

Abstract
Long-distance dispersal (LDD) plays an important role in many population processes like colonization, range expansion, and epidemics. LDD of small particles like fungal spores is often a result of turbulent wind dispersal and is best described by functions with power-law behavior in the tails (“fat tailed”). The influence of fat-tailed LDD on population genetic structure is reported in this article. In computer simulations, the population structure generated by power-law dispersal with exponents in the range of −2 to −1, in distinct contrast to that generated by exponential dispersal, has a fractal structure. As the power-law exponent becomes smaller, the distribution of individual genotypes becomes more self-similar at different scales. Common statistics like GST are not well suited to summarizing differences between the population genetic structures. Instead, fractal and self-similarity statistics demonstrated differences in structure arising from fat-tailed and exponential dispersal. When dispersal is fat tailed, a log–log plot of the Simpson index against distance between subpopulations has an approximately constant gradient over a large range of spatial scales. The fractal dimension D2 is linearly inversely related to the power-law exponent, with a slope of ∼ −2. In a large simulation arena, fat-tailed LDD allows colonization of the entire space by all genotypes whereas exponentially bounded dispersal eventually confines all descendants of a single clonal lineage to a relatively small area.
THE importance of long-distance dispersal (LDD) for the distribution and evolution of organisms has long been recognized and was acknowledged as early as 1859 by Darwin (Darwin 1859). Until recently, however, population genetic and evolutionary studies have concentrated mainly on short-distance dispersal that is easier to measure but nonetheless has important consequences for local population dynamics. Owing to better methodology for assessing LDD and increased awareness of its importance, interest in it has risen again in the last 15 years (Nathan et al. 2003).
LDD plays an important role in colonization of islands (Cain et al. 2000 and references therein; Gittenberger et al. 2006), in range expansion (Cain et al. 1998; Clark 1998), and in the rates of population expansion and spread of epidemics (Shaw 1995; Kot et al. 1996). However, LDD is rare and difficult to analyze in detail in the field. Modeling of LDD has thus become an instrument to investigate its importance in evolutionary and ecological processes. One example is the range expansion of oak trees to the north during the postglacial recolonization of Europe. LDD plays an important role in explaining the speed of the expansion (Le Corre et al. 1997; Austerlitz and Garnier-Géré 2003; Davies et al. 2004; Bialozyt et al. 2006). Computer models of the process explained the patchy genetic patterns, observed in modern oak populations, by LDD founder events (Davies et al. 2004; Bialozyt et al. 2006).
Dispersal by wind is a major mechanism of LDD. Fungal spores causing severe agricultural diseases are dispersed in rare events over hundreds or even thousands of kilometers (Brown and Hovmøller 2002). Transport through the air is profoundly affected by turbulence over a wide range of spatial scales (Aylor 2003; Nathan et al. 2005).
There is an ongoing debate about what kind of dispersal kernel, the function that describes the probability that a propagule will be deposited at a given distance, is best suited to describe LDD. Several studies have modeled LDD of insects or seeds by a mixture of two exponential dispersal distributions, one with a short median dispersal distance and one with a very long one (e.g., Nichols and Hewitt 1994; Bialozyt et al. 2006).
Inferring true dispersal curves from small, wind-dispersed biological objects like spores or pollen is difficult. Measured dispersal distributions are frequently leptokurtic or fat tailed, meaning that they have greater density in their shoulders and tails than a Gaussian distribution with the same variance (references in Kot et al. 1996). Many pollen dispersal data are best fitted with an inverse power-law function (Bullock and Clarke 2000; Austerlitz et al. 2004; Devaux et al. 2005; Klein et al. 2006; Shaw et al. 2006). For fungal spores, the question of the best-fitting dispersal function is hindered by the necessity of large experimental plots free from too much background infection. A recent study addressing the above problems showed that dispersal of the wheat stripe or yellow rust fungus (Puccinia striiformis) fitted a power-law model well if enough sufficiently distant spore traps were used (Sackett and Mundt 2005). Moreover, although several physical processes underlie wind dispersal, theoretical arguments strongly propose that LDD of small objects can be modeled by a single function that will have inverse power-law behavior in the tails (Shaw 1995; Kot et al. 1996; Stockmarr 2002; Aylor 2003; Shaw et al. 2006). Uplift is the most important factor for heavier propagules but many factors are equally important for smaller objects such as spores (Nathan et al. 2005). A simplification of the stochastic dispersal process by a single negative power-law dispersal function is a useful basis for theoretical modeling.
Recent simulation studies have addressed either the influence of LDD, modeled as a dual exponential function, on population genetic structure (Bialozyt et al. 2006) or the influence of power-law dispersal function on spatial distribution of species (Cannas et al. 2006).
This article investigates the influence of LDD, modeled as a negative power law, on population genetic structure of populations in quasi-equilibrium. Inverse power-law functions with exponents in the range of 1 < b ≤ 2 were used as dispersal functions to simulate fat-tailed dispersal. The resulting population structures were compared to those generated by a negative exponential dispersal function or a global dispersal (uniform random) function. Widely used statistics from ecology and population genetics were applied to the resulting populations. Some of them were more suitable than others to describe the population structures and to distinguish the outcomes of different modes of dispersal.
The simulations reported here used an arena several orders of magnitude larger than the median dispersal distance of the dispersal function. We used a novel simulation strategy that allowed us to investigate a range of spatial scales covering nine orders of magnitude. This usage of a large arena may be of paramount importance for a thorough investigation of the consequences of LDD as it is hypothesized that the population structure of organisms with small wind-dispersed propagules is influenced over scales ranging from centimeters to several hundred or even thousands of kilometers (Pedgrey 1986; O'hara and Brown 1998; Brown and Hovmøller 2002).
MATERIALS AND METHODS
Computer simulations:
An individual-based, spatially explicit model of haploid individuals without sex in a continuous habitat was used, developing the models of Shaw (1995, 1996). The simulation aimed at investigating the spatial genetic structure of populations with a roughly stable population size. This scenario corresponds to a natural population that is restricted in growth, e.g., by limited space or limited nutrients. The simulation arena used was very large and so the occupying population should be very large as well. As computer memory was inevitably limited, only sample lineages were simulated. A large population was assumed to be present in the background of these individuals, competing for resources and thus limiting the expansion of the simulated individuals. A birth process with Poisson-distributed progeny number of one individual per parent and a fixed death age of one generation were used to simulate these sample lineages in a fluctuating, nonexpanding population.
General model settings:
Simulations were initiated with 30,000 individuals, initially all of different genotypes, each represented by a 32-bit number, in effect 32 biallelic loci. The initial individuals were placed randomly in a simulation arena of 108 × 108 square units in size, with 1 unit corresponding to the closest distance allowed between two individuals. Individuals gave birth to offspring at the beginning of each time step, which were immediately dispersed according to the chosen dispersal function. The genotype of a new individual was either the same as that of the parent or mutated by conversion of one random bit of the 32-bit genotype. Mutation took place at random with a frequency set by the mutation rate μ = 10−4. Individuals died after 1 time step and thus had the chance to produce progeny only once in their lifetime. Offspring were not placed outside of the simulation arena or closer to other offspring than the minimal interaction distance. If the simulation generated such an event, a new location was calculated until a legitimate one was found. Individuals possibly adjacent to a given point were found quickly using the indexing algorithm in Shaw (1996). The simulations were assumed to be a part of a huge population of uniform density. All “background” lineages were assumed to disperse in the same way as the simulated lineages and thus result in a similar population genetic structure. The main simulations were aimed to run for 50,000 generations. Simulations that were used mainly to calculate the fractal dimensions were run for 10,000 generations only.
Dispersal functions:
The dispersal of the spores was modeled by an inverse power-law probability density function with the spore concentration t(r | θ) at distance r from the source along a given bearing θ given by
 |
(1) |
with b > 1 (Shaw et al. 2006). Of special interest were values of b ≤ 2. Theoretical and experimental results suggest this is the relevant range in wind dispersal of small particles like fungal spores and pollen (Mccartney 1987; Mccartney and Bainbridge 1987; Ferrandino 1993; Sackett and Mundt 2005; Klein et al. 2006; Shaw et al. 2006). If not stated otherwise, simulations used values of 1.2 ≤ b ≤ 4. The main simulations were performed with values of b = 2.5, 2.0, and 1.5. The case of b = 2 corresponds approximately to the well-known Cauchy distribution. Quasi-random power-law variates were generated as described in Shaw (1996). Two rational function approximations were derived using the algorithm in Mathematica 4.0, calculating the median of a power-law distribution with any exponent by numerical integration. For b between 1.2 and 2,
 |
(2) |
For b ≥ 2,
 |
(3) |
For comparison, simulations were also done following the commonly used dispersal model of an exponential decline of spore concentration with a characteristic scale of k:
 |
(4) |
Simulations with global or uniform random dispersal were performed for further comparison, in which offspring were placed randomly in the arena, with both x and y coordinates drawn from a uniform distribution. Simulations were repeated 20 times for the negative exponential dispersal and 12 times for other dispersal functions.
Parameter settings:
Distances in our model were chosen to fit experimental data, especially on wind dispersal of powdery mildew (Blumeria graminis) and yellow rust (P. striiformis) of cereals. The minimal interaction distance between individuals, 1 unit, corresponding to the minimum distance between two distinct rust or mildew lesions on one leaf, was assumed for the sake of simplicity to be 10 cm. Other authors have assumed it of similar magnitude (2 cm was used as the size of a single yellow rust lesion by Lett and Østergård 2000). The median dispersal distance was set to 30 units for the exponentially bounded function. The power-law functions were adjusted to approximate the same median value, corresponding to 3 m in real-world dimensions. The median dispersal for powdery mildew conidiospores is estimated to be 3.1 m for field disease gradients (half the distance of the exponential model; Fitt et al. 1987) or 1 m to several meters (modeled from deposition velocities and impaction efficiency, with 50% deposition rate; Mccartney 1987). Modeled estimates for wheat yellow rust are 2.8 m–6.5 m (calculated from exponential fits to the three largest data sets for upwind dispersal in Sackett and Mundt 2005). Estimates by O'hara and Brown (1998) are similar. The median dispersal distance setting was thus in concordance with field data. The extension of our simulation arena of 108 units can then be regarded as a distance of ∼104 km, e.g., spanning further than the whole of Europe.
Software and hardware:
Simulation software was written in Kylix (Borland Delphi for Linux, version 14.5, Open Edition) using the Free Pascal Compiler (version 1.9.8). Simulations were run on a GNU/Linux platform with four 2.8-GHz processors. Statistical analysis of populations and graphical output were performed using the free R software suite (version 2.1.0; R Development Core Team 2004).
Characterization of populations:
Statistical characterization of populations was performed at different time steps. Results are shown for generation 6000 and in some cases for generation 40,000. If not stated otherwise, results from later generations did not notably differ from those from generation 6000. Results of repeated runs were summarized as box plots.
The spatial genetic structure of the simulated lineages was analyzed using graphs of genetic dissimilarity against genetic distance. For this, 20,000 individuals were drawn randomly from the population and grouped into pairs. The geographic distance and the genetic dissimilarity (1, dissimilar; 0, similar) of each pair were determined. The distance range was divided into 100 intervals and the percentage of dissimilar pairs in each interval was plotted against the logarithm of the midpoint of the distance interval.
Spatial statistics:
Several statistics were calculated at different spatial scales. These were fractal dimensions D0 and D2, conditional incidence IC, conditional Simpson incidence IC2, and a measure for population subdivision GST. Analysis was done for a square area a little smaller than the simulation arena with side length of 226 (≈6.7 × 107) units, randomly placed within the simulation arena. The analysis area was divided into smaller and smaller squares by repeated equal divisions into four. The smallest square had a side length of 2 units. Each step of this subdivision formed one scale of analysis. The units of analysis were the squares of subdivision (“boxes”), which can also be regarded as subpopulations. If boxes contained one or more individuals they were counted as “occupied” and were analyzed, and if they had none they were counted as empty.
A fractal distribution of individuals is reflected by a relationship of the type
 |
(5) |
with N(s) the number of boxes needed to cover all individuals at the scale s, the side length of the boxes (Hastings 1993). The exponent D0 is the box-counting dimension. By introducing a constant k0 (5) can be linearized to
 |
(6) |
A practical approach to determine D0 was to cover the arena of the simulated populations with grids of different box sizes and do box counts for the different scales (Halley et al. 2004), counting the number of boxes, Nj(s), needed to cover individuals of a given genotype j and at each scale (Figure 1). This was done for all common genotypes of frequency of at least 10, partly to reduce the computational demands and partly because rare genotypes will most likely not be detected in a population study. Moreover, rare genotypes contribute little to calculation of D0 as their box-occupation pattern is inevitably similar at most scales. D0 was then estimated by the negative slope of all log(Nj(s)) on log(s) by applying a regression analysis to the linear region of the graph (judged by eye).
Figure 1.—

Example of the box-counting procedure. (A) The big squares represent the simulation arena with grids of different box sizes superimposed. The arena is populated by individuals of two genotypes (solid and shaded dots). A box count for each genotype and each box-side length, or scale, is performed. The box count is the number of boxes containing one or more individuals of a given genotype. In the above example, all boxes that contribute to the box counts for the “solid” individuals are shaded. Scales and counts for those individuals are listed under the arenas and used for the plot in B. (B) The results from the box counting are plotted as log(box number) against log(scale). If the resulting graph, or parts of it, shows a linear region with a measurable gradient, the negative of this gradient is the box-counting dimension D0 of that genotype over the extent of the linear region. D0 is an indication of a fractal distribution of that genotype.
The correlation dimension D2 is related to D0 and either equal to or smaller than it (Grassberger 1983). It detects a power-law relationship between the concentration of individuals in the boxes and the scale. The concentration is measured by the Simpson index, IS, calculated from the proportion of individuals that fall in each box, pi, as 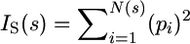 (Simpson 1949), with N(s) the number of boxes at that scale. The relationship is thus
(Simpson 1949), with N(s) the number of boxes at that scale. The relationship is thus
 |
(7) |
By introducing a constant k2 (7) can be linearized by analogy to (6) as
 |
(8) |
D2 was determined from the linear region of the graph of  against log(s) for all common genotypes j (Halley et al. 2004).
against log(s) for all common genotypes j (Halley et al. 2004).
The conditional incidence IC is a measure of self-similarity, effectively the derivative of D0 (Shaw 1995; Shaw et al. 2006). IC is calculated as the ratio of two incidences at neighboring scales,
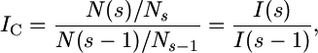 |
(9) |
with I(s) the proportion of occupied boxes N(s) among total boxes Ns at scale s. IC is constant for a truly self-similar distribution. For constant IC and each scale being half the length of the next larger one (which results in a subdivision into quarters), the relation between D0 and IC is thus
 |
(10) |
(Shaw 1995).
The conditional Simpson incidence IC2 was calculated in a similar way but using the Simpson index divided by the total number of boxes as a measure of incidence at a scale:
 |
(11) |
IC and IC2 were calculated separately for each common genotype at each scale.
A common way of summarizing population genetic structure over a wide range of spatial scales is to use F-statistics (Wright 1951), especially the coefficient of inbreeding, FST. GST, the coefficient of gene differentiation, is used as an analog of FST (Nei 1973) and often used for haploid organisms and was applied here by regarding the occupied boxes as subpopulations at different scales. The GST values estimated from the simulated lineages are proportional to those that would be found by unbiased sampling from the whole populations.
RESULTS
Quasi-equilibrium:
The simulation assumed a large background population of which only 30,000 lineages were followed. The simulated individuals were restricted in growth, as the total population size was assumed to be regulated in a density-dependent manner. Natural populations generally fluctuate in size, modeled here simplistically by a fixed mean birth rate and death after 1 time step. The numbers of individuals fluctuated during the course of the simulations and in a considerable number of cases all lineages died out. This was expected because all branching processes with one expected descendant per individual are certain to die out (Feller 1968). For further analysis simulations were chosen that ran for 50,000 time steps with individual numbers between 15,000 and 85,000 to make results from different dispersal functions and time steps comparable. The mean population size varied slightly between sets of these “long runs” with different dispersal functions: ∼39,400 ± 13,200 for power-law (PL) r−1.5, 42,800 ± 12,900 for PL r−2.0, 40,700 ± 12,800 for PL r−2.5, 42,400 ± 13,500 for exponential, and 40,900 ± 15,000 for global dispersal (mean ± standard deviation); the differences of the means were statistically significant. These numbers were products of large numbers of calls of the Poisson generator. Since the Poisson generator and the dispersal module were tested independently and had no shared variables or parameters, variations in population size must have been due to slight remaining long-range correlations in the underlying random-number generator, coupled with differences in the number of calls made to it. The generator, Knuth's subtractive method to generate uniform random deviates (Press et al. 1996), is widely recommended and has passed all the usual tests. However, no generator is truly random and there will always be some remaining signatures of the underlying deterministic nature of the algorithm. Since the imbalance between large and small random Poisson deviates was of the order of only 1 in 105, we believe the general patterns in the data were robust. This belief was supported by the observation that in general results from simulations that went extinct were very similar to the long-run results. Exceptions were runs where the population size dropped dramatically under 10,000 and recovered, causing a dramatic drop in genotype number to near uniformity.
Initially, all 30,000 individuals were of different genotypes. Most of the genotypes died out within the first 4000 generations in a process similar to exponential decay as is expected for a Poisson birth process with constant death rate. Without mutation, all genotypes but one died out (data not shown). With mutation, the number of genotypes stabilized after 4000 generations at a mean value of 62 ± 20 (Figure 2), indicating an equilibrium between mutation and drift. In addition to similar genotype numbers, the distribution of genotype frequencies was also similar between time steps and dispersal functions.
Figure 2.—

Decrease of genotype number with time. The logarithm of genotype number of inverse power-law [PL r−1.5 (black), PL r−2.0 (dark shading), and PL r−2.5 (medium shading)] and exponentially (light shading) and globally (very light shading) dispersed lineages is plotted against time. Box plots correspond to the statistics of 20 (exponential) or 12 replicates (all others).
Spatial patterns:
The different dispersal functions generated different spatial population patterns (Figure 3). Differences became more prominent over time. Initially all individuals were randomly distributed. In the case of global dispersal, that distribution persisted (not shown). For the other dispersal functions, the combination of dispersal and the Poisson birth process led to an aggregation of individuals in clusters. Individuals of the same genotype tended to cluster together because the dispersal patterns were inversely related to distance. This clustering process was strongest for the exponential simulations, in which tight clusters arose over time, of which most died out (Figure 3, bottom row) and only one cluster survived in the analyzed simulations after 40,000 generations. The exponential dispersal pattern did not enable new clusters to form as the maximum distance over which dispersal was at all probable was in the same range as the spatial size of an individual cluster. The structures that arose from PL r−2.5 and PL r−2.0 dispersal also led to a few distinct clusters, but the process of reducing the number of clusters was slower for PL r−2.5 and even slower for the PL r−2.0 dispersal. The resulting clusters were less dense; the PL r−2.5 dispersed lineages were clustered more densely than the PL r−2.0 dispersed ones (Figure 3, middle rows). LDD was sufficiently frequent with PL r−1.5 dispersal to counteract the concentrating effect of the Poisson birth process (Figure 3, top row). After an initial loss of single individuals and small clusters within the first 100 generations, the lineages remained scattered in clusters of a wide range of sizes over the entire arena. The number and sizes of clusters appeared stable in that state over the whole duration of the simulations (Figure 3, top row, different time steps). It was hard to define a cluster unambiguously with PL r−1.5 dispersal, so it was not possible to analyze this situation quantitatively. However, the number of “clusters” observed was determined by two processes. The first process was the rate at which individuals within the cluster radius became extinct and the second one the rate at which new lineages reached a distance larger than the cluster radius and were able to multiply to form a new cluster. The second rate depended strongly on the probability of LDD and was much greater for smaller b. The difference between dispersal functions was greater the larger the cluster radius being considered. Cluster numbers were highest with PL r−1.5 dispersal of all reasonable values for the cluster radius.
Figure 3.—

Examples of spatial structures of simulated lineages. Top three rows, inverse power-law dispersal (PL r−1.5, PL r−2.0, and PL r−2.5, respectively); bottom row, exponential dispersal. From left to right: generations 6000, 40,000, and a close-up of 40,000 (magnifications of factor 10 for PL r−1.5, 100 for PL r−2.0, 1000 for PL r−2.5, and 10,000 for exponential). Numbers on the arena scale (left and bottom) are multiples of 107. Individuals are shown as dots; different colors indicate different genotypes in that simulation.
Spatial genetic structure:
Population genetic theory states that limited dispersal results in individuals located close to each other being genetically more similar than individuals further apart (Malécot 1969). However, many of the presented simulations were using fat-tailed LDD. The relationship of genetic dissimilarity and geographic distance at quasi-equilibrium was thus of interest and investigated as the percentage of genetic dissimilar pairs found at different distances (Figure 4). Individuals were initially genetically dissimilar and were distributed randomly over the whole arena with wide spacing between them. Over time, genetically similar individuals appeared as particular lineages grew by chance. In lineages dispersing with an exponential dispersal function these individuals were found near one another. In these lineages genetically dissimilar individuals appeared near one another as well, as mutants arose within lineages of spatially adjacent individuals. The graphs of genetic dissimilarity reflect the same clustering tendencies of genotypes as discussed above for the whole population (Figure 3). The strong clustering tendency of the exponential dispersal resulted in the dissimilarity graph having a short extension on the log(geographic distance) axis (Figure 4). The extension of the graphs was longer for the power-law dispersal functions, reaching higher geographic distances. This trend increased with decreasing b. The extremely fat-tailed distribution PL r−1.5 dispersed sufficient individuals to distances comparable to the arena size to cause pairs of similar and dissimilar individuals to be found at far distances at all time steps. With all dispersal functions but the global dispersal an increase of genetic dissimilarity with geographic distance was found (Figure 4).
Figure 4.—

Percentages of genetically dissimilar pairs in intervals of geographic distance. Percentages calculated for typical simulations of PL r−1.5, PL r−2.0, PL r−2.5, and exponentially dispersed lineages after 6000 (top row) and 40,000 (bottom row) generations are shown. Geographic distance scale is given as log10 (1 = 101, 2 = 102, . . . ).
Fractal dimensions D0 and D2:
Many natural objects have relevant features on a variety of different scales. If their properties are similar across a wide range of scales, they are self-similar over that range and have a fractal structure. Natural objects are often not ideal fractals, but are sufficiently fractal that the tools of fractal geometry, including characterization by noninteger dimensions, can be used to describe them. Fractal measures, such as the box-counting dimension, have been used for describing the spatial distribution of species (e.g., Kunin 1998) and power laws, which are closely connected with fractals, are a major descriptive tool in ecology (Halley et al. 2004).
The most basic fractal dimension is the box-counting dimension (Equation 6). This box count was applied separately to individuals of the same genotype in the simulated samples at different scales and summed up in the graph of log(D0) against log(s) (Figure 5). For the exponentially dispersed lineages the maximum scale, the point where the regression line meets the log(s) axis, was a little above 104 and much smaller than those for the inverse power-law dispersed lineages. This simply reflects the limited spatial extent of any genotype with exponential dispersal. In PL r−2.5 dispersed lineages, the maximum spatial scale was ∼105, in PL r−2.0 it was ∼106, while in PL r−1.5 dispersed lineages, the maximum scale was larger than the simulation arena (Figure 5). This was confirmed with a limited number of simulations in a larger arena where still the line did not meet the log(s) axis (data not shown).
Figure 5.—

Estimates of fractal dimensions. Box-counting dimension D0 (left column) and correlation dimension D2 (right column) of clonal descendants of inverse power-law (PL r−1.5, PL r−2.0, and PL r−2.5) and exponentially and globally dispersed lineages at generation 6000. D0: log–log plot of the number of “occupied” boxes against scale. D2: log–log plot of the Simpson index of occupied boxes against scale. Box plots correspond to the statistics of 20 (exponential) or 12 replicates (all others).
A regression analysis for the box-counting graphs of the exponentially dispersed lineages produced a coefficient of determination of R2 = 0.87. The linear region stretched over a spatial range of about two orders of magnitude only, which is too short to merit description as a truly fractal structure (Halley et al. 2004). By contrast, broader linear regions, spanning three orders of magnitude and three to four orders of magnitude, were found in PL r−2.5 and PL r−2.0 dispersed lineages, indicating fractal spatial patterns over several orders of magnitude with fractal dimensions of D0 = 0.994 ± 0.008 (R2 = 0.95) and D0 = 0.760 ± 0.006 (R2 = 0.93), respectively. As ranges were limited these can be classified only as a quasi-fractal spatial pattern. The regression analysis of the box-counting curve of PL r−1.5 dispersed lineages found a poorer fit with R2 = 0.85 but the linear region stretched over a much wider scale, six orders of magnitude. Hence PL r−1.5 populations had a more extended fractal structure than PL r−2.0 populations but the pattern seemed to be less homogeneous. The fractal dimension was D0 = 0.439 ± 0.004 (Figure 5).
However, the box-counting dimension does not consider the number of individuals in each box, which can vary enormously. To capture this aspect of population structure, the correlation dimension D2 was applied (Equation 8). This uses the Simpson index (Simpson 1949) that quantifies the evenness of occurrence between subpopulations (boxes).
In PL r−1.5 dispersed lineages, a fractal dimension of D2 = 0.534 ± 0.004 was calculated (R2 = 0.90). The linear region stretched over nearly six orders of magnitude (Figure 5). The distribution of genotypes therefore shows a very strongly fractal pattern, over most of the observed area. In PL r−2.0 dispersed lineages, the fractal pattern is a little weaker, D2 = 0.777 ± 0.006 (R2 = 0.92), and the linear region stretched over four orders of magnitude, which, though less than for PL r−1.5, is still quite large by comparison with reports in natural ecological systems (Halley et al. 2004). Results for the PL r−2.5 dispersed lineages indicate an even weaker fractal structure with D2 = 1.008 ± 0.07 (R2 = 0.94). The linear region of the log(Simpson index) against the log(s) plot for the exponentially dispersed lineages stretched over only two orders of magnitude and so did not indicate a fractal genotype distribution. As expected, globally dispersed lineages did not show patterns of any kind as D0 and D2 were both zero.
The fractal dimensions were determined for five further power-law dispersal functions (1.2 ≤ b ≤ 4) at generation 6000. Linear relationships between the fractal dimensions and the inverse of the power-law exponent were found (Figure 6). The regression equations of fractal dimension on 1/b were 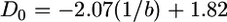 (R2 = 0.99) and
(R2 = 0.99) and  (R2 = 0.99).
(R2 = 0.99).
Figure 6.—

Relationship between fractal dimensions and 1/b (b: power-law exponent). Data points were plotted as solid circles (D0) or open squares (D2) and regression lines were fitted to the two fractal dimension types (solid line, D0; dashed line, D2).
Conditional incidence (IC) and conditional Simpson incidence (IC2):
The self-similarity measures were applied to individuals of the same genotypes of the simulated lineages. Stable patterns of IC and IC2, respectively, against log(s) were found, which were characteristic of the dispersal function (Figure 7). As found by Shaw (1995) the IC values describing exponentially dispersed lineages were much larger than those in PL r−2.0 dispersed lineages.
Figure 7.—

Self-similarity statistics. Conditional incidence (IC, left column) and conditional Simpson incidence (IC2, right column) of inverse power-law (PL r−1.5, PL r−2.0, and PL r−2.5) and exponentially and globally dispersed lineages at generation 6000 plotted against log10 of scale. Box plots correspond to the statistics of 20 (exponential) or 12 replicates (all others).
The IC graphs of the exponentially dispersed lineages were characterized by a sharp rise from 0.25, which is the minimum possible IC value for a grid based on a division of each square into four smaller squares, to a maximum value around 0.75 over one to two orders of magnitude followed by a little less steep decline (Figure 7). The fat-tailed power-law dispersed lineages showed flatter IC graphs, with a rise extended over more scales, around two and three orders of magnitude for PL r−2.5 and PL r−2.0 dispersal functions, respectively, and more or less covering all observed scales for PL r−1.5 dispersal. This confirms that the spatial genetic structure generated by fat-tailed inverse power-law dispersal had a much larger extent than that produced by exponential dispersal. However, IC graphs for PL r−4.0 dispersed lineages looked identical to those for exponential dispersed lineages and thus had the same spatial extent (graphs not shown). IC graphs for globally dispersed lineages were, as expected, uninformative.
For the very fat-tailed inverse power-law dispersal function PL r−1.5, the rise of the IC against the log(s) curve was very shallow over about two to three orders of magnitude. A flat IC curve signifies a self-similar structure, so the underlying genetic structure of PL r−1.5 lineages was close to self-similar over a limited but substantial range.
The results from the IC2 analysis were in most respects similar to those from the IC analysis. IC2 against log(s) graphs had the same modes as the IC graphs. The IC2 graphs of the global and exponential dispersal were nearly identical to the IC graphs. However, the two extreme fat-tailed dispersal functions under investigation resulted in the IC2 graphs having a plateau, reflecting a self-similar structure in the lineages. For PL r−2.0 the plateau stretched about two orders of magnitude. For PL r−1.5 the plateau stretched to all scales above 102. Although IC2 analysis results were very similar to the IC results, the IC2 was more sensitive to the self-similar structure present in the lineages.
Gene differentiation GST values can be interpreted as characterizing the population structure by the proportion of total gene diversity between subpopulations. High GST values near 1 were found for all dispersal functions at small scales (Figure 8). This means that almost all the variation in the populations occurred between subpopulations at small scales. Subpopulations contained only one main genotype. In the PL r−2.0 and PL r−1.5 dispersed populations, GST values stayed at very high values over many scales, approaching zero only at very large scales. The population structure seemed quite homogeneous over a wide range of scales. For exponentially dispersed populations the GST values fell much earlier and reached a plateau around scale 104, the same maximum scale discovered before and again simply showing that the sample was concentrated in a few areas with distinct genotypes. The exact value of GST and the length of the plateau depends on the number of genotypes present in the population and the location of population clusters. GST graphs from the PL r−2.5 dispersed lineages showed an intermediate behavior between PL r−1.5 and exponential GST graph trends. In the case of global dispersal, GST values were either 1 for small and medium scales where only single individuals were present per box or 0 at large scales, revealing perfect mixing at those scales. A transition phase with intermediate GST values is very short.
Figure 8.—

GST at different scales. GST of inverse power-law (PL r−1.5, PL r−2.0, and PL r−2.5) and exponentially dispersed lineages at generation 6000 plotted against log10 of scale is shown. Box plots correspond to the statistics of 20 (exponential) or 12 replicates (all others).
DISCUSSION
Several experiments and theoretical arguments suggest that LDD dispersal of small propagules by wind is best described by inverse power-law dispersal functions. Here, the importance of such LDD dispersal events on population genetic structure in quasi-equilibrium is investigated by computer simulation of lineages of clonal haploid organisms, which were dispersed following inverse power-law dispersal functions with an exponent 1 < b ≤ 4. Our most important finding is a fractal distribution of genotypes in the resulting populations for small b, which is best detected by the D2 fractal dimension, applying the Simpson index to subpopulations at different scales (Figure 5). In contrast, if dispersal is modeled by an exponentially bounded function, no fractal population genetic structure is produced. The D2 statistic therefore is well suited to differentiate populations dispersed by inverse power-law functions from those dispersed by exponentially bounded functions.
These results corroborate results from studies of focal expansion with fat-tailed dispersal, which showed that LDD produces a patchy, fractal-like, spatial population structure very different from the structure of a homogeneous front found with exponentially bounded dispersal (Shaw 1995; Lett and Østergård 2000; Cannas et al. 2006). With the present quasi-equilibrium setting we were able to quantify differences in fractal structure.
It was furthermore possible to find a simple relationship between the fractal dimension of the population structure and the migration rate (Figure 6). The linear relationship between the D2 fractal dimension and the inverse power-law exponent was ∼D2 = 1.8 − 2.0 × 1/b. If the necessary data were available, it should be possible to estimate the rate of migration, including the power-law exponent, from the fractal structure observed in field studies. The factor of 2 appearing in this equation suggests that the relationship has a simple mathematical explanation, but we cannot deduce it.
The power-law dispersal distribution used for this study is in essence very similar to Lévy-flight process (Shlesinger 1996). The resulting genotype distribution should have a Lévy-stable distribution (scale free) in good agreement with the fractal population structures we report here. The difference between power-law and exponentially dispersed populations is also demonstrated by self-similarity statistics IC and IC2 (Figure 7), which are effectively derivatives of D0 and D2, respectively. The IC statistic was suggested by Shaw (1995) and a very similar statistic but for the factor of subdivision, the species-level self-similarity parameter α, was recently suggested by Ostling et al. (2003) as helpful for characterizing species distribution in ecology. In particular, if IC2 is applied to PL r−1.5 dispersed populations, it is constant over several orders of magnitude of scale and reveals the highly self-similar structure of those populations (Figure 7). IC2 values for exponentially dispersed populations are very different in showing a distinctive peak and no self-similar structure is detected. The self-similarity statistics can pick up other patterns besides self-similarity in the spatial structures. Peaks are a sign of visual patchiness, they are caused by areas with more occupied boxes, and the scale immediately greater than the peak is the scale of greatest patchiness (Cousens et al. 2004). IC2 may be the most suitable tool if applied to noisy field data to detect fractal structures or self-similarity. The expected graphs may indicate differences stronger than the fractal dimension analysis. In theory, IC2 values are interchangeable with the fractal dimension by the relation D2 = 2 + log2(IC2). However, it seems difficult to estimate IC2 from the graphs as plateaus are short for all values of b ≥ 2. From the PL r−1.5 simulations, the value for IC2 can be estimated as 0.36 (Figure 7), implying D2 = 0.52, which is near the value of 0.54 from D2 statistics. However, it seems preferable to use the IC2 graphs for detecting self-similarity only and determine the correlation dimension in case self-similarity was found. To test the distinctness of the self-similarity statistics, some preliminary simulations with different median dispersal distances were performed. The mode of the IC or IC2 curves moved to larger scales for higher median dispersal distances. However, the shape of the graph was not changed as long as the mode still lay within the arena. An exponentially bounded distribution induced a different pattern from that of an inverse power-law distribution with b ≤ 2. Even if the median dispersal distance of the exponential function was very long range the IC graph still showed a distinctive peak, underlining the potential of this statistic to discriminate between fat-tailed LDD and exponential dispersal.
Applying our statistics to field data in the future will show how robust they are to noise and sparsity of data at larger scales. Data at many different scales are necessary for using the new statistics and to detect the different structures we predict. We therefore suggest that sampling schemes for population genetic analyses in which the nature of dispersal between subpopulations may be significant should routinely include as wide a range of spatial scales as possible. Sampling on a wider range of spatial scale does not necessarily mean that more samples are needed but that the sampling scheme has to be optimized for this task.
With all dispersal functions except global dispersal, an increased genetic dissimilarity with geographic distance was found (Figure 4). Some population studies include the estimation of mean dispersal distances from the genetic structure of the population. This strategy is problematic for power-law dispersal with b ≤ 2 because mean and variance are not defined for these dispersal functions. As this is a substantial problem it cannot be dealt with in the frame of this work.
In contrast to the fractal or self-similar measures, the common population genetic statistic GST is not very useful for differentiating between different dispersal types (Figure 8). Patterns of change were rather similar for all dispersal functions. To apply the statistic to field data to determine the underlying dispersal function would be difficult, as clear differences are small and apparent only at very long scales, at distances from the source where data will normally be very scarce.
The emergence of fractal or scale-invariant patterns in simulations of power-law dispersed biological objects was shown previously by Cannas et al. (2006). They investigated the expansion of an invading species from a central focus, such as a newly established weedy species. Using exponents of 2 < b ≤ 3, the resulting spatial structure showed a fractal character, but only at some times and at the border of the main patch. In contrast, our study showed a fractal pattern of genotype distribution developing from an initially uniform random distribution of genotypes for a population with power-law dispersal and stable size. Once developed, the fractal pattern remained present during the whole remaining time of simulation because mutation in our model acts as a continual source of new invasions superimposed on the existing population. Moreover, fractal patterns appeared most strongly with small power law coefficients, of magnitude <2.
Fat-tailed dispersal of haploid individuals results in a fractal distribution of haploid genotypes over several scales. However, true nonsexual propagation is not very frequent in nature. Even organisms that use clonal reproduction for the rapid production of many descendants generally have occasional sexual reproduction, such as many ascomycete and basidiomycete fungi. The sexual phase leads to an uncoupling of genes and loss of linkage disequilibrium. We predict, therefore, that our main conclusions will apply equally to alleles of a single gene in a sexual population. In particular, we predict that power-law dispersal will result in a fractal distribution of alleles of a gene in a sexual population, just as it does of genotypes in a clonal population.
This study investigates LDD in a simulation arena that is large enough to reflect the dispersal of wind-blown small particles like fungal spores at scales ranging from leaves to continents. Such LDD is observed in nature, e.g., the spread of recently mutated genotypes of P. striiformis (wheat yellow rust) from the United Kingdom to Denmark, France, and Germany (Brown and Hovmøller 2002; Hovmøller et al. 2002) or the regular reinfection of wheat-growing areas in northern China with wheat yellow rust from more southerly regions (Brown and Hovmøller 2002). The ability of the PL r−1.5 dispersal to generate a wide-ranging distribution of an individual genotype over the whole arena was clear. The observations of LDD of wheat yellow rust are so far in accordance with those of our model. Our study is consistent with the presence of fractal structure in natural rust or mildew populations but more data on several scales than are usually collected for a population genetic study for confirmation would be needed.
The fractal analysis of the PL r−1.5 dispersed populations does not determine a maximum scale of dispersal as there may be none with this kind of dispersal function. Realistically, the range of dispersal will be finite, because of processes such as deposition or death, but nevertheless very large (Shaw et al. 2006). This was not considered in the present study as the population genetic structure will be dominated by the effective dispersal function at scales well below the arena size.
For larger wind-dispersed particles like seeds, a power-law dispersal function with b > 2 will apply (Cannas et al. 2006). However, inverse power-law distribution functions may also be useful to model nonwind LDD, as, e.g., the dispersal of small birds, tits, which followed an inverse-square (Cauchy) law (Paradis et al. 2002). Larger negative power-law exponents than b > 4 may not be very useful for simulations of biological processes because the spatial pattern produced cannot be distinguished from that arising from a negative exponential and thus the less computationally expensive model of negative exponential dispersal can be chosen.
From the present study it appears to be inappropriate ever to use exponentially bounded dispersal functions with a scale parameter much smaller than the size of the available arena to model biological dispersal processes. With exponential dispersal alone a lineage of a dispersing clonal organism (or, we propose, an allele in a sexual organism) will inevitably stay in an area a few orders of magnitude larger than the median dispersal distance (Figure 3). In a nongrowing population this leads to a concentration of each genotype in a single location. Such populations would be much more prone to extinction in the case of changes in the environment. If the only means of dispersal is an exponentially bounded process, any species is in fact evolving as strongly isolated populations, even if it occupies a large homogeneous, suitable habitat. It therefore seems likely that the majority of organisms have a LDD means of dispersal, which will have allowed their ancestors to avoid restriction to a limited area. This argument may seem to depend on the short median dispersal distances we assumed. However, long-distance (e.g., 105 km) exponential dispersal is inconsistent with all observations of dispersal of spores, pollen, or seeds. If a mixture of two exponential functions is used as in Nichols and Hewitt (1994), the model still exhibits a distinct scale, albeit the larger one, with quite distinct isolated patches evolving independently. Assuming a high proportion of long-distance exponential dispersal in the mixture conflicts with the lack of such observations in field data, as stated before.
This inappropriateness of exponentially bounded functions for modeling airborne dispersal processes is of importance for risk assessment studies that aim at long-term, macroscale predictions. The dispersal potential is a key uncertainty in predicting the risk of an airborne disease (Yang 2006) and the same should apply to the spread of pollen of genetically modified plants or droplet-borne diseases such as foot-and-mouth disease. The use of exponentially bounded functions for predicting dispersal of organisms that in fact follows a small-exponent power law in this context is inappropriate because such models would tend to seriously underestimate risks.
The extensive simulations described here show a characteristic population genetic pattern generated by LDD in a homogeneous landscape. This is a simplification of real conditions but is partly met in the modern type of agriculture that favors monoculture. However, fragmented habitats or barriers like mountains or oceans may influence population genetic structure. Extensions of the present model to accommodate for inhomogeneous landscapes will be necessary to investigate this in detail. Simulations using long-range exponential functions have shown such LDD resulting in faster bridging between niches over unsuitable habitats and greater genetic diversity in remote subpopulations (Le Corre et al. 1997; Davies et al. 2004; Bialozyt et al. 2006). It is to be expected that fat-tailed LDD will result in an even faster niche colonization and a further increase in genetic diversity of remote subpopulations. Predictions on the presence of fractal structure are more difficult as this may depend on the niche distribution.
Acknowledgments
We thank two helpful anonymous reviewers. This research was supported by the Biotechnology and Biological Sciences Research Council and was part of the BioExploit project in the European Union Framework 6 Programme.
References
- Austerlitz, F., and P. H. Garnier-Géré, 2003. Modelling the impact of colonisation on genetic diversity and differentiation of forest trees: interaction of life cycle, pollen flow and seed long-distance dispersal. Heredity 90: 282–290. [DOI] [PubMed] [Google Scholar]
- Austerlitz, F., C. W. Dick, C. Dutech, E. K. Klein, S. Oddou-Muratorio et al., 2004. Using genetic markers to estimate the pollen dispersal curve. Mol. Ecol. 13: 937–954. [DOI] [PubMed] [Google Scholar]
- Aylor, D. E., 2003. Spread of plant disease on a continental scale: role of aerial dispersal of pathogens. Ecology 84: 1989–1997. [Google Scholar]
- Bialozyt, R., B. Ziegenhagen and R. J. Petit, 2006. Contrasting effects of long distance seed dispersal on genetic diversity during range expansion. J. Evol. Biol. 19: 12–20. [DOI] [PubMed] [Google Scholar]
- Brown, J. K. M., and M. S. Hovmøller, 2002. Aerial dispersal of pathogens on the global and continental scales and its impact on plant disease. Science 297: 537–541. [DOI] [PubMed] [Google Scholar]
- Bullock, J. M., and R. T. Clarke, 2000. Long distance seed dispersal by wind: measuring and modelling the tail of the curve. Oecologia 124: 506–521. [DOI] [PubMed] [Google Scholar]
- Cain, M. L., H. Damman and A. Muir, 1998. Seed dispersal and the Holocene migration of woodland herbs. Ecol. Monogr. 68: 325–347. [Google Scholar]
- Cain, M. L., B. G. Milligan and A. E. Strand, 2000. Long-distance seed dispersal in plant populations. Am. J. Bot. 87: 1217–1227. [PubMed] [Google Scholar]
- Cannas, S. A., D. E. Marco and M. A. Montemurro, 2006. Long range dispersal and spatial pattern formation in biological invasions. Math. Biosci. 203: 155–170. [DOI] [PubMed] [Google Scholar]
- Clark, J. S., 1998. Why trees migrate so fast: confronting theory with dispersal biology and the paleorecord. Am. Nat. 152: 204–224. [DOI] [PubMed] [Google Scholar]
- Cousens, R., J. Wallinga and M. Shaw, 2004. Are the spatial patterns of weeds scale-invariant? Oikos 107: 251–264. [Google Scholar]
- Darwin, C., 1859. The Origin of Species. John Murray, London.
- Davies, S., A. White and A. Lowe, 2004. An investigation into effects of long-distance seed dispersal on organelle population genetic structure and colonization rate: a model analysis. Heredity 93: 566–576. [DOI] [PubMed] [Google Scholar]
- Devaux, C., C. Lavigne, H. Falentin-Guyomarc'h, S. Vautrin, J. Lecomte et al., 2005. High diversity of oilseed rape pollen clouds over an agro-ecosystem indicates long-distance dispersal. Mol. Ecol. 14: 2269–2280. [DOI] [PubMed] [Google Scholar]
- Feller, W., 1968. An Introduction to Probability Theory and Its Applications, Ed. 3, pp. 295–298. Wiley, New York.
- Ferrandino, F. J., 1993. Dispersive epidemic waves: I. Focus expansion within a linear planting. Phytopathology 83: 795–802. [Google Scholar]
- Fitt, B. D. L., P. H. Gregory, A. D. Todd, H. A. McCartney and O. C. MacDonald, 1987. Spore dispersal and plant disease gradients: a comparison between two empirical models. J. Phytopathol. 118: 227–242. [Google Scholar]
- Gittenberger, E., D. S. J. Groenenberg, B. A. S. Kokshoorn and R. C. Preece, 2006. Molecular traits from hitch-hiking snails. Nature 439: 409. [DOI] [PubMed] [Google Scholar]
- Grassberger, P., 1983. Generalized dimensions of strange attractors. Phys. Lett. A 97: 227–230. [Google Scholar]
- Halley, J. M., S. Hartley, A. S. Kallimanis, W. E. Kunin, J. J. Lennon et al., 2004. Uses and abuses of fractal methodology in ecology. Ecol. Lett. 7: 254–271. [Google Scholar]
- Hastings, H. M., 1993. Fractals: A User's Guide for the Natural Sciences. Oxford University Press, London/New York/Oxford.
- Hovmøller, M. S., A. F. Justesen and J. K. M. Brown, 2002. Clonality and long-distance migration of Puccinia striiformis f.sp. tritici in north-west Europe. Plant Pathol. 51: 24–32. [Google Scholar]
- Klein, E. K., C. Lavigne, H. Picault, M. Renard and P.-H. Gouyon, 2006. Pollen dispersal of oilseed rape: estimation of the dispersal function and effects of field dimension. J. Appl. Ecol. 43: 141–151. [Google Scholar]
- Kot, M., M. A. Lewis and P. van den Driessche, 1996. Dispersal data and the spread of invading organisms. Ecology 77: 2027–2042. [Google Scholar]
- Kunin, W. E., 1998. Extrapolating species abundance across spatial scales. Science 281: 1513–1515. [DOI] [PubMed] [Google Scholar]
- Le Corre, V., N. Machon, R. J. Petit and A. Kremer, 1997. Colonization with long-distance seed dispersal and genetic structure of maternally inherited genes in forest trees: a simulation study. Genet. Res. 69: 117–125. [Google Scholar]
- Lett, C., and H. Østergård, 2000. A stochastic model simulating the spatiotemporal dynamics of yellow rust on wheat. Acta Phytopathol. Entomol. Hung. 35: 287–293. [Google Scholar]
- Malécot, G., 1969. The Mathematics of Heredity. W. H. Freeman, San Francisco.
- McCartney, H. A., 1987. Deposition of Erysiphe graminis conidia on a barley crop. 2. Consequences for spore dispersal. J. Phytopathol. 118: 258–264. [Google Scholar]
- McCartney, H. A., and A. Bainbridge, 1987. Deposition of Erysiphe graminis conidia on a barley crop. 1. Sedimentation and impaction. J. Phytopathol. 118: 243–257. [Google Scholar]
- Nathan, R., G. Perry, J. T. Cronin, A. E. Strand and M. L. Cain, 2003. Methods for estimating long-distance dispersal. Oikos 103: 261–273. [Google Scholar]
- Nathan, R., N. Sapir, A. Trakhtenbrot, G. G. Katul, G. Bohrer et al., 2005. Long-distance biological transport processes through the air: Can nature's complexity be unfolded in silico? Divers. Distrib. 11: 131–137. [Google Scholar]
- Nei, M., 1973. Analysis of gene diversity in subdivided populations. Proc. Natl. Acad. Sci. USA 70: 3321–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols, R. A., and G. M. Hewitt, 1994. The genetic consequences of long distance dispersal during colonization. Heredity 72: 312–317. [Google Scholar]
- O'Hara, R. B., and J. K. M. Brown, 1998. Movement of barley powdery mildew within field plots. Plant Pathol. 47: 394–400. [Google Scholar]
- Ostling, A., J. Harte, J. Green and A. Kinzig, 2003. A community-level fractal property produces power-law species-area relationships. Oikos 103: 218–224. [Google Scholar]
- Paradis, E., S. R. Baillie and W. J. Sutherland, 2002. Modeling large-scale dispersal distances. Ecol. Modell. 151: 279–292. [Google Scholar]
- Pedgrey, D. E., 1986. Long distance transport of spores, pp. 346–365 in Plant Disease Epidemiology, Population Dynamics and Management, edited by K. J. Leonard and W. E. Fry. Macmillan, New York/London.
- Press, W. H., S. A. Teukolsky, W. T. Vetterling and B. P. Flannery, 1996. Numerical Recipes in C, Ed. 2, pp. 274–286. Cambridge University Press, Cambridge/London/New York.
- R Development Core Team, 2004. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria.
- Sackett, K. E., and C. C. Mundt, 2005. Primary disease gradients of wheat stripe rust in large field plots. Phytopathology 95: 983–991. [DOI] [PubMed] [Google Scholar]
- Shaw, M. W., 1995. Simulation of population expansion and spatial pattern when individual dispersal distributions do not decline exponentially with distance. Proc. R. Soc. Lond. Ser. B 259: 243–248. [Google Scholar]
- Shaw, M. W., 1996. Simulating dispersal of fungal spores by wind, and the resulting patterns. Asp. Appl. Biol. 46: 165–172. [Google Scholar]
- Shaw, M. W., T. D. Harwood, M. J. Wilkinson and L. Elliott, 2006. Assembling spatially explicit landscape models of pollen and spore dispersal by wind for risk assessment. Proc. R. Soc. B 273: 1705–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlesinger, M. F., 1996. Lévy walks versus Lévy flights, pp. 279–283 in On Growth and Form, edited by H. E. Stanley and N. Ostrowsky. Nijhoff, Dordrecht, The Netherlands.
- Simpson, E. H., 1949. Measurement of species diversity. Nature 163: 688. [Google Scholar]
- Stockmarr, A., 2002. The distribution of particles in the plane dispersed by a simple 3-dimensional diffusion process. J. Math. Biol. 45: 461–469. [DOI] [PubMed] [Google Scholar]
- Wright, S., 1951. The genetical structure of populations. Ann. Eugen. 15: 323–354. [DOI] [PubMed] [Google Scholar]
- Yang, X. B., 2006. Framework development in plant disease risk assessment and its application. Eur. J. Plant Pathol. 115: 25–34. [Google Scholar]