

Abstract
A new method is developed for estimating rates of somatic mutation in vivo. The stop-enhanced green fluorescent protein (EGFP) transgenic mouse carries multiple copies of an EGFP gene with a premature stop codon. The gene can revert to a functional form via point mutations. Mice treated with a potent mutagen, N-ethyl-N-nitrosourea (ENU), and mice treated with a vehicle alone are assayed for mutations in liver cells. A stochastic model is developed to model the mutation and gene expression processes and maximum-likelihood estimators of the model parameters are derived. A likelihood-ratio test (LRT) is developed for detecting mutagenicity. Parametric bootstrap simulations are used to obtain confidence intervals of the parameter estimates and to estimate the significance of the LRT. The LRT is highly significant (α < 0.01) and the 95% confidence interval for the relative effect of the mutagen (the ratio of the rate of mutation during the interval of mutagen exposure to the rate of background mutation) ranges from a minimum 200-fold effect of the mutagen to a maximum 2000-fold effect.
SOMATIC mutation is a process of fundamental importance in many human diseases such as cancer (Hanahan and Weinberg 2000; Ponder 2001; Yang et al. 2003); it may also play a role in biological processes such as aging, although this is controversial (Sedelnikova et al. 2004; Gorbunova and Seluanov 2005). Many factors influence the rates (and patterns) of somatic mutation. Some influences are environmental (e.g., exposure to chemicals and UV-radiation, etc.) and others are genetic (e.g., GC content of DNA sequences, mutations in DNA repair genes, etc.). To uncover the important factors influencing rates of somatic mutation (and by extension, rates of cancer, etc.) in a mammalian system, improved mutation detection systems are required for estimating rates of somatic mutation in cells exposed to potential environmental, or genetic, risk factors.
In the early 1990s, transgenic mouse mutation detection systems were developed by inserting either the lacZ or the lacI bacterial transgene into mice (Gossen et al. 1989; Kohler et al. 1991). Such transgenic mutation detection systems have been widely used for measuring mutant frequency (i.e., the relative number of mutants in a population of cells) after treatment with various mutagenic agents in different tissues (Dean et al. 1999; Suzuki et al. 1999; Thybaud et al. 2003). The validity of these systems for studying somatic mutations in mammals has been questioned, however, because many features of the bacterial lacZ and lacI genes, the target genes for mutations, are typically prokaryotic and the systems therefore may not represent the characteristic patterns of mutations one would expect to see in mammalian genes (Skopek 1998). Another shortcoming is that overall mutant frequency can be influenced by various nonmutagenic factors such as the clonal expansion of mutant cells, the sampling time following treatment when the mutation assay is performed, etc. (Heddle 1999a; Sun and Heddle 1999; Thybaud et al. 2003). Estimation of the mutation rate (i.e., the rate at which mutations arise, rather than the frequency of mutants) is desirable in mutation studies because the mutation rate better reflects the underlying mutational mechanisms and addresses mutagenicity questions more directly than the mutant frequency (Drake 1970; Thompson et al. 1998).
Recently, a novel transgenic mouse system has been developed that carries an enhanced green fluorescent protein (EGFP) gene containing a premature stop codon (referred to as the stop-EGFP gene) and the wild-type enhanced blue fluorescent protein (EBFP) gene. This system has been applied to trace clonal cell lineages in vivo (Ro 2004; Ro and Rannala 2004, 2005). In the stop-EGFP mouse, a cell having undergone a mutation at the premature stop codon within the stop-EGFP gene (and its descendant cells) expresses functional revertant EGFP, thus allowing clonal cell lineages to be traced using fluorescence imaging. Because the stop-EGFP gene can function as a reporter for mutation and green fluorescent mutant cells generated by revertant mutations at the premature stop codon within the stop-EGFP gene can be easily detected using fluorescence imaging, the stop-EGFP system has the potential to be utilized as an in vivo mutation detection system. Several characteristics of the stop-EGFP mouse are anticipated to be advantageous for in vivo mutation studies. First, the EGFP gene has been genetically modified to enhance mammalian characteristics (Yang et al. 1996) and thus the stop-EGFP gene is expected to reveal mutational characteristics typical of mammalian genes. Second, the stop-EGFP gene is transcribed in cells (Ro and Rannala 2004), which will further enhance the similarity to mammalian endogenous genes. Third, because the EBFP gene, colocalized with the EGFP gene, provides a target nucleotide for GC → AT transitions, the stop-EGFP system allows all possible point mutations to be detected that may arise by either transitions (AT → GC and GC → AT) or transversions (AT → TA, AT → CG, GC → TA, and GC → CG) (see Ro and Rannala 2004).
Finally, independent mutations in a tissue of the stop-EGFP mouse can be counted by detecting green fluorescent colonies (clonal cell lineages originating from mutant cells), facilitating estimation of mutation rate. Here, we use the stop-EGFP system to study somatic mutation rates. We treated the stop-EGFP mouse with N-ethyl-N-nitrosourea (ENU) at a dose of 150 mg/kg body weight and examined the mutagenicity of the substance in the liver. Mutagenesis in live mammals is a complex process that involves DNA damage incurred by exposure to a mutagen, fixation of mutations, transcription from a mutant gene, expression of mutant phenotypes, etc. (Heddle 1999b). A mutation assay carried out at an earlier time point might underestimate the actual mutagenic potential of a substance because of insufficient expression of the mutant phenotype from some mutant cells (Sun and Heddle 1999). To better evaluate the mutagenic potential of a substance using the stop-EGFP mouse system we carried out a time-course study of mutations observed in the left caudal liver lobe after exposure to ENU. On the basis of estimation by maximum likelihood using a novel statistical model and inference procedure developed in this study, we compared the rate of mutation (in the liver) induced by ENU with the background rate and also estimated the waiting time until a revertant phenotype is expressed in a cell.
MATERIALS AND METHODS
Preparation of ENU solution:
The ENU solution was prepared in a type IIB2 biosafety cabinet that exhausts 100% to the outside. A vial containing ∼1 g of ENU powder was purchased (ISOPAC; Sigma, St. Louis). A 10-ml quantity of 95% ethanol was injected into a vial through the rubber injection port and ENU was dissolved by gentle shaking of the vial. A 90-ml quantity of phosphate–citrate (0.1 m sodium phosphate and 0.05 m sodium citrate, pH 5.0) was added to the ENU solution and mixed by inverting and shaking the vial.
Animal experiments:
All experiments using live mice were performed in compliance with the recommendations of the Canadian Council on Animal Care and have been approved by the Health Sciences Animal Policy and Welfare Committee of the University of Alberta.
Animal treatment:
The optical density (OD) of the ENU solution at 398 nm was measured immediately prior to injection to precisely determine the concentration of the ENU solution. A concentration of 1 mg/m1 of ENU corresponds to OD 0.72 at 398 nm (Justice et al. 2000). Only male mice were used for this study. Mice were anesthetized by inhalation of isoflurane gas prior to injection. ENU was administered intraperitoneally at a dose of 150 mg/kg body weight. Control mice were injected intraperitoneally with vehicle (9.5% ethanol in phosphate–citrate buffer, see above) in a volume of 20 ml/kg body weight.
Mutation assay:
For mutation assay, a mouse was killed by CO2 asphyxiation and perfused with 10 ml of saline followed by 10 ml of 4% paraformaldehyde. The liver was removed from the mouse and stored in 4% paraformaldehyde at 4° with gentle agitation for 11 hr. After fixation, the organ was transferred to PBS with 1 mm MgCl2 and stored at 4° overnight. The organ was sectioned into slices (100 μm in thickness) using a vibratome (VT1000S; Leica, Deerfield, IL). Each slice was then transferred to a 24-well plate containing PBS with 1 mm MgCl2 and stored until the imaging experiment. At least 4 hr before imaging, slices were transferred to microscopic slides for mounting. Ultrapure glycerol (Invitrogen, San Diego) and 10× PBS were mixed with a ratio of 9:1 and used as mounting media. Slices were illuminated by use of a 50-W mercury lamp and scanned using a Zeiss Axiovert 200M inverted microscope with a 10× F-Fluar lens (NA 0.5) and LP 520 emission filter (Carl Zeiss, Thornwood, NY). Images of green fluorescent cells were collected with a confocal laser scanning microscope (LSM 510 NLO, software version 3.2; Carl Zeiss) mounted on the Zeiss Axiovert 200M inverted microscope with a 25× multi-immersion F-Fluar lens (NA 0.8). EGFP was excited with the 488-nm laser line and a band-pass filter (505–530 nm wavelength) was used for detecting emissions from EGFP.
4′,6-Diamidino-2-phenylindole staining and colocalization of nuclei:
Several slices containing EGFP signals were selected and used to verify colocalization of 4′,6-diamidino-2-phenylindole (DAPI)-stained nuclei and bright EGFP signals within mutant cells. Slices were incubated for 30 min at room temperature in a PBS solution containing 1 μg/ml of DAPI. Slices were then briefly washed with PBS and dehydrated by storing them in 30, 50, 60, 70, 90, 100% ethanol, serially for 5 min at each step. Slices were then mounted in methyl salicylate. The DAPI-stained EGFP-expressing cells were imaged with a 25× multi-immersion F-Fluar lens (NA 0.8) on a confocal laser scanning microscope (LSM 510 NLO, software version 3.2; Carl Zeiss), using the 488-nm laser line to activate EGFP and a two-photon laser of 760 nm to activate DAPI-stained DNA.
Estimation of the total number of cells in each lobe:
The total number of cells in the left caudal liver lobe of each mouse was estimated by multiplying the volume of each lobe by the total cell number in a unit volume. To calculate the volume, the whole area of each slice (100 μm in thickness, see above) of the lobe was imaged by the “Tile scan” function using a motorized scanning stage of a confocal laser scanning microscope (LSM 510 NLO, software version 3.2; Carl Zeiss) with a 2.5× Fluar lens (NA 0.12). Autofluorescence in slices was activated with the 488-nm laser line and detected using a band-pass filter (535- to 590-nm wavelength). The total area of each slice was then measured using MetaMorph software (version 6.26; Molecular Devices, Menlo Park, CA). To calculate the total cell number in a unit volume, randomly chosen 100 × 100 μm areas (15 different areas of a liver slice stained with DAPI) were scanned along the z-axis and all nuclei contained in this volume were imaged with a 25× multi-immersion F-Fluar lens (NA 0.8) on a confocal laser scanning microscope (LSM 510 NLO, software version 3.2; Carl Zeiss) using 760 nm as excitation light. The number of total nuclei in the specified volume was counted using the spot-counting module of Imaris software (version 4.2, Bitplane AG).
THEORY
To model the process of somatic mutation in untreated mice and mice treated with a potential mutagen, we develop a simple three-parameter model. Let the observed data be represented as a vector X = {Xi}, where Xi is the number of independent stop-EGFP mutations observed for mouse i and i = 1,…, n. The observed (and/or experimentally fixed) model parameters (for mouse i) are the total number of cells examined, Ni, the age of the mouse when the mutation assay is performed, 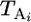 , and the age at time of exposure (to either the mutagen or the vehicle),
, and the age at time of exposure (to either the mutagen or the vehicle), 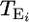 , both in units of days. The unobserved parameters to be estimated are the background rate of mutation (per cell, per day), μ, the rate of mutation during exposure to the mutagen, δ, and the expected time (in units of days) until a mutant cell begins expressing a revertant protein, 1/λ.
, both in units of days. The unobserved parameters to be estimated are the background rate of mutation (per cell, per day), μ, the rate of mutation during exposure to the mutagen, δ, and the expected time (in units of days) until a mutant cell begins expressing a revertant protein, 1/λ.
It is natural to assume that mutation processes in individual mice are independent, and thus we first develop the probabilistic model for a single mouse (dropping subscripts for simplicity). The stochastic model we develop assumes background mutations arise according to a homogenous Poisson process with rate μ per cell; the expected number of mutations per unit time is therefore μN. Time-dependent mutation rates could be easily incorporated, but the data are likely insufficient to estimate the parameters of a more complex model. At time-point TE, a spike in the mutation rate occurs so that the expected number of mutations during the period of mutagen exposure is δN.
Background mutation:
We first consider the model of background mutation. Let Y be the total number of mutations arising on the time interval (0, TA). The expectation of Y is 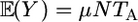 . We assume that no more than one mutation occurs in a cell, which is reasonable when
. We assume that no more than one mutation occurs in a cell, which is reasonable when  is very small. The marginal distribution of the age at which any given mutation arises is uniform on (0, TA). We assume that the waiting time until a revertant protein is expressed in a cell, given that a mutation has occurred, follows an exponential distribution with mean 1/λ. Conditional on a mutation having occurred in a cell, the probability that it is expressing the revertant protein at time TA is
is very small. The marginal distribution of the age at which any given mutation arises is uniform on (0, TA). We assume that the waiting time until a revertant protein is expressed in a cell, given that a mutation has occurred, follows an exponential distribution with mean 1/λ. Conditional on a mutation having occurred in a cell, the probability that it is expressing the revertant protein at time TA is
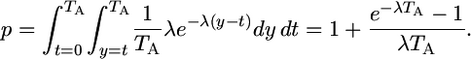 |
The probability that at time TA, nB cells are expressing revertant proteins that arose via the background mutation process is then
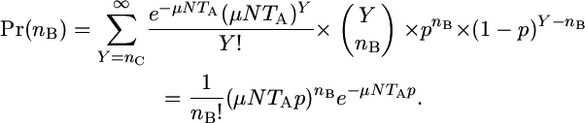 |
In deriving this result, we assume that the number of cells is stationary, but this can be easily relaxed by allowing the number of cells to be a function of time N(t) and using a nonhomogenous Poisson model (to account for changes in cell number in early development, for example).
Exposure to a mutagen:
It is assumed that the period of exposure to a mutagenic substance is of short duration (e.g., a few days), which is true for the mutagen (ENU) that we use in this study. This implies that mutations induced by the mutagen arise only at time TE (e.g., the day on which the treatment is applied). The probability that a mutation arising at time TE is expressed as a revertant protein at time TA (assuming an exponential distribution for the waiting time to expression) is
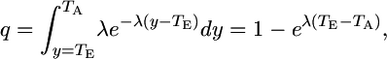 |
and the probability that nE mutations are induced by the mutagen and are expressing revertant protein at time TA is
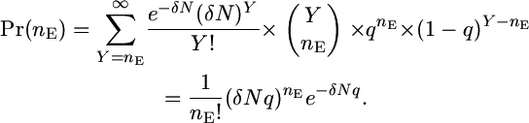 |
The likelihood:
The observed data for mouse i are the total number of mutations, which are the sum of background mutations, 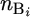 , and mutagen-induced mutations,
, and mutagen-induced mutations, 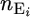 , namely
, namely 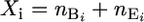 . This is a convolution of two Poisson distributions, which is also a Poisson distribution,
. This is a convolution of two Poisson distributions, which is also a Poisson distribution,
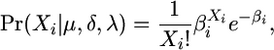 |
but with an expectation, βi, that is the sum of the expectations of the two variables,
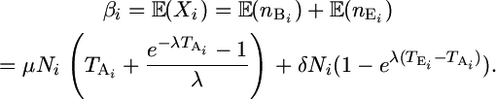 |
Now let k be the number of mice treated with the mutagen and n − k be the number of mice treated with the vehicle alone. Order the labels of the mice such that if mouse i is treated with the vehicle alone then i ≤ n − k. For the mice treated only with the vehicle (controls), we assume that δ = 0. The joint likelihood is
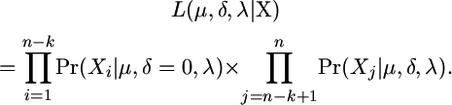 |
The log-likelihood was numerically maximized using the R statistics package (http://r-project.org) to obtain joint maximum-likelihood estimates (MLEs) of parameters μ, δ, and λ. It is possible to analytically calculate the gradients of the likelihood and the Hessian matrix (the matrix of partial second derivates) and we therefore used the R package “trust” that uses gradients and Hessians to carry out optimization (http://www.stat.umn.edu/geyer/trust/). Functions were written in R to simulate samples from this likelihood allowing the parametric bootstrap to be applied to generate confidence intervals (and a variance–covariance matrix) for the estimates without appealing to asymptotic theory. An R script implementing these procedures is freely available from http://rannala.org.
Likelihood-ratio test of mutagenicity:
To test the mutagenicity of a substance we propose a likelihood-ratio test (LRT). Under the null hypothesis, exposure to a substance does not produce an increased rate of mutation and δ = 0 in both control and treated mice. Under the alternative hypothesis, δ = 0 in control mice and δ > 0 for treated mice. The LRT statistic is −2log Λ, where Λ is
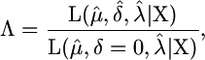 |
where the numerator is the likelihood of the data with the MLEs obtained by maximizing over the three parameters jointly and the denominator is the likelihood of the data with MLEs obtained jointly for μ and λ with δ = 0. The test statistic should follow an asymptotic χ2-distribution with 1 d.f. However, because δ = 0 is a boundary condition this is not guaranteed and so we also calculated significance using a parametric bootstrap approach (simulating data under the null hypothesis). The estimate of μ appears to be insensitive to the value of λ for biologically reasonable values of the waiting time to expression (e.g., λ > 0.01 and λ < 100). Fixing λ it is possible to obtain an analytical expression for the MLE of μ,
 |
This expression was used to obtain MLEs of μ under the null model for the LRT and the C.I. for μ was obtained by parametric bootstrap simulations.
RESULTS
Detection of mutations in the left caudal liver lobe:
Mutations in the liver of the stop-EGFP mouse were directly observed in situ using fluorescence imaging. The assay is based on identifying mutant cells having undergone a revertant mutation at the premature stop codon within the stop-EGFP gene, which would express a functional EGFP revertant. For mutation assay, slices from the left caudal liver lobe were scanned using a fluorescence microscope. Mutant cells exhibited bright green fluorescence when illuminated with the excitation light of 488 nm and were easily identified (Figure 1). Mutant cells contained what appeared to be brighter nuclei. The bright spots were confirmed to be nuclei by use of DAPI staining (Figure 1, A and B).
Figure 1.—

Green fluorescent mutant cells in the liver. (A and B) Merged images of EGFP signals and DAPI signals of mutant cells detected in the liver. The bright green fluorescent signals within these cells are colocalized with the DAPI-stained nuclei. (C and D) Images of EGFP signals of mutant hepatocytes. Bars, 10 μm.
A cell having undergone a mutation at the premature stop codon and its clonal descendant cells will constitute a green fluorescent patch (i.e., a mutant colony), which can be regarded as a single mutation. Thus, the number of independent mutations in the left caudal liver lobe of each mouse can be determined by counting the number of green fluorescent colonies in the lobe.
Time-course study of mutations in livers of ENU-exposed and control mice:
We performed a mutation assay on the livers of 10 stop-EGFP mice at 5 months postvehicle treatment (see materials and methods) as a control. The number of mutations and estimated total number of cells in the liver lobe of each mouse are shown in Table 1 (ID nos. 1–10) as well as the ages of mice at the time of mutation assay. The data were used to estimate the rate of spontaneous mutation in the liver. Consistent with our previous data, a mutant colony carrying more than two nuclei has not been detected in the liver (Ro and Rannala 2004), and this may reflect the slow rate of cell proliferation in this tissue.
TABLE 1.
Mutation counts
| Mouse ID | TE | TA | No. mutations | No. cells | Treatment/control |
|---|---|---|---|---|---|
| 1 | 69 | 220 | 0 | 2.07 × 108 | Control |
| 2 | 231 | 382 | 0 | 1.52 × 108 | Control |
| 3 | 82 | 233 | 0 | 1.60 × 108 | Control |
| 4 | 57 | 208 | 0 | 1.43 × 108 | Control |
| 5 | 204 | 355 | 1 | 1.80 × 108 | Control |
| 6 | 57 | 208 | 1 | 1.21 × 108 | Control |
| 7 | 82 | 233 | 2 | 1.74 × 108 | Control |
| 8 | 69 | 220 | 2 | 1.41 × 108 | Control |
| 9 | 284 | 435 | 2 | 1.58 × 108 | Control |
| 10 | 284 | 435 | 4 | 1.71 × 108 | Control |
| 11 | 158 | 188 | 0 | 1.16 × 108 | Treatment |
| 12 | 55 | 85 | 2 | 1.06 × 108 | Treatment |
| 13 | 55 | 85 | 0 | 1.02 × 108 | Treatment |
| 14 | 158 | 188 | 0 | 1.23 × 108 | Treatment |
| 15 | 71 | 163 | 2 | 1.25 × 108 | Treatment |
| 16 | 139 | 231 | 3 | 1.70 × 108 | Treatment |
| 17 | 101 | 193 | 7 | 1.53 × 108 | Treatment |
| 18 | 62 | 276 | 0 | 1.31 × 108 | Treatment |
| 19 | 71 | 285 | 2 | 1.39 × 108 | Treatment |
| 20 | 71 | 285 | 1 | 1.30 × 108 | Treatment |
| 21 | 96 | 310 | 12 | 1.61 × 108 | Treatment |
| 22 | 144 | 358 | 6 | 1.62 × 108 | Treatment |
Mutation counts were obtained by imaging the left caudal liver lobe of mice treated with a mutagen ENU (treatment) or the vehicle alone (control). TE is the time of exposure (to ENU or vehicle) in days since birth, TA is the time at which a mouse was imaged (in days since birth). The number of mutations (column 4) is the total number of mutant EGFP-expressing colonies observed and the number of cells (column 5) is the total number of liver cells scanned.
We also performed a mutation assay in the liver at different time points following exposure to ENU to investigate how the number of observed mutations changes. Twelve stop-EGFP mice were treated with ENU at a dose of 150 mg/kg body weight and mice were killed at 1, 3, and 7 months post-ENU administration. The number of observed mutations and estimated total number of cells in the left caudal liver lobe of each mouse are shown in Table 1 (see ID nos. 11–22). These data were used to estimate the rate of mutation induced by ENU in the liver.
The data suggest that a longer waiting time between exposure and the mutation assay results in more mutations (on average) presumably because some cells carrying a mutation may not exhibit the mutant phenotype due to insufficient time to express a revertant protein. At 1 month post-ENU administration, few mutations were found in the ENU-treated liver (Table 1). We speculate the time period of 1 month after treatment with ENU might be too short for mutant cells in the liver to commonly exhibit the mutant phenotype. Expression of the mutant phenotype in the liver of a stop-EGFP mouse is a complex multistage process that requires fixation of a mutation, transcription from a mutant copy of the stop-EGFP gene, accumulation of mutant mRNA containing no premature stop codon, and protein synthesis from the mutant mRNA at a level sufficient for detection under a fluorescence microscope (Heddle 1999b). These processes may take months in the liver (Douglas et al. 1996; Wang et al. 2004).
Estimates of parameters:
On the basis of the experimental data given in Table 1 and the model developed in this study (see theory), we estimated the background mutation rate per cell per day (μ), a spike in the mutation rate (per cell per day) due to the treatment with ENU (δ), the ratio of the two parameters (R = δ/μ) that quantifies the relative effect of the mutagen, and the waiting time until a revertant protein is expressed in a cell (1/λ). The marginal log-likelihoods of each of the three parameters (obtained by setting the remaining two parameters at their MLE values) are shown in Figure 2. The MLEs and associated 95% confidence intervals of the parameters are given in Table 2. The confidence intervals are based on a parametric bootstrap using 1000 simulated data sets. The LRT statistic is −2 log Λ = 21.04, which is significant at the α = 0.01 level based on either a χ2-distribution with 1 d.f. or a parametric bootstrap analysis (see Figure 3). Thus, the treatment with ENU appears to elevate the mutation rate significantly over the rate of spontaneous mutation. This is also clear from the 95% confidence interval of R that ranges from a minimum 200-fold effect to a maximum 2000-fold effect of the mutagen on mutation rates. It is interesting to note that although the mutation rate estimates themselves are quite imprecise, the relative mutagenic effect of ENU is very strongly supported by the data.
Figure 2.—

Marginal log-likelihood (logL) curves for the three parameters λ, δ, and μ obtained using the stop-EGFP mouse data given in Table 1. The marginal logL of each parameter is plotted with the remaining two parameters fixed at their maximum-likelihood estimates.
TABLE 2.
Parameter estimates
| Model | Parameter | MLE | 95% C.I. | logL |
|---|---|---|---|---|
| H0: δ = 0 | μ | 5.6 × 10−11 | (3.9 × 10−11, 7.1 × 10−11) | −8.174 |
| H1: δ > 0 | μ | 3.7 × 10−11 | (1.6 × 10−11, 1.3 × 10−7) | 2.346 |
| δ | 2.7 × 10−8 | (1.3 × 10−8, 1.1 × 10−4) | ||
| R = δ/μ | 737 | (311, 1956) | ||
| 1/λ | 106 | (13, 8 × 106) |
Maximum-likelihood estimates of parameters and 95% confidence intervals (obtained using a parametric bootstrap procedure) are shown. The parameters are μ, the spontaneous rate of mutation per site, δ, the rate of mutation per site induced by treatment, and 1/λ, the average waiting time (in units of days) until an EGFP mutant cell expressed revertant protein. Note that because proportionality constants that do not affect inferences have been removed from the log-likelihood function, logL may be positive.
Figure 3.—

Sampling distribution of likelihood-ratio test statistic 2 log(L1 − L0) for a mutagenic effect of ENU under the null hypothesis that ENU has no effect on mutation rate (e.g., δ = 0). The histogram bars represent the sampling distribution estimated by parametric bootstrap simulations and the smooth curve represents a χ2-distribution with 1 d.f. (the asymptotic sampling distribution). The observed value of 2 log(L1 − L0) was 21.04, which is highly significant.
DISCUSSION
In this study, we have applied the stop-EGFP mouse to quantify the numbers of independent mutations having arisen in vivo and to estimate rates of somatic mutation in the liver. Our analysis using stop-EGFP transgenic mice allowed us to determine the mutagenic effect of ENU on liver cells. The study suggests that an ∼1000-fold increase over the spontaneous mutation rate was induced by treatment with ENU (2.7 × 10−8 vs. 3.7 × 10−11 per cell per day). Previously, mutation studies using transgenic mouse systems carrying a bacterial transgene showed only modest increases in mutant frequencies in ENU-treated liver over control (Collaborative Study Group for the Transgenic Mouse Mutation Assay Mammalian Mutagenesis Study Group of the Environmental Mutation Society of Japan 1996; Yamada et al. 1999). In many cases, such studies have simply compared mutant frequencies between treated and control groups at the time of the experimental end point. This approach can underestimate the specific mutagenic effect of ENU because mutant cells having arisen due to spontaneous mutations over the lifetime of a mouse might outnumber mutants generated by ENU treatment during a very short time period. To specifically determine the mutagenic effect of ENU we estimated the rate of mutation induced by ENU during the time interval of the ENU administration and compared this rate with the background mutation rate based on the model developed in this study. ENU is very unstable under physiological conditions with a half-life of <1 hr at pH 6.8–7.3 (Winter and Gearhart 2001) and thus it is expected that most of the mutagen will have degraded within ∼1 day and mutations induced by treatment with ENU will most likely arise within a single day following ENU administration. This is explicitly accommodated in our model.
One might argue, on biological grounds, that the preferred units for measuring mutation rate are “per cell division” rather than “per cell per day” as in our study. Although it would be interesting to estimate the per cell division mutation rate induced by ENU treatment and compare it with the spontaneous mutation rate per cell division, there are technical difficulties with precise determination of the number of cell divisions during the period of ENU administration and thus estimates of per cell division rates were not attempted in this study. However, as opposed to base analogs, ENU, an alkylating agent, is mutagenic regardless of whether DNA is replicating (Snustad and Simmons 2006). By transferring the ethyl group to oxygen or nitrogen radicals at a number of reactive sites, ENU can be mutagenic to nonreplicating DNA (Balling 2001). Thus, it is questionable whether mutation rate per cell division would be any more informative than mutation rate per cell per day in the case of ENU or other alkylating agents.
The waiting time until the mutant phenotype is expressed in the liver is estimated to be 106 days (∼3.5 months) after ENU treatment. The estimated average waiting time is slightly longer than the observed time of maximum mutant frequency achieved in the liver of the transgenic mouse carrying a bacterial transgene (∼1–2 months after ENU treatment) (Douglas et al. 1996; Wang et al. 2004). In the stop-EGFP system, to exhibit the mutant phenotype, mutant cells must express revertant EGFP at a level sufficient for detection under a fluorescence microscope. However, in bacterial transgene-based systems such as the lacZ or lacI transgenic mouse, mutant phenotypes are observed in an in vitro bacterial assay system and therefore gene expression from the mutant gene in vivo is not required for the assay (Gossen et al. 1989; Kohler et al. 1991). Thus, it is reasonable to speculate that more waiting time may be required to detect a mutant phenotype from mutant cells in the stop-EGFP system than in the lacZ or lacI transgenic mouse.
A similar approach to that used in this study could be applied to identify other potential mutagens or to quantify their relative mutagenic effects. As well, several strains of knockout mice with a deficient DNA repair gene have already been generated (Winter and Gearhart 2001; Wijnhoven and Van Steeg 2003) and rates of somatic mutation in these strains of knockout mice could be estimated using the stop-EGFP system after crossing the knockout mice with stop-EGFP mice. It would also be interesting to see whether an increase in mutation rate (i.e., genomic instability) is observed in cancerous tissues using the stop-EGFP mouse (Nowell 1976; Beckman and Loeb 2006). Various types of tumors can be induced in stop-EGFP mice by either treating them with a carcinogen or crossing them with tumor-prone mice such as mice with a viral oncogene (Jakubczak et al. 1996) or a deficient p53 (Buettner et al. 1996). Mutation rates then could be estimated in tumor samples and compared with those of normal cells, quantifying levels of hypermutability in various cancers.
The stop-EGFP system carrying the stop-EGFP gene and the EBFP gene can detect all possible point mutations involving base pair substitutions (BPSs). One type of transition (AT → GC) and all types of transversions (AT → TA, AT → CG, GC → TA, and GC → CG) can be detected at the premature stop codon (TAG) of the stop-EGFP gene, while the other type of transition (GC → AT) can be detected at codon 66 of the EBFP gene (see Ro and Rannala 2004). Mutations involving insertions or deletions (“indels”) are unlikely to be detected using this system except when a deletion mutation removes three (or a multiple of three) nucleotides containing at least one nucleotide of the premature stop codon of the stop-EGFP gene. If the aim were instead to specifically detect frameshift mutations, a new type of “frameshifted” stop-EGFP gene could instead be generated with nucleotides [e.g., a poly(G) tract] inserted within the coding region of the functional EGFP gene. A double transgenic mouse that carries both the stop-EGFP gene and the frameshifted EGFP gene could, in principle, be used to detect indels or frameshift mutations as well as BPSs.
Acknowledgments
Support for this research was provided by Canadian Institutes of Health Research (CIHR) grant MOP 44064 and National Institutes of Health grant HG01988 (to B.R.). Salary support for B.R. was provided by the Alberta Heritage Foundation for Medical Research and the CIHR/Peter Lougheed Scholar Award.
References
- Balling, R., 2001. ENU mutagenesis: analyzing gene function in mice. Annu. Rev. Genomics Hum. Genet. 2: 463–492. [DOI] [PubMed] [Google Scholar]
- Beckman, R., and L. A. Loeb, 2006. Efficiency of carcinogenesis with and without a mutator mutation. Proc. Natl. Acad. Sci. USA 103: 14140–14145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buettner, V. L., K. A. Hill, H. Nishino, D. J. Schaid, C. S. Frisk et al., 1996. Increased mutation frequency and altered spectrum in one of four thymic lymphomas derived from tumor prone p53/Big Blue double transgenic mice. Oncogene 13: 2407–2413. [PubMed] [Google Scholar]
- Collaborative Study Group for the Transgenic Mouse Mutation Assay Mammalian Mutagenesis Study Group of the Environmental Mutation Society of Japan, 1996. Organ variation in the mutagenicity of ethylnitrosourea in MutaTM mouse: results of the collaborative study on the transgenic mutation assay by JEMS/MMS. Environ. Mol. Mutagen. 28: 363–375. [DOI] [PubMed] [Google Scholar]
- Dean, S. W., T. M. Brooks, B. Burlinson, J. Mirsalis, B. Myhr et al., 1999. Transgenic mouse mutation assay systems can play an important role in regulatory mutagenicity testing in vivo for the detection of site-of-contact mutagens. Mutagenesis 14: 141–151. [DOI] [PubMed] [Google Scholar]
- Douglas, G. R., J. Jiao, J. D. Gingerich, L. M. Soper and J. A. Gossen, 1996. Temporal and molecular characteristics of lacZ mutations in somatic tissues of transgenic mice. Environ. Mol. Mutagen. 28: 317–324. [DOI] [PubMed] [Google Scholar]
- Drake, J., 1970. The Molecular Basis of Mutation. Holden–Day, San Francisco.
- Gorbunova, V., and A. Seluanov, 2005. Making ends meet in old age: dsb repair and aging. Mech. Ageing Dev. 126: 621–628. [DOI] [PubMed] [Google Scholar]
- Gossen, J. A., W. J. de Leeuw, C. H. Tan, E. C. Zwarthoff, F. Berends et al., 1989. Efficient rescue of integrated shuttle vectors from transgenic mice: a model for studying mutations in vivo. Proc. Natl. Acad. Sci. USA 86: 7971–7975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan, D., and R. A. Weinberg, 2000. The hallmarks of cancer. Cell 100: 57–70. [DOI] [PubMed] [Google Scholar]
- Heddle, J. A., 1999. a Mutant manifestation: the time factor in somatic mutagenesis. Mutagenesis 14: 257–260. [DOI] [PubMed] [Google Scholar]
- Heddle, J. A., 1999. b Mutant manifestation: the time factor in somatic mutagenesis. Mutagenesis 14: 1–3. [DOI] [PubMed] [Google Scholar]
- Jakubczak, J. L., G. Merlino, J. E. French, W. J. Muller, B. Paul et al., 1996. Analysis of genetic instability during mammary tumor progression using a novel selection-based assay for in vivo mutations in a bacteriophage lambda transgene target. Proc. Natl. Acad. Sci. USA 93: 9073–9078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justice, M. J., D. A. Carpenter, J. Favor, A. Neuhauser-Klaus, M. Hrabe de Angelis et al., 2000. Effects of ENU dosage on mouse strains. Mamm. Genome 11: 484–488. [DOI] [PubMed] [Google Scholar]
- Kohler, S. W., G. S. Provost, A. Fieck, P. L. Kretz, W. O. Bullock et al., 1991. Spectra of spontaneous and mutagen-induced mutations in the laci gene in transgenic mice. Proc. Natl. Acad. Sci. USA 88: 7958–7962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowell, P. C., 1976. The clonal evolution of tumor cell populations. Science 194: 23–28. [DOI] [PubMed] [Google Scholar]
- Ponder, B. A., 2001. Cancer genetics. Nature 411: 336–341. [DOI] [PubMed] [Google Scholar]
- Ro, S., 2004. Magnifying stem cell lineages: the stop-EGFP mouse. Cell Cycle 3: 1246–1249. [DOI] [PubMed] [Google Scholar]
- Ro, S., and B. Rannala, 2004. A stop-EGFP transgenic mouse to detect clonal cell lineages generated by mutation. EMBO Rep. 5: 914–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ro, S., and B. Rannala, 2005. Evidence from the stop-EGFP mouse supports a niche-sharing model of epidermal proliferative units. Exp. Dermatol. 14: 838–843. [DOI] [PubMed] [Google Scholar]
- Sedelnikova, O. A., I. Horikawa, D. B. Zimonjic, N. C. Popescu, W. M. Bonner et al., 2004. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat. Cell Biol. 6: 168–170. [DOI] [PubMed] [Google Scholar]
- Skopek, T. R., 1998. Transgenic mutation models: research, testing, and reality checks. Environ. Mol. Mutagen. 32: 104–105. [DOI] [PubMed] [Google Scholar]
- Snustad, D., and M. Simmons, 2006. Principles of Genetics. John Wiley & Sons, Hoboken, NJ.
- Sun, B., and J. A. Heddle, 1999. The relationship between mutant frequency and time in vivo: simple predictions for any tissue, cell type, or mutagen. Mutat. Res. 425: 179–183. [DOI] [PubMed] [Google Scholar]
- Suzuki, T., S. Itoh, M. Nakajima, N. Hachiya and T. Hara, 1999. Target organ and time-course in the mutagenicity of five carcinogens in MutaMouse: a summary report of the second collaborative study of the transgenic mouse mutation assay by JEMS/MMS. Mutat. Res. 444: 259–268. [DOI] [PubMed] [Google Scholar]
- Thompson, J. N. J., R. C. Woodruff and H. Huai, 1998. Mutation rate: a simple concept has become complex. Environ. Mol. Mutagen. 32: 292–300. [DOI] [PubMed] [Google Scholar]
- Thybaud, V., S. Dean, T. Nohmi, J. de Boer, G. R. Douglas et al., 2003. In vivo transgenic mutation assays. Mutat. Res. 540: 141–151. [DOI] [PubMed] [Google Scholar]
- Wang, J., X. Liu, R. H. Heflich and T. Chen, 2004. Time course of cII gene mutant manifestation in the liver, spleen, and bone marrow of N-ethyl-N-nitrosourea-treated Big Blue transgenic mice. Toxicol. Sci. 82: 124–128. [DOI] [PubMed] [Google Scholar]
- Wijnhoven, S. W. P., and H. van Steeg, 2003. Transgenic and knockout mice for DNA repair functions in carcinogenesis and mutagenesis. Toxicology 193: 171–187. [DOI] [PubMed] [Google Scholar]
- Winter, D. B., and P. J. Gearhart, 2001. Altered spectra of hypermutation in DNA repair-deficient mice. Philos. Trans. R. Soc. Lond. B Biol. Sci. 356: 5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada, T. R., R. Yamamoto, H. Kaneko and A. Yoshitake, 1999. Ethylnitrosourea-induced mutation and molecular analysis of transgenic mice containing the gpt shuttle vector. Mutat. Res. 441: 59–72. [DOI] [PubMed] [Google Scholar]
- Yang, T. T., L. Cheng and S. R. Kain, 1996. Optimized codon usage and chromophore mutations provide enhanced sensitivity with the green fluorescent protein. Nucleic Acids Res. 24: 4592–4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z., S. Ro and B. Rannala, 2003. Likelihood models of somatic mutation and codon substitution in cancer genes. Genetics 165: 695–705. [DOI] [PMC free article] [PubMed] [Google Scholar]