of DNA replication. Nat. Cell Biol. 5: 668–674. [DOI] [PubMed] [Google Scholar]
- Bachant, J., A. Alcasabas, Y. Blat, N. Kleckner and S. J. Elledge, 2002. The SUMO-1 isopeptidase Smt4 is linked to centromeric cohesion through SUMO-1 modification of DNA topoisomerase II. Mol. Cell 9: 1169–1182. [DOI] [PubMed] [Google Scholar]
- Bacher, C. P., M. Guggiari, B. Brors, S. Augui, P. Clerc et al., 2006. Transient colocalization of X-inactivation centres accompanies the initiation of X inactivation. Nat. Cell Biol. 8: 293–299. [DOI] [PubMed] [Google Scholar]
- Bantignies, F., C. Grimaud, S. Lavrov, M. Gabut and G. Cavalli, 2003. Inheritance of Polycomb-dependent chromosomal interactions in Drosophila. Genes Dev. 17: 2406–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bantignies, F., C. Grimaud and G. Cavalli, 2005. Two-colour fluorescent in situ DNA hybridization on whole mount Drosophila embryos and larval imaginal discs (PROT07). (http://www.epigenome-noe.net/researchtools/protocol.php?protid=5).
- Brasset, E., and C. Vaury, 2005. Insulators are fundamental components of the eukaryotic genomes. Heredity 94: 571–576. [DOI] [PubMed] [Google Scholar]
- Cairns, J., 2006. Cancer and the immortal strand hypothesis. Genetics 174: 1069–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capelson, M., and V. G. Corces, 2004. Boundary elements and nuclear organization. Biol. Cell 96: 617–629. [DOI] [PubMed] [Google Scholar]
- Chang, C. J., S. Goulding, W. C. Earnshaw and M. Carmena, 2003. RNAi analysis reveals an unexpected role for topoisomerase II in chromosome arm congression to a metaphase plate. J. Cell Sci. 116: 4715–4726. [DOI] [PubMed] [Google Scholar]
- Chen, J. D., and V. Pirrotta, 1993. Stepwise assembly of hyperaggregated forms of Drosophila zeste mutant protein suppresses white gene expression in vivo. EMBO J. 12: 2061–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherbas, L., 2007. Drosophila Genomics Resource Center (DGRC) protocols. (http://dgrc.cgb.indiana.edu/cells/protocols.html).
- Cherbas, L., and P. Cherbas, 1997. “Parahomologous” gene targeting in Drosophila cells: an efficient, homology-dependent pathway of illegitimate recombination near a target site. Genetics 145: 349–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens, J. C., C. A. Worby, N. Simonson-Leff, M. Muda, T. Maehama et al., 2000. Use of double-stranded RNA interference in Drosophila cell lines to dissect signal transduction pathways. Proc. Natl. Acad. Sci. USA 97: 6499–6503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb, J., R. K. Reddy, C. Park and M. A. Handel, 1997. Analysis of expression and function of topoisomerase I and II during meiosis in male mice. Mol. Reprod. Dev. 46: 489–498. [DOI] [PubMed] [Google Scholar]
- Coelho, P. A., J. Queiroz-Machado and C. E. Sunkel, 2003. Condensin-dependent localisation of topoisomerase II to an axial chromosomal structure is required for sister chromatid resolution during mitosis. J. Cell Sci. 116: 4763–4776. [DOI] [PubMed] [Google Scholar]
- Colaiacovo, M. P., 2006. The many facets of SC function during C. elegans meiosis. Chromosoma 115: 195–211. [DOI] [PubMed] [Google Scholar]
- Conboy, M. J., A. O. Karasov and T. A. Rando, 2007. High incidence of non-random template strand segregation and asymmetric fate determination in dividing stem cells and their progeny. PLoS Biol. 5: e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csink, A. K., and S. Henikoff, 1998. Large-scale chromosomal movements during interphase progression in Drosophila. J. Cell Biol. 143: 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debec, A., 1978. Haploid cell cultures of Drosophila melanogaster. Nature 274: 255–256. [DOI] [PubMed] [Google Scholar]
- Debec, A., 1984. Evolution of karyotype in haploid cell lines of Drosophila melanogaster. Exp. Cell Res. 151: 236–246. [DOI] [PubMed] [Google Scholar]
- Dej, K. J., and A. C. Spradling, 1999. The endocycle controls nurse cell polytene chromosome structure during Drosophila oogenesis. Development 126: 293–303. [DOI] [PubMed] [Google Scholar]
- Dernburg, A., 2000. In situ hybridization to somatic chromosomes, pp. 24–55 in Drosophila Protocols, edited by W. Sullivan, M. Ashburner and R. S. Hawley. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [DOI] [PubMed]
- Dernburg, A. F., and J. W. Sedat, 1998. Mapping three-dimensional chromosome architecture in situ. Methods Cell Biol. 53: 187–233. [DOI] [PubMed] [Google Scholar]
- Diaz-Perez, S. V., D. O. Ferguson, C. Wang, G. Csankovszki, C. Wang et al., 2006. A deletion at the mouse Xist gene exposes trans-effects that alter the heterochromatin of the inactive X chromosome and the replication time and DNA stability of both X chromosomes. Genetics 174: 1115–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon, N., 2006. Gene regulation and large-scale chromatin organization in the nucleus. Chromosome Res. 14: 117–126. [DOI] [PubMed] [Google Scholar]
- Donohoe, M. E., L. F. Zhang, N. Xu, Y. Shi and J. T. Lee, 2007. Identification of a Ctcf cofactor, Yy1, for the X chromosome binary switch. Mol. Cell 25: 43–56. [DOI] [PubMed] [Google Scholar]
- Dorsett, D., 2007. Roles of the sister chromatid cohesion apparatus in gene expression, development, and human syndromes. Chromosoma 116: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan, I. W., 2002. Transvection effects in Drosophila. Annu. Rev. Genet. 36: 521–556. [DOI] [PubMed] [Google Scholar]
- Earnshaw, W. C., and M. M. Heck, 1985. Localization of topoisomerase II in mitotic chromosomes. J. Cell Biol. 100: 1716–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earnshaw, W. C., B. Halligan, C. A. Cooke, M. M. Heck and L. F. Liu, 1985. Topoisomerase II is a structural component of mitotic chromosome scaffolds. J. Cell Biol. 100: 1706–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echalier, G., 1997. Drosophila Cells in Culture. Academic Press, San Diego.
- Echalier, G., and A. Ohanessian, 1969. Isolation, in tissue culture, of Drosophila melangaster cell lines. C. R. Acad. Sci. Hebd. Seances Acad. Sci. D 268: 1771–1773. [PubMed] [Google Scholar]
- Faro-Trindade, I., and P. R. Cook, 2006. Transcription factories: structures conserved during differentiation and evolution. Biochem. Soc. Trans. 34: 1133–1137. [DOI] [PubMed] [Google Scholar]
- Fraser, P., 2006. Transcriptional control thrown for a loop. Curr. Opin. Genet. Dev. 16: 490–495. [DOI] [PubMed] [Google Scholar]
- Fritsch, C., G. Ploeger and D. J. Arndt-Jovin, 2006. Drosophila under the lens: imaging from chromosomes to whole embryos. Chromosome Res. 14: 451–464. [DOI] [PubMed] [Google Scholar]
- Fung, J. C., W. F. Marshall, A. Dernburg, D. A. Agard and J. W. Sedat, 1998. Homologous chromosome pairing in Drosophila melanogaster proceeds through multiple independent initiations. J. Cell Biol. 141: 5–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser, S. M., T. Laroche, J. Falquet, E. Boy de la Tour and U. K. Laemmli, 1986. Metaphase chromosome structure. Involvement of topoisomerase II. J. Mol. Biol. 188: 613–629. [DOI] [PubMed] [Google Scholar]
- Gaszner, M., and G. Felsenfeld, 2006. Insulators: exploiting transcriptional and epigenetic mechanisms. Nat. Rev. Genet. 7: 703–713. [DOI] [PubMed] [Google Scholar]
- Gemkow, M. J., P. J. Verveer and D. J. Arndt-Jovin, 1998. Homologous association of the Bithorax-Complex during embryogenesis: consequences for transvection in Drosophila melanogaster. Development 125: 4541–4552. [DOI] [PubMed] [Google Scholar]
- Gerton, J. L., and R. S. Hawley, 2005. Homologous chromosome interactions in meiosis: diversity amidst conservation. Nat. Rev. Genet. 6: 477–487. [DOI] [PubMed] [Google Scholar]
- Gloor, G. B., 2002. The role of sequence homology in the repair of DNA double-strand breaks in Drosophila. Adv. Genet. 46: 91–117. [DOI] [PubMed] [Google Scholar]
- Golic, M. M., and K. G. Golic, 1996. A quantitative measure of the mitotic pairing of alleles in Drosophila melanogaster and the influence of structural heterozygosity. Genetics 143: 385–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant-Downton, R. T., and H. G. Dickinson, 2004. Plants, pairing and phenotypes–Two's company? Trends Genet. 20: 188–195. [DOI] [PubMed] [Google Scholar]
- Grimaud, C., F. Bantignies, M. Pal-Bhadra, P. Ghana, U. Bhadra et al., 2006. RNAi components are required for nuclear clustering of Polycomb group response elements. Cell 124: 957–971. [DOI] [PubMed] [Google Scholar]
- Gubb, D., J. Roote, J. Trenear, D. Coulson, and M. Ashburner, 1997. Topological constraints on transvection between white genes within the transposing element TE35B in Drosophila melanogaster. Genetics 146: 919–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haars, R., H. Zentgraf, E. Gateff and F. A. Bautz, 1980. Evidence for endogenous reovirus-like particles in a tissue culture cell line from Drosophila melanogaster. Virology 101: 124–130. [DOI] [PubMed] [Google Scholar]
- Halfer, C., and C. Barigozzi, 1973. Prophase synapsis in somatic cells of Drosophila melanogaster. Chromosomes Today 4: 181–186. [Google Scholar]
- Holt, C. M., 1917. Multiple complexes in the alimentary tract of Culex pipiens. J. Morphol. 29: 607–627. [Google Scholar]
- Hossain, M. S., K. Kurokawa, N. Akimitsu and K. Sekimizu, 2004. DNA topoisomerase II is required for the G0-to-S phase transition in Drosophila Schneider cells, but not in yeast. Genes Cells 9: 905–917. [DOI] [PubMed] [Google Scholar]
- Iwabata, K., A. Koshiyama, T. Yamaguchi, H. Sugawara, F. N. Hamada et al., 2005. DNA topoisomerase II interacts with Lim15/Dmc1 in meiosis. Nucleic Acids Res. 33: 5809–5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju, B. G., V. V. Lunyak, V. Perissi, I. Garcia-Bassets, D. W. Rose et al., 2006. A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science 312: 1798–1802. [DOI] [PubMed] [Google Scholar]
- Kassis, J. A., 2002. Pairing-sensitive silencing, polycomb group response elements, and transposon homing in Drosophila. Adv. Genet. 46: 421–438. [DOI] [PubMed] [Google Scholar]
- Kavi, H. H., H. R. Fernandez, W. Xie and J. A. Birchler, 2006. Polycomb, pairing and PIWI–RNA silencing and nuclear interactions. Trends Biochem. Sci. 31: 485–487. [DOI] [PubMed] [Google Scholar]
- Kennison, J. A., and J. W. Southworth, 2002. Transvection in Drosophila. Adv. Genet. 46: 399–420. [DOI] [PubMed] [Google Scholar]
- Kopczynski, C. C., and M. A. Muskavitch, 1992. Introns excised from the Delta primary transcript are localized near sites of Delta transcription. J. Cell Biol. 119: 503–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kravchenko, E., E. Savitskaya, O. Kravchuk, A. Parshikov, P. Georgiev et al., 2005. Pairing between gypsy insulators facilitates the enhancer action in trans throughout the Drosophila genome. Mol. Cell. Biol. 25: 9283–9291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn, E. J., and P. K. Geyer, 2003. Genomic insulators: connecting properties to mechanism. Curr. Opin. Cell Biol. 15: 259–265. [DOI] [PubMed] [Google Scholar]
- Larsen, A. K., A. E. Escargueil and A. Skladanowski, 2003. Catalytic topoisomerase II inhibitors in cancer therapy. Pharmacol. Ther. 99: 167–181. [DOI] [PubMed] [Google Scholar]
- LaSalle, J. M., and M. Lalande, 1996. Homologous association of oppositely imprinted chromosomal domains. Science 272: 725–728. [DOI] [PubMed] [Google Scholar]
- Lee, D. J., A. Wynveen and A. A. Kornyshev, 2004. DNA-DNA interaction beyond the ground state. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 70: 051913. [DOI] [PubMed] [Google Scholar]
- Ling, J. Q., T. Li, J. F. Hu, T. H. Vu, H. L. Chen et al., 2006. CTCF mediates interchromosomal colocalization between Igf2/H19 and Wsb1/Nf1. Science 312: 269–272. [DOI] [PubMed] [Google Scholar]
- Lomvardas, S., G. Barnea, D. J. Pisapia, M. Mendelsohn, J. Kirkland et al., 2006. Interchromosomal interactions and olfactory receptor choice. Cell 126: 403–413. [DOI] [PubMed] [Google Scholar]
- Lupo, R., A. Breiling, M. E. Bianchi and V. Orlando, 2001. Drosophila chromosome condensation proteins Topoisomerase II and Barren colocalize with Polycomb and maintain Fab-7 PRE silencing. Mol. Cell 7: 127–136. [DOI] [PubMed] [Google Scholar]
- Maeshima, K., and U. K. Laemmli, 2003. A two-step scaffolding model for mitotic chromosome assembly. Dev. Cell 4: 467–480. [DOI] [PubMed] [Google Scholar]
- Maeshima, K., M. Eltsov and U. K. Laemmli, 2005. Chromosome structure: improved immunolabeling for electron microscopy. Chromosoma 114: 365–375. [DOI] [PubMed] [Google Scholar]
- Marahrens, Y., 1999. X-inactivation by chromosomal pairing events. Genes Dev. 13: 2624–2632. [DOI] [PubMed] [Google Scholar]
- Marshall, W. F., A. F. Dernburg, B. Harmon, D. A. Agard and J. W. Sedat, 1996. Specific interactions of chromatin with the nuclear envelope: positional determination within the nucleus in Drosophila melanogaster. Mol. Biol. Cell 7: 825–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClintock, B., 1933. The association of non-homologous parts of chromosomes in the midprophase of meiosis in Zea mays. Z. Zellforsch. Mikrok. Anat. 19: 191–237. [Google Scholar]
- McKee, B. D., 2004. Homologous pairing and chromosome dynamics in meiosis and mitosis. Biochim. Biophys. Acta 1677: 165–180. [DOI] [PubMed] [Google Scholar]
- Metz, C. W., 1916. Chromosome studies on the Diptera. II. The paired association of chromosomes in the Diptera and its significance. J. Exp. Zool. 21: 213–279. [Google Scholar]
- Metz, C. W., 1922. Association of homologous chromosomes in tetraploid cells of Diptera. Biol. Bull. 43: 369–373. [Google Scholar]
- Metz, C. W., 1925. Prophase chromosome behavior in triploid individuals of Drosophila melanogaster. Genetics 10: 345–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlynarczyk-Evans, S., M. Royce-Tolland, M. K. Alexander, A. A. Andersen, S. Kalantry et al., 2006. X chromosomes alternate between two states prior to random X-inactivation. PLoS Biol. 4: e159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabirochkin, S., M. Ossokina and T. Heidmann, 1998. A nuclear matrix/scaffold attachment region co-localizes with the gypsy retrotransposon insulator sequence. J. Biol. Chem. 273: 2473–2479. [DOI] [PubMed] [Google Scholar]
- Nichols, W. W., C. Bradt and W. Bowne, 1971. V. Cytogenetic studies on cells in culture from the class Insecta. Curr. Top. Microbiol. Immunol. 55: 61–69. [DOI] [PubMed] [Google Scholar]
- Osman, K., E. Sanchez-Moran, J. D. Higgins, G. H. Jones and F. C. Franklin, 2006. Chromosome synapsis in Arabidopsis: analysis of the transverse filament protein ZYP1 reveals novel functions for the synaptonemal complex. Chromosoma 115: 212–219. [DOI] [PubMed] [Google Scholar]
- Pawlowski, W. P., and W. Z. Cande, 2005. Coordinating the events of the meiotic prophase. Trends Cell Biol. 15: 674–681. [DOI] [PubMed] [Google Scholar]
- Peel, D. J., and M. J. Milner, 1991. Karyotype analysis of imaginal disc cell lines. Dros. Inf. Serv. 70: 176–177. [Google Scholar]
- Peel, D. J., S. A. Johnson and M. J. Milner, 1990. The ultrastructure of imaginal disc cells in primary cultures and during cell aggregation in continuous cell lines. Tissue Cell 22: 749–758. [DOI] [PubMed] [Google Scholar]
- Perrimon, N., 2007. RNAi protocols. (http://flyrnai.org/RNAi_protocols.html).
- Pirrotta, V., 1999. Transvection and chromosomal trans-interaction effects. Biochim. Biophys. Acta 1424: M1–M8. [DOI] [PubMed] [Google Scholar]
- Pommier, Y., 2006. Topoisomerase I inhibitors: camptothecins and beyond. Nat. Rev. Cancer 6: 789–802. [DOI] [PubMed] [Google Scholar]
- Porter, A. C., and C. J. Farr, 2004. Topoisomerase II: untangling its contribution at the centromere. Chromosome Res. 12: 569–583. [DOI] [PubMed] [Google Scholar]
- Riesselmann, L., and T. Haaf, 1999. Preferential S-phase pairing of the imprinted region on distal mouse chromosome 7. Cytogenet. Cell Genet. 86: 39–42. [DOI] [PubMed] [Google Scholar]
- Rong, Y. S., and K. G. Golic, 2003. The homologous chromosome is an effective template for the repair of mitotic DNA double-strand breaks in Drosophila. Genetics 165: 1831–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronshaugen, M., and M. Levine, 2004. Visualization of trans-homolog enhancer-promoter interactions at the Abd-B Hox locus in the Drosophila embryo. Dev. Cell 7: 925–932. [DOI] [PubMed] [Google Scholar]
- Sass, G. L., and S. Henikoff, 1999. Pairing-dependent mislocalization of a Drosophila brown gene reporter to a heterochromatic environment. Genetics 152: 595–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savvidou, E., N. Cobbe, S. Steffensen, S. Cotterill and M. M. Heck, 2005. Drosophila CAP-D2 is required for condensin complex stability and resolution of sister chromatids. J. Cell Sci. 118: 2529–2543. [DOI] [PubMed] [Google Scholar]
- Schneider, I., 1972. Cell lines derived from late embryonic stages of Drosophila melanogaster. J. Embryol. Exp. Morphol. 27: 353–365. [PubMed] [Google Scholar]
- Silahtaroglu, A., H. Pfundheller, A. Koshkin, N. Tommerup and S. Kauppinen, 2004. LNA-modified oligonucleotides are highly efficient as FISH probes. Cytogenet. Genome Res. 107: 32–37. [DOI] [PubMed] [Google Scholar]
- Silahtaroglu, A. N., N. Tommerup and H. Vissing, 2003. FISHing with locked nucleic acids (LNA): evaluation of different LNA/DNA mixmers. Mol. Cell. Probes 17: 165–169. [DOI] [PubMed] [Google Scholar]
- Smiley, R. D., T. R. Collins, G. G. Hammes and T. S. Hsieh, 2007. Single-molecule measurements of the opening and closing of the DNA gate by eukaryotic topoisomerase II. Proc. Natl. Acad. Sci. USA 104: 4840–4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solier, S., A. Lansiaux, E. Logette, J. Wu, J. Soret et al., 2004. Topoisomerase I and II inhibitors control caspase-2 pre-messenger RNA splicing in human cells. Mol. Cancer Res. 2: 53–61. [PubMed] [Google Scholar]
- Sondermeijer, P. J., J. W. Derksen and N. H. Lubsen, 1980. New cell line: established cell lines of Drosophila hydei. In Vitro 16: 913–914. [DOI] [PubMed] [Google Scholar]
- Spilianakis, C. G., M. D. Lalioti, T. Town, G. R. Lee and R. A. Flavell, 2005. Interchromosomal associations between alternatively expressed loci. Nature 435: 637–645. [DOI] [PubMed] [Google Scholar]
- Stevens, N., 1907. The chromosomes in Drosophila ampelophila. Proceedings of the 7th International Zoological Congress. Cambridge University Press, Cambridge, UK.
- Stevens, N., 1908. A study of the germ cells of certain Diptera, with reference to the heterochromosomes and phenomena of synapsis. J. Exp. Zool. 5: 359–374. [Google Scholar]
- Thatcher, K. N., S. Peddada, D. H. Yasui and J. M. Lasalle, 2005. Homologous pairing of 15q11–13 imprinted domains in brain is developmentally regulated but deficient in Rett and autism samples. Hum. Mol. Genet. 14: 785–797. [DOI] [PubMed] [Google Scholar]
- Tsubouchi, T., and G. S. Roeder, 2005. A synaptonemal complex protein promotes homology-independent centromere coupling. Science 308: 870–873. [DOI] [PubMed] [Google Scholar]
- Ui, K., S. Nishihara, M. Sakuma, S. Togashi, R. Ueda et al., 1994. Newly established cell lines from Drosophila larval CNS express neural specific characteristics. In Vitro Cell Dev. Biol. Anim. 30A: 209–216. [DOI] [PubMed] [Google Scholar]
- Valenzuela, L., and R. T. Kamakaka, 2006. Chromatin insulators. Annu. Rev. Genet. 40: 107–138. [DOI] [PubMed] [Google Scholar]
- Van Hooser, A. A., P. Yuh and R. Heald, 2005. The perichromosomal layer. Chromosoma 114: 377–388. [DOI] [PubMed] [Google Scholar]
- Vazquez, J., A. S. Belmont and J. W. Sedat, 2002. The dynamics of homologous chromosome pairing during male Drosophila meiosis. Curr. Biol. 12: 1473–1483. [DOI] [PubMed] [Google Scholar]
- Vazquez, J., M. Muller, V. Pirrotta and J. W. Sedat, 2006. The Mcp element mediates stable long-range chromosome-chromosome interactions in Drosophila. Mol. Biol. Cell 17: 2158–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veile, R., 1990. Chromosome analysis methods (http://hg.wustl.edu/hdk_lab_manual/cam/camcontents.html).
- Wang, X., and W. Dai, 2005. Shugoshin, a guardian for sister chromatid segregation. Exp. Cell Res. 310: 1–9. [DOI] [PubMed] [Google Scholar]
- Watanabe, Y., 2005. Sister chromatid cohesion along arms and at centromeres. Trends Genet. 21: 405–412. [DOI] [PubMed] [Google Scholar]
- Wu, C. T., and J. R. Morris, 1999. Transvection and other homology effects. Curr. Opin. Genet. Dev. 9: 237–246. [DOI] [PubMed] [Google Scholar]
- Wyman, C., D. Ristic and R. Kanaar, 2004. Homologous recombination-mediated double-strand break repair. DNA Repair 3: 827–833. [DOI] [PubMed] [Google Scholar]
- Xu, N., C. L. Tsai and J. T. Lee, 2006. Transient homologous chromosome pairing marks the onset of X inactivation. Science 311: 1107–1109. [DOI] [PubMed] [Google Scholar]
- Yanagawa, S., J. S. Lee and A. Ishimoto, 1998. Identification and characterization of a novel line of Drosophila Schneider S2 cells that respond to wingless signaling. J. Biol. Chem. 273: 32353–32359. [DOI] [PubMed] [Google Scholar]
- Zickler, D., 2006. From early homologue recognition to synaptonemal complex formation. Chromosoma 115: 158–174. [DOI] [PubMed] [Google Scholar]