

SUMMARY
The regulation of bone remodeling by an adipocyte-derived hormone implies that bone may exert a feedback control of energy homeostasis. To test this hypothesis we looked for genes expressed in osteoblasts, encoding signaling molecules and affecting energy metabolism. We show here that mice lacking the protein tyrosine phosphatase OST-PTP are hypoglycemic and protected from obesity and glucose intolerance because of an increase in β-cell proliferation, insulin secretion and insulin sensitivity. In contrast, mice lacking the osteoblast-secreted molecule osteocalcin display decreased β-cell proliferation, glucose intolerance and insulin resistance. Removing one Osteocalcin allele from OST-PTP-deficient mice corrects their metabolic phenotype. Ex vivo, osteocalcin can stimulate CyclinD1 and Insulin expression in β-cells and Adiponectin, an insulin-sensitizing adipokine, in adipocytes; in vivo osteocalcin can improve glucose tolerance. By revealing that the skeleton exerts an endocrine regulation of sugar homeostasis this study expands the biological importance of this organ and our understanding of energy metabolism.
INTRODUCTION
The prevailing paradigm in skeletal biology is that differentiation and functions of the two bone-specific cell types, osteoblasts and osteoclasts, are determined by secreted molecules that can either be cytokines acting locally, or hormones acting systemically (Harada and Rodan, 2003; Teitelbaum and Ross, 2003). A remarkable feature of most hormonal regulations is that they are controlled by feedback loops such that a cell type affected by a hormone sends signals influencing the hormone-producing cell. When applied to skeletal biology the concept of feedback regulation suggests that bone cells may exert an endocrine function.
Bone remodeling, the process whereby bones renew themselves, is regulated by multiple hormones. That obesity protects mammals from osteoporosis led us to propose that bone remodeling and energy metabolism could be regulated by the same hormone(s) (Ducy et al., 2000a). Verifying this hypothesis we showed that leptin, an adipocyte-derived hormone that appears during evolution with bony skeleton, is a major regulator of bone remodeling by acting on osteoblasts through two different neural pathways (Karsenty, 2006). Regardless of the molecular complexity of this novel neuroendocrine regulation, if indeed bone cells determine the level of activity of hormone-producing cells, then osteoblasts should affect energy metabolism.
Osteocalcin, one of the very few osteoblast-specific proteins, has several features of a hormone. It is, for instance, a cell-specific molecule, synthesized as a pre-pro-molecule and secreted in the general circulation (Hauschka et al., 1989; Price, 1989). Because of their exquisite cell-specific expression the Osteocalcin genes have been intensively studied to identify osteoblast-specific transcription factors and to define molecular bases of bone physiology (Harada and Rodan, 2003). In the course of the latter study we generated Osteocalcin−/ − mice (Ducy et al., 1996). While analyzing these mutant mice we noticed that they had an abnormal amount of visceral fat (P. D. and G. K., unpublished observation). This was the first evidence suggesting that skeleton may regulate energy metabolism.
Osteocalcin undergoes an unusual post-translational modification whereby glutamic acid residues are carboxylated to form γ-carboxyglutamic acid (Gla) residues, hence its other name, bone Gla protein (Hauschka et al., 1989). Gla residues usually confer to proteins high affinity for mineral ions, yet loss- and gain-of function experiments have failed to identify a function for osteocalcin in extracellular matrix mineralization in vivo (Ducy et al., 1996; Murshed et al., 2004). Thus at the present time the biological role, if any, of osteocalcin γ-carboxylation remains unknown.
A characteristic of osteoblasts is their paucity of cell-specific gene expression. We took advantage of this property and, with the goal of identifying osteoblast-enriched genes affecting energy metabolism, generated mutant mouse strains lacking genes encoding signaling molecules expressed only or preferentially in osteoblasts. Through this effort we inactivated, via classical means and in an osteoblast-specific manner, Esp also known as Ptprv, a gene expressed in osteoblasts and Sertoli cells that encodes a receptor-like protein tyrosine phosphatase termed OST-PTP (Mauro et al., 1994). Remarkably, mice lacking Esp in osteoblasts only display an increase in β-cell proliferation, insulin secretion and sensitivity that protects them from induced obesity and diabetes; all these phenotypes are corrected by deleting one allele of Osteocalcin. Accordingly, Osteocalcin−/ − mice are glucose intolerant and fat; genetic and cell-based assays show that osteocalcin can favor proliferation of pancreatic β-cells, Insulin and Adiponectin expression in β-cells and adipocytes. To our knowledge this study provides the first in vivo evidence that skeleton exerts an endocrine regulation of energy metabolism and thereby may contribute to the onset and severity of metabolic disorders.
RESULTS
Generation and perinatal lethality of Esp−/ − mouse models
We further established that Esp expression was restricted to bone and testes by making use of a LacZ allele knocked into the Esp locus and performing in situ hybridization and real time PCR studies. All analyses verified that Esp is expressed in osteoblasts but not in β-cells of the pancreas or in adipocytes (Figures 1A, 1B, and S5A).
Figure 1. Increased insulin secretion and β-cell proliferation in Esp −/− mice.
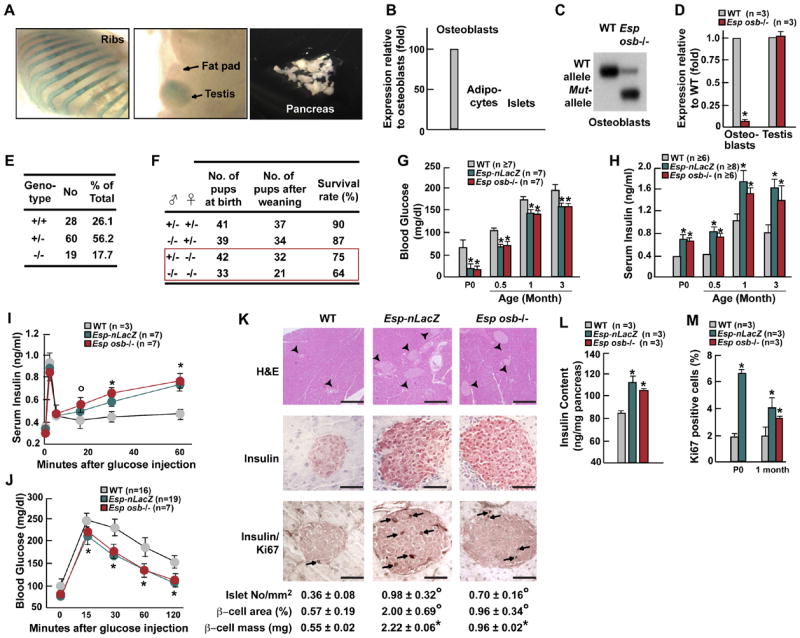
(A) LacZ stained tissues from newborn Esp −/ − mice demonstrating Esp locus activity in bone and testis but not in pancreas or fat pads. (B) Expression of Esp in osteoblasts, adipocytes, and pancreatic islets by real time PCR in 1 month-old mice. (C) Southern blot analysis showing efficient recombination at the Esp locus in osteoblasts of Esposb −/ − mice. (D) Using real time PCR Esp expression is 90% decreased in osteoblasts but not altered in testis of Esposb −/ − mice. (E) Decreased percentage at weaning of Esp −/ − pups born from crosses between Esp+/ − mice. (F) Lower survival at birth and at weaning of Esp −/ − pups born from Esp+/ − and Esp −/ − mothers. (G and H) Blood glucose levels (G) and serum insulin levels (H) in WT and Esp −/ − newborn before feeding (P0) or after random feeding at indicated ages. (I-J) GSIS (I) and GTT (J) test in 1 month-old WT and Esp −/ − mice. (K) H&E staining, insulin immunostaining and insulin/Ki67 double immunostaining showing larger islets and increased β-cell proliferation in pancreas of WT and 1 month-old Esp −/ − mice. Arrowheads indicate islets and arrows point at Ki67 positive cells. Scale bars, 100μm except upper panels, 800μm. Histomorphometric comparisons of islet number, size and β-cell mass between 1 month-old WT and Esp −/ − mice (lowest panel). (L) Pancreas insulin content in 1 month-old WT and Esp −/ − mice. (M) Quantification of the number of Ki67 immunoreactive cells in pancreatic islets of P5 and 1 month-old WT and Esp −/ − mice. All panels except I and J, °p<0.05 and *p<0.01 vs WT (Student’s t test). Panels I and J, °p<0.05 vs WT and *p 0.001 vs WT (ANOVA followed by post hoc analysis).
Esp was disrupted in a classical way (Esp-nLacZ) (Dacquin et al., 2004) and in an osteoblast-specific manner (Esposb−/ −) by deleting exons encoding the phosphatase domain, using the LoxP/Cre recombinase technology (Figure S1A). Mice harboring Esp floxed alleles were crossed with α1(1) collagen-Cre mice (Dacquin et al., 2002) to generate osteoblast-specific Esp-deficient mice (Esposb−/ −) (Figure S1B). Recombination occurred at high frequency at the Esp locus in osteoblasts. Accordingly, Esp expression was reduced nearly 90% in Esposb−/ − osteoblasts and unaffected in testes (Figures 1C and 1D). For the sake of clarity we will refer in the rest of the text to Esp−/ − mice when both Esp-nLacZ and Esposb−/ − mice were studied.
When analyzed at weaning, intercrosses of Esp+/− mice never yielded more than 20% of Esp−/ − pups although they were of normal appearance (Figures 1E, S1C, and S1D). Analysis of skeletal preparations of newborn wild-type (WT) and Esp−/ − pups failed to detect any abnormality of skeletogenesis that could explain a perinatal lethality (Figures S1E and S1F). Thus, we asked whether Esp−/ − pups lethality could be due to a humoral abnormality. In that case mutant pups born from homozygous mutant mothers should die at a higher frequency than those born from heterozygous mothers. While lethality of Esp−/ − pups born from Esp+/− mothers never reached 15% up to 35% of Esp−/ − pups born from Esp−/ − mothers died before weaning (Figure 1F) indicating that Esp−/ − pups lethality was caused, at least in part, by a humoral abnormality.
Increased β-cell proliferation and insulin secretion in Esp−/ − mice
Regardless of genetic background, sex, and type of deletion performed, the only humoral abnormality observed in Esp−/ − pups was a 3-fold reduction of blood glucose levels at birth before milk ingestion (Figure 1G). In some mutant pups this level was even too low to be detected. Blood glucose level remained abnormally low in adult Esp−/ − mice after feeding (Figure 1G). Explaining this hypoglycemia there was a marked hyperinsulinemia in newborn and adult Esp−/ − mice (Figure 1H). Serum level of C-peptide was also increased in Esp−/ − mice (Figure S2A) while pancreas content and serum level of glucagon, a hormone secreted by pancreatic α-cells in response to hypoglycemia, was normal in Esp−/ − mice (Figure S2B). Esp−/ − mice display a severe hyperinsulinemia, a feature known to inhibit glucagons secretion (Maruyama et al., 1984; Raju and Cryer, 2005) and that in all likelihood, antagonized the increase in glucagon secretion that should have been triggered by their hypoglycemia. Serum levels of IGF-1 and PYY were similar in WT and Esp−/ − mice. Surprisingly, serum levels of amylin, a protein synthesized by β-cells, was decreased 25% in mutant mice (Figure S2C-E).
The existence of an increase in insulin secretion in Esp−/ − mice was demonstrated by intraperitoneal (IP) glucose stimulated insulin secretion tests (GSIS, Figure 1I). To assess how this increase in insulin secretion affects the ability of the mice to dispose of a glucose load we performed glucose tolerance tests following IP injection of glucose (2g/kg of body weight) after an overnight fast (GTT). These tests revealed that Esp−/ − mice had a significantly higher tolerance to glucose than WT mice (Figure 1J). Histological and immunochemical analyses showed an increase in pancreas insulin content, number of islets, islets size and of β-cell mass in Esp−/ − pancreas (Figures 1K and 1L). While TUNEL assay failed to detect any abnormal apoptosis, Ki67 immunostaining showed that β-cell proliferation was increased 60 to 300% in 5 day-old (P5) and 1 month-old Esp−/ − mice (data not shown and Figure 1M).
Increased insulin sensitivity in Esp −/ − mice
To determine whether the enhanced ability of Esp−/ − mice to dispose of a glucose load was also caused by an increase in insulin sensitivity, we performed insulin tolerance tests (ITT). Despite their hyperinsulinemia insulin sensitivity was significantly increased in Esp−/ − compared to WT mice (Figures 2A). Hyperinsulinemic euglycemic clamps analysis verified the existence of an increase in insulin sensitivity by showing that steady-state glucose infusion rates were increased more than 50% in Esp−/ − mice compared to WT littermates (Figure 2B). This was due to an increase in insulin-stimulated glucose uptake in muscle, brown and white fat and in liver (Table S7). We also performed molecular and morphological analyses in skeletal muscle and liver. Expression of Pgc1α, a target gene of insulin, and of Nrf1 and Mcad, two target genes of Pgc1α, was significantly increased in Esp−/ − compared to WT muscles (Figure 2C). These results suggested that mitochondrial activity was enhanced in absence of Esp. Consistent with this hypothesis mitochondrial area was larger in Esp−/ − than in WT muscles while muscle mass over body mass ratio was normal (Figures 2D and S2F). In liver, expression of Foxa2 was increased while Pepck expression was decreased; fat content was also decreased in Esp−/ − liver, a feature consistent with an increase in insulin sensitivity (Figure 2E). In all analyses Esp+/− mice behaved like WT littermates (data not shown).
Figure 2. Increased insulin sensitivity and Adiponectin expression in Esp −/ − mice.
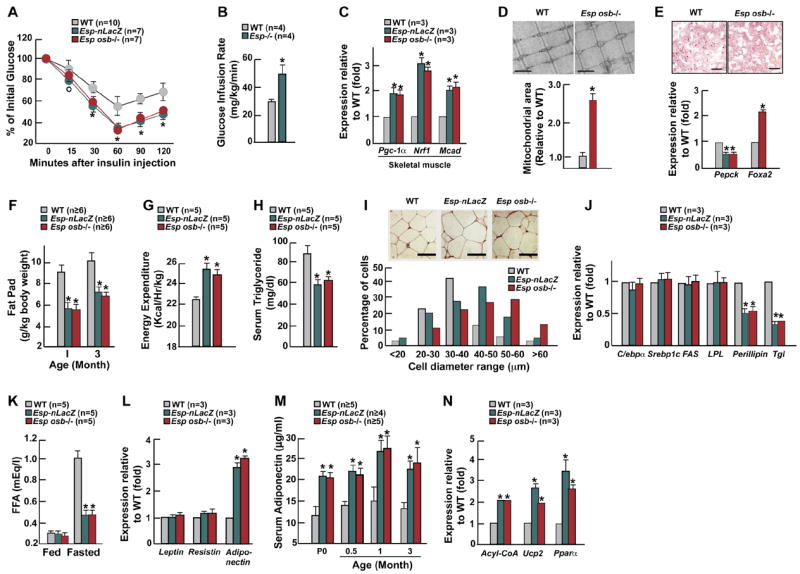
All experiments compare 1 month-old mice WT and Esp −/ − unless otherwise indicated. (A) ITT. (B) Glucose infusion rate during hyperinsulinemic-euglycemic clamp. (C) Expression of markers of insulin sensitivity in skeletal muscle measured by real-time PCR. (D) Electron microscopy images (upper panel, 20,000X) and corresponding quantification (lower panel) of mitochondrial area in gastrocnemius muscle. Scale bars, 1μm .(E) Decreased number of lipid droplets on oil red O stained liver sections (upper panel) and modified expression of insulin target genes by real time PCR (lower panel) in Esposb −/ − mice. Scale bars, 50 μm. (F) Fat pad mass (fat pad weight over body weight). (G) Energy expenditure. (H) Serum triglyceride levels after an overnight fast. (I) H&E staining of adipose tissues of WT and Esp −/ − mice (upper panel) and respective distribution of diameters for 100 measured adipocytes per slide (lower panel). Scale bars, 50μm. (J) Expression of markers of adipogenesis, lipogenesis, fat uptake, and lipolysis in fat. (K) Serum free fatty acid (FFA) in fed and overnight-fasted mice. (L) Expression of Leptin, Resistin and Adiponectin in fat. (M) Serum levels of adiponectin in newborn mice before feeding (P0) and after random feeding at other indicated ages. (N) Expression of adiponectin target genes in tissues of WT and Esp −/ − mice. Panel A, °p<0.05 vs WT and *p 0.001 vs WT (ANOVA followed by post hoc analysis); panels B-N, *p<0.01 vs WT (Student’s t test).
Adult Esp−/ − mice displayed another phenotype; their gonadal fat pads were significantly lighter than the ones of WT littermates despite being hyperinsulinemic (Figure 2F). This decrease in fat mass was restricted to visceral fat (Figure S2G). Contributing to explaining this phenotype, energy expenditure was increased in Esp−/ − mice while food intake was not affected (Figures 2G and S2H). Serum triglyceride levels were also lower in Esp−/ − than in WT mice (Figure 2H). Although there were fewer adipocytes in Esp−/ − than in WT mice (WT, 93.2±10.7x103 adipocytes/fat pad (n=5); Esp−/ −, 37±5.1x103 adipocytes/fat pad (n=3)) they were larger (Figure 2I). To understand this phenotype we studied expression of multiple molecular markers. C/EBPα, Srebp1c, Fatty acid synthase (Fas) and Lipoprotein lipase (LPL) were similarly expressed in Esp−/ − and WT adipocytes suggesting that adipogenesis, lipogenesis and fat uptake were not affected by the mutation (Figure 2J). In contrast, expression of Perilipin and Triglyceride lipase (Tgl), two lipolytic genes whose expression is inhibited by insulin, was markedly decreased in Esp−/ − adipocytes (Figure 2J) indicating that lipolysis is inhibited in Esp−/ − mice. Accordingly, serum level of free fatty acid did not increase following an overnight fast in Esp−/ − mice as it did in WT littermates (Figure 2K). There was no inflammation in fat since Tnfα and IL-6 expression and serum levels were low in Esp−/ − mice (Figures S2I and S2J).
Increased Adiponectin expression in Esp−/ − mice
To uncover the mechanism leading to an increase in insulin sensitivity in Esp−/ − mice we studied various adipokines. Expression and serum levels of resistin, an adipokine mediating insulin resistance, were virtually not affected by Esp deletion; the same was true for leptin, an insulin-sensitizing hormone (Friedman and Halaas, 1998; Steppan et al., 2001) (Figures 2L and S2K). On the other hand, expression and serum level of adiponectin, an adipokine enhancing insulin sensitivity (Yamauchi et al., 2001), were respectively increased 3- and 2-fold in Esp−/ − mice (Figures 2L and 2M). Accordingly, expression of the adiponectin target genes Acyl-CoA Oxidase, Pparα and Ucp2 was increased in Esp−/ − mice (Figure 2N) (Kadowaki and Yamauchi, 2005).
In summary, Esp inactivation causes hypoglycemia with decreased adiposity as a result of increased pancreatic β-cell proliferation, enhanced insulin secretion and improved insulin sensitivity. That these abnormalities were observed both in Esp-nLacZ−/ − and Esposb−/ − mice demonstrates that osteoblasts regulate glucose homeostasis.
Esp−/ − mice are protected from obesity and glucose intolerance
The increase in insulin secretion and sensitivity characterizing Esp−/ − mice raised the prospect that these mutant mice could be protected from obesity and diabetes. Esp-nLacZ−/ − and Esposb−/ − showing identical metabolic and molecular abnormalities we tested this hypothesis in Esp-nLacZ −/ − mice through three different assays.
First, we injected gold thioglucose (GTG) in 1 month-old mice to lesion the ventromedial hypothalamus (Brecher et al., 1965). GTG induced ventromedial hypothalamic lesions (Figure S3) and hyperphagia (Figure 3A) in both WT and Esp −/ − mice. When analyzed 3 months after injection GTG-treated WT mice were obese, glucose intolerant and insulin resistant, their serum triglyceride levels were also significantly increased (Figures 3B-3F). In contrast, GTG-treated Esp −/ − mice remained lean, had fat pad mass and serum triglyceride levels similar to the ones of PBS-treated WT mice and displayed no evidence of glucose intolerance or of insulin insensitivity (Figures 3B-3F). Second, we fed WT and Esp −/ − mice a high fat diet (HFD) for 6 weeks. As shown in Figures 3G-I, Esp −/ − fed a HFD gained significantly less weight than WT mice and did not develop glucose intolerance or insulin resistance as WT mice did. Third, we asked whether the increase in insulin sensitivity could protect Esp −/ − mice from pancreatic β-cell failure. To that end we injected mice with streptozotocin (STZ) to provoke oxidative stress in βcells and cell death (Le May et al., 2006). STZ markedly decreased pancreas insulin content and insulin serum level in both genotypes (Figures 3J and 3K). Eight days after STZ injection 3 of the 7 STZ-treated WT mice had died and all the surviving ones had serum glucose levels above 500 mg/dl (Figures 3L and 3M). On the other hand, only one STZ-treated Esp −/ − mouse died during this period and blood glucose level of the surviving ones did not exceed 250mg/dl. Unlike what was the case for STZ-treated WT mice, glucose could not be detected in urine of STZ-injected Esp −/ − mice (Figure 3N). Since both STZ-treated WT and Esp −/ − mice had a similar decrease in islets insulin content the absence of an overt diabetic phenotype in STZ-treated Esp −/ − mice suggests that their increase in insulin sensitivity protected them from diabetes. Results of these three experiments establish that Esp function is required for the development of obesity and glucose intolerance in mice.
Figure 3. Esp −/− mice are protected from obesity and glucose intolerance.
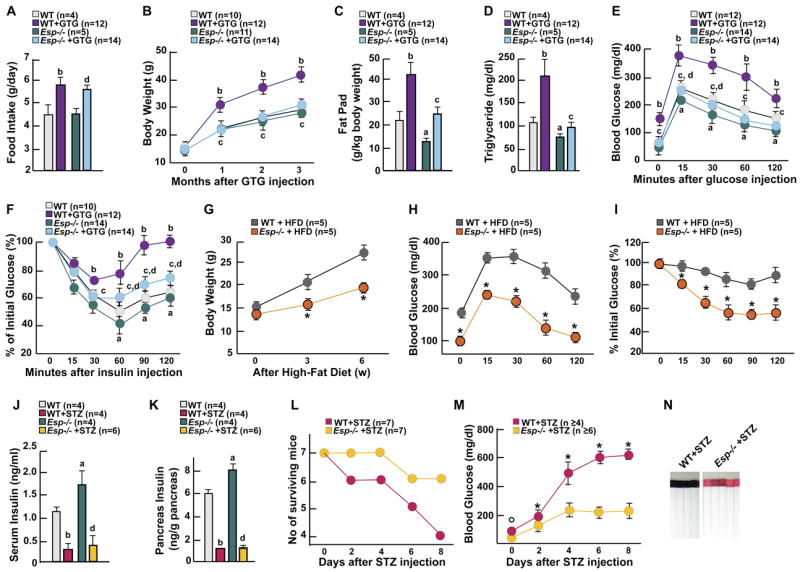
(A-F) Food intake per day (A), body weight curve (B), fat pad mass (C), serum triglyceride levels (D), GTT (E) and ITT (F) in 4 month-old WT and Esp −/ − mice 3 months after GTG or vehicle injection. (G-I) Body weight curve (G), GTT (H), and ITT (I) in 3 month-old WT and Esp −/ − mice fed a high fat diet for 6 weeks. (J and K) Serum insulin levels (J) and pancreatic insulin content (K) in 1 month-old WT and Esp −/ − mice 8 days after STZ or vehicle injection. (L and M) Survival of mice (L) and change of blood glucose levels (M) in 1 month-old WT and Esp −/ − mice during the 8 days following STZ injection. (N) Urinary glucose assays in 1 month-old WT and Esp −/ − mice 8 days after STZ injection. Panels A-F, J and K: a, WT vs Esp −/ −; b, WT+GTG(or STZ) vs WT+vehicle; c, WT+GTG(or STZ) vs Esp −/ − +GTG(or STZ); d, Esp −/ − +GTG(or STZ) vs Esp −/ − +vehicle. Panels G-I and M, *p<0.05 WT vs Esp −/ −. Panels A, C, D, J and K: Student’s t test, p<0.05 for a-d; panels B, E-I, L and M: ANOVA followed by post hoc analysis when number of groups>2, p 0.001 for a-d.
Esp influences the bioactivity of osteoblast-secreted molecule(s)
To further establish that it is through its osteoblastic expression that Esp regulates glucose metabolism we next relied on gain-of-function experiments. Transgenic mice overexpressing full-length Esp cDNA selectively in osteoblasts (α1(I)-Esp mice) displayed decreased β-cell proliferation, lower β-cell mass, hypoinsulinemia in the fed state and impaired insulin secretion in response to glucose (Figures 4A-C). They also showed lower adiponectin serum concentrations (Figure 4B). As a result, α1(I)-Esp mice on a regular chow developed hyperglycemia, glucose intolerance and insulin resistance (Figures 4B, 4D and 4E). That it is observed in mice overexpressing Esp in osteoblasts supports the notion that OST-PTP regulates the bioactivity of an osteoblast-derived secreted molecule regulating glucose homeostasis. That it is only observed in transgenic mice overexpressing full-length Esp suggests that the phosphatase activity of OST-PTP is required to affect glucose homeostasis.
Figure 4. Osteoblasts secrete a factor regulating Insulin and Adiponectin expression.
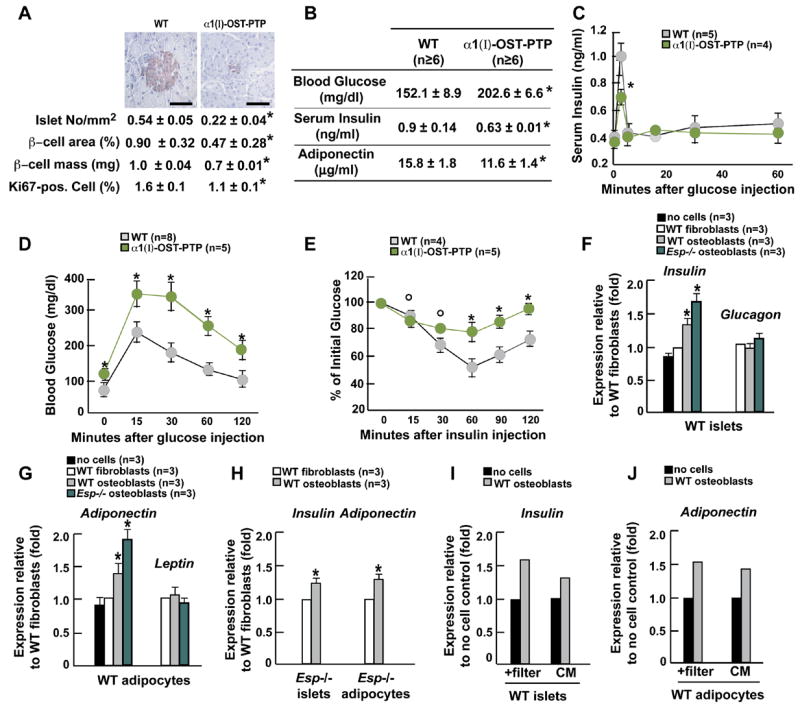
(A-E) All experiments compare 1 month-old WT and α1(I)–Esp mice. (A) Insulin immunostaining (upper panel) and histomorphometric comparisons of islet number, size, β-cell mass and Ki67 immunoreactive cells in pancreas (lower panel). Scale bars, 100μm. (B) Blood glucose, and serum insulin and adiponectin levels. (C) GSIS test. (D) GTT. (E) ITT. (G) Expression of Insulin and Glucagon in WT islets co-cultured with fibroblasts or osteoblasts. (H) Expression of Adiponectin and Leptin in WT adipocytes co-cultured with fibroblasts or osteoblasts. (I) Expression of Insulin and Adiponectin in Esp −/ − indicated cells co-cultured with fibroblasts or osteoblasts. (J and K) Expression of Insulin (J) and Adiponectin (K) in WT indicated cells co-cultured with or without osteoblasts in presence of a filter preventing cell-cell contact or in presence of conditioned medium (CM) collected from osteoblast cultures. Panels A, B and F-J: *p<0.05 vs WT (Student’s t test); panels C-E: °p<0.05 vs WT and *p 0.001 vs WT (ANOVA).
Next we co-cultured osteoblasts, which are adherent cells, with either pancreatic islets or adipocytes, which are not adherent. Co-culture of differentiated WT osteoblasts with islets isolated from WT mice increased Insulin expression 40% (Figure 4F). This enhancement of Insulin expression by osteoblasts was specific since fibroblasts did not have this ability. In agreement with the increase in insulin secretion observed in Esp −/ − mice, Esp −/ − osteoblasts enhanced Insulin expression to a higher extent than WT osteoblasts (Figure 4F). Expression of Glucagon, a gene expressed in a different cell type in the islets, was not affected by osteoblasts (Figure 4F), further suggesting that Glucacon expression is not affected by Esp inactivation. We also co-cultured osteoblasts or fibroblasts with adipocytes. WT osteoblasts, but not fibroblasts, increased expression of Adiponectin and Esp −/ − osteoblasts were 2-fold more potent in this action; Adiponectin was the only tested adipokine whose expression was affected (Figure 4G). Control experiments using WT osteoblasts co-cultured with Esp −/ − islets or adipocytes showed the same increase in Insulin and Adiponectin expression than when using WT islets or adipocytes (Figure 4H). Study of Leptin and Adiponectin expression in osteoblasts and of osteoblast markers in adipocytes did not detect misexpression of any of these genes at the end of this experiment thus excluding a transdifferentiation event (Figure S4A-D).
To establish that the regulation of Insulin and Adiponectin expression by osteoblasts occurs via the release of secreted molecule(s) we first co-cultured osteoblasts with either islets or adipocytes using a filter preventing cell-cell contact. Second, islets and adipocytes were cultured in the presence of supernatant of osteoblast cultures. In both cases we observed a significant increase in Insulin and Adiponectin expression (Figures 4I and 4J).
The osteoblast secreted molecule osteocalcin favors β-cell proliferation, insulin secretion and sensitivity
In search of osteoblast-specific secreted molecule(s) regulating glucose homeostasis under the control of Esp we focused our attention on mice lacking osteocalcin (Ocn −/ − mice) since we noticed upon their generation that they were abnormally fat.
Ocn −/ − mice had higher blood glucose level and lower insulin serum level than WT mice (Figures 5A and 5B). Insulin secretion and sensitivity as well as glucose tolerance analyzed by GSIS, GTT, ITT and hyperinsulinemic euglycemic clamps were all decreased in Ocn −/ − mice, as was energy expenditure (Figures 5C-G and Table S7). Expression of insulin target genes was decreased in skeletal muscle and liver while Pepck expression was increased (Figure 5H). Islets size and number, β-cell mass, pancreas insulin content and insulin immunoreactivity were all markedly decreased in Ocn −/ − mice (Figure 5I). β-cell proliferation measured by Ki67 immunostaining was decreased 2-fold in Ocn −/ − pancreas (Figure 5I). There was also in Ocn −/ − mice an increase in fat mass, adipocytes number (WT, 93.2±10.7 x103 adipocytes/fat pad (n=5), Ocn −/ −, 125.6±10.6 x103 adipocytes/fat pad (n=3)) and serum triglyceride levels (Figures 5J and 5K). Adiponectin expression and serum levels were significantly lower in Ocn −/ − than in WT mice while expression of other adipokines was not affected (Figures 5L and 5M). Expression of molecular targets of adiponectin action was decreased in Ocn −/ − mice (Figure 5N). Ocn+/ − mice were indistinguishable from WT littermates (Figures 7A-G). All these abnormalities were observed in mice fed a normal diet. We verified that Osteocalcin was not expressed in islets or adipocytes (Figures S5A and S5B).
Figure 5. Osteocalcin regulatesβ-cell proliferation, insulin secretion and insulin sensitivity.
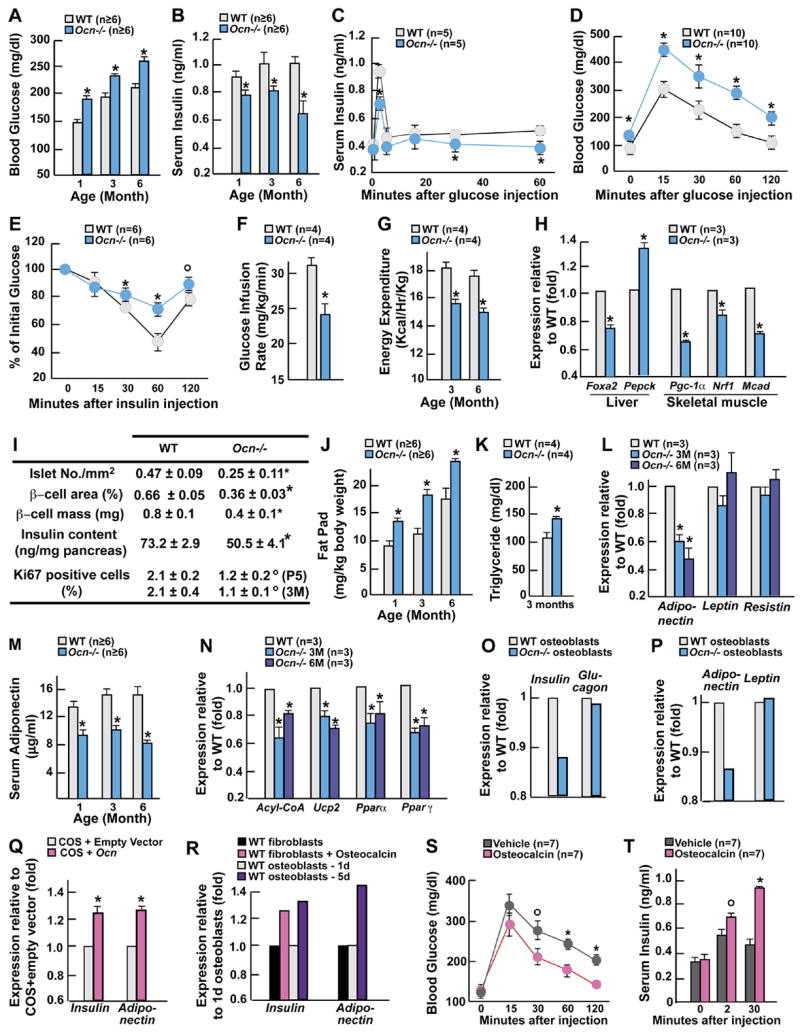
All experiments compare 3 month-old mice WT and Ocn −/ − mice unless otherwise indicated. (A) Blood glucose levels after random feeding. (B) Insulin levels. (C) GSIS test. (D) GTT. (E) ITT. (F) Glucose infusion rate during hyperinsulinemic-euglycemic clamp. (G) Energy expenditure. (H) Expression of insulin target genes by real time PCR. (I) Histomorphometric comparisons of islet numbers, islet size, β-cell mass, insulin content in pancreas and Ki67 immunoreactive cells in pancreatic islets. P5, 5 day-old pups; 3M, 3 month-old mice. (J) Fat pad mass (fat pad weight over body weight). (K) Serum triglyceride levels after an overnight fast. (L and M) serum levels (L) and gene expression (M) of adiponectin. (N) Expression of adiponectin target genes by real time PCR. (O) Expression of Insulin and Glucagon in WT pancreatic islets co-cultured with osteoblasts of indicated genotypes. (P) Expression of Adiponectin and Leptin in WT adipocytes co-cultured with osteoblasts of indicated genotypes. (Q) Expression of Insulin and Adiponectin in WT indicated cells cultured in presence of conditioned media from COS cells transfected with an Osteocalcin expression vector or its empty counterpart. (R) Expression of Insulin and Adiponectin in WT islets and adipocytes co-cultured with fibroblasts in presence of recombinant osteocalcin (3ng/ml) or vehicle, or with osteoblasts expressing (5d) or not (1d) Osteocalcin. (S and T) Dynamic of glucose (S) and insulin levels (T) in Ocn −/ − mice injected simultaneously with glucose and 20ng of recombinant osteocalcin or vehicle. Panels A, B, F-R: *p<0.05 vs WT (Student’s t test); panels C-E, S and T, °p 0.01 vs WT and *p 0.001 vs WT (ANOVA).
Figure 7. Esp −/− mice are a model of increased osteocalcin bioactivity.
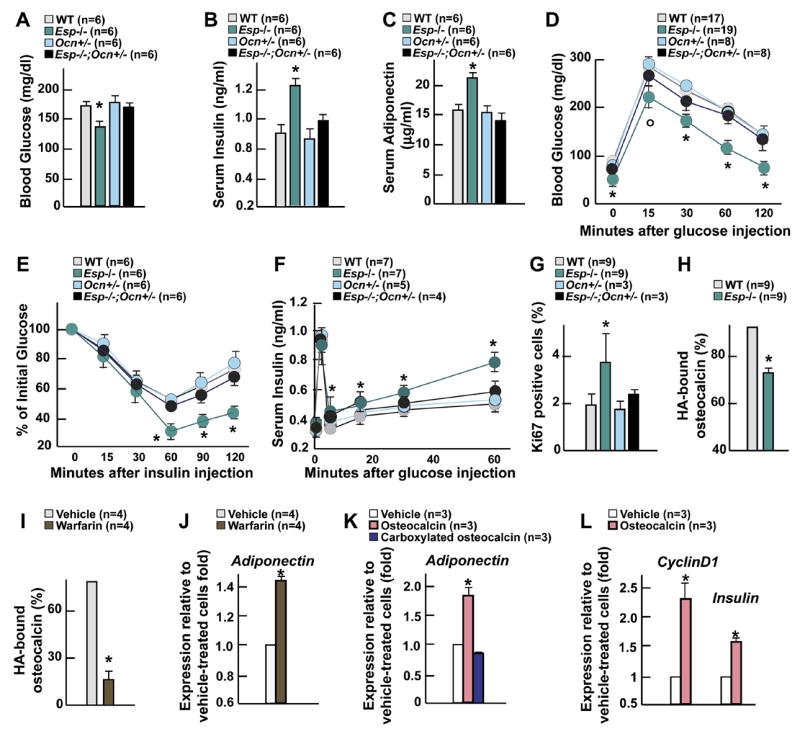
(A-G) Comparison between 6 week-old WT, Esp −/ −, Ocn+/ −, and Esp −/ −;Ocn+/ − mice. (A) Blood glucose levels. (B) Serum insulin levels. (C) Serum adiponectin levels. (D) GTT. (E) ITT. (F) GSIS test. (G) Quantification of the number of Ki67 immunoreactive cells in pancreatic islets. (H and I) Quantification of the percentage of osteocalcin bound to hydroxyapatite (HA) resin after a 15 min. incubation of serum of 1 month-old mice of indicated genotypes (H) or of conditioned medium from osteoblast cultures treated with warfarin or vehicle (I). (J) Expression of Adiponectin in WT adipocytes co-cultured with osteoblasts treated with warfarin or vehicle. (K) Expression of Adiponectin in WT adipocytes cultured in presence of vehicle or of 1ng/ml of commercially available carboxylated osteocalcin (Immunotopics) or bacterially produced uncarboxylated osteocalcin. (L) Expression of Insulin and CyclinD1 in WT islets cultured in presence of 3ng/ml of bacterially produced uncarboxylated osteocalcin or vehicle. Panels A-C and G-L: *p<0.05 vs WT (Student’s t test); panels D-F, °p<0.05 vs WT and *p 0.001 vs WT (ANOVA followed by post hoc analysis).
In co-culture assays, Ocn −/ − osteoblasts could not enhance expression of Insulin and Adiponectin (Figures 5O and 5P). Likewise, WT immature osteoblasts, that do not express Osteocalcin (Ducy et al., 2000b), could not induce Insulin or Adiponectin expression (Figure 5R). In contrast, forced expression of Osteocalcin in COS cells allowed these cells to enhance Insulin and Adiponectin expression (Figure 5Q). We also added bacterially produced recombinant osteocalcin (3ng/ml, Figure S6C) to a co-culture of WT fibroblasts and β-cells and observed an induction of Insulin expression, a phenomenon that fibroblasts could not trigger otherwise (Figure 5R).
To add further credence to these cell-based results we tested whether recombinant osteocalcin could affect glucose metabolism in vivo. To that end Ocn −/ − mice were subjected to a GTT in which half the mice received glucose alone while the other half received glucose and recombinant osteocalcin (20ng). Osteocalcin decreased significantly blood glucose levels at the 30, 60 and 120 minutes time points of this assay (Figure 5S). Osteocalcin also increased insulin secretion in Ocn −/ − mice in a GSIS test setting (Figure 5T). These experiments are consistent with the notion that osteocalcin is a molecule secreted by osteoblasts that can increase Insulin and Adiponectin expression.
Osteocalcin regulates insulin sensitivity through adiponectin
To determine whether insulin and adiponectin both contribute to the metabolic phenotype of the Ocn −/ − mice we asked two related questions. Does osteocalcin regulate Adiponectin expression independently of its action on insulin secretion? And if it is the case, does the decrease in Adiponectin expression noted in Ocn −/ − mice explain their decrease in insulin sensitivity? To address these questions we generated compound heterozygote Ocn+/ −;Adiponectin(Adipo)+/ − mice. Insulin sensitivity was markedly decreased in these mice while blood glucose levels, insulin serum levels and insulin secretion as determined by GSIS remained within the normal range (Figures 6A-D). Adiponectin serum levels were also significantly decreased in Ocn+/ −;Adipo+/ −compared to WT or single heterozygote mice (Figure 6E). These observations are consistent with the hypothesis that osteocalcin regulates insulin sensitivity independently of its effect on insulin secretion and that this regulation of insulin sensitivity occurs, at least in part, through adiponectin.
Figure 6. Osteocalcin regulates insulin sensitivity via adiponectin.
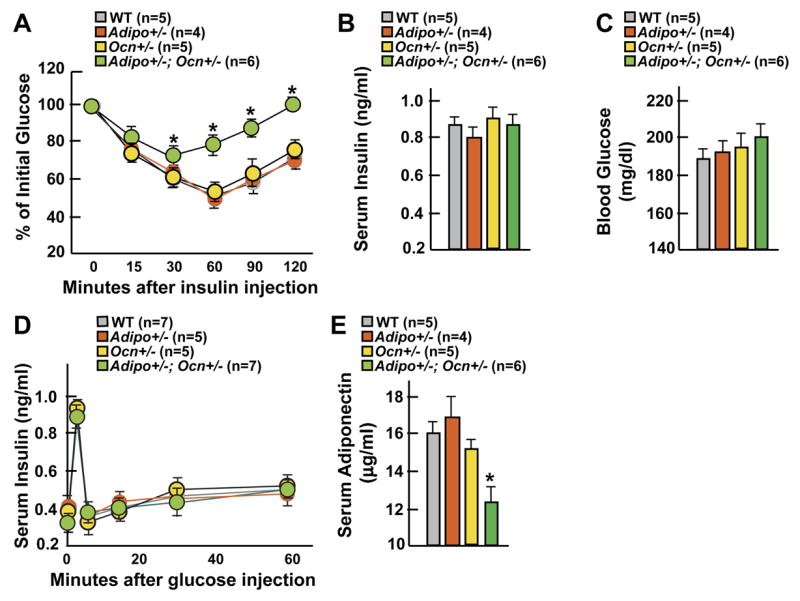
(A-E) Comparison between 6 week-old WT, Adiponectin+/ − (Adipo+/ −), Osteocalcin+/ − (Ocn+/ −) , and Ocn+/ −; Adipo+/ − mice. (A) ITT. (B) Insulin serum levels. (C) Blood glucose levels. (D) GSIS test. (E) Adiponectin serum levels. Panels A and D, *p 0.001 vs WT (ANOVA followed by post hoc analysis); panels B, C and E: *p<0.05 vs WT (Student’s t test).
OST-PTP regulates osteocalcin bioactivity
The metabolic phenotype of Ocn −/ − mice is the mirror image of the one observed in Esp −/ − mice suggesting that in the latter there is a gain of osteocalcin activity. If this is the case then metabolic abnormalities of Esp −/ − mice should be corrected by reducing Osteocalcin expression. Indeed, Esp −/ − mice lacking one allele of Osteocalcin showed a remarkable reversal of all their metabolic abnormalities (Figures 7A-F). In addition, Ki67 staining showed that β-cell proliferation was also reduced in these mutant mice (Figure 7G). Osteocalcin expression and serum levels were normal in Esp −/ − mice thus ruling out that OST-PTP regulates Osteocalcin expression (Figures S6A and S6B). These results provide genetic evidence that Esp and Osteocalcin lie in the same regulatory pathway and infer that Esp −/ − mice metabolic phenotype is caused by a gain-of-activity of this hormone.
Is the level of γ-carboxylation, the main post-translational modification of osteocalcin, different in WT and Esp −/ − mice? Carboxylated osteocalcin has a higher affinity for hydroxyapatite (HA) than uncarboxylated osteocalcin (Hauschka et al., 1989; Price, 1989). As shown in Figure 7H following a 15 min. incubation period 90% of osteocalcin present in the serum of WT mice was bound to HA whereas only 74% using serum from Esp −/ − mice. This experiment suggested that OST-PTP influences osteocalcin function by regulating its degree of γ-carboxylation and that it was the uncarboxylated form of osteocalcin that regulates glucose homeostasis. To test this latter hypothesis we performed two additional experiments. First, WT osteoblasts were treated with warfarin, an inhibitor of γ-carboxylation (Berkner, 2005) prior to and during co-culture assays. This treatment resulted in a marked decrease in the percentage of osteocalcin bound to HA (Figure 7J) and warfarin-treated osteoblasts induced Adiponectin expression to a significantly higher extent than vehicle-treated osteoblasts (Figure 7K). Second, we used carboxylated osteocalcin and bacterially produced, and therefore uncarboxylated, osteocalcin (Figure S6C) in cell-based assays. Only uncarboxylated osteocalcin could induce expression of Adiponectin in adipocytes, and of Insulin and CyclinD1, a molecular marker of cell proliferation, in islets (Kushner et al., 2005) (Figures 7K and 7L).
DISCUSSION
The results presented in this study uncover a novel pathway taking place in osteoblasts and resulting in the secretion of a hormone improving glucose homeostasis. Our results expand the spectrum of functions of skeleton, add further credence to the concept that bone and energy metabolisms regulate each other (Ducy et al., 2000a) and also suggest that the pathogenesis of some degenerative diseases of energy metabolism may be more complex than anticipated.
Mouse genetic to reveal novel cross talk between organs
Genetic manipulation in mice has been a powerful tool to reach a better understanding of vertebrate physiology. It has shown repeatedly for instance that organs not known to exert any regulatory influences were in fact fulfilling important endocrine functions. This is the case for instance of fat through the secretion of adipokines, of liver through bile acids and of vascular smooth muscle cells through synthesis of Emilin, a regulator of blood pressure (Spiegelman and Flier, 2001; Watanabe et al., 2006; Zacchigna et al., 2006). This genetic approach to whole animal physiology had unforeseen consequences. For instance, it linked together organs not thought previously to affect each other, and also showed that we do not know all the functions of most major organs. This is the case for skeleton.
Osteoblasts regulation ofβ-cell and adipocyte biology
That adipocytes regulate bone mass by acting on osteoblasts suggested that osteoblasts may affect adipocyte biology (Ducy et al., 2000a). Several observations reported here demonstrate that, indeed, osteoblasts secrete hormones influencing energy metabolism albeit in unanticipated yet important manners. For instance, Esp deletion in osteoblasts results in an increase in β-cell proliferation; given the current emphasis on the stimulation of β-cell proliferation this function of osteoblasts is of great potential from a therapeutic point of view. WT and even more so Esp −/ − osteoblasts can enhance Insulin expression within a short time in isolated islets indicating that this function occurs independently of their ability to promote cell proliferation. Paradoxically given their effect on insulin secretion, WT and even more so Esp −/ − osteoblasts enhance expression in adipocytes of Adiponectin, an adipokine whose overexpression enhances Insulin sensitivity (Otabe et al., 2007). This in turn explains why Esp −/ − mice have an increase in insulin sensitivity. Thus, all metabolic functions of the osteoblasts described here tend to improve glucose handling in vivo. It has been proposed that Esp is a pseudogene in human (Cousin et al., 2004). However, 2 close homologs of Esp are expressed in human osteoblasts suggesting that they may fulfill its function (data not shown).
Mediation of osteoblasts metabolic functions
What are the genetic and molecular bases of osteoblasts metabolic functions? Co-culture assays using filters separating each cell types demonstrated that it is through the release of one or several hormones that these functions are achieved. Mice lacking the osteoblast-specific secreted molecule osteocalcin develop a series of phenotypic abnormalities such as decreased insulin secretion, decreased β-cell proliferation, insulin resistance, increased adiposity and serum triglyceride levels that mirror those observed in Esp −/ − mice. Removing one allele of Osteocalcin corrects Esp −/ − mice metabolic abnormalities. In cell culture osteocalcin stimulates CyclinD1 expression in β-cells and replicates the effect of osteoblasts on Insulin and Adiponectin expression. Reintroducing purified osteocalcin in Osteocalcin −/ − mice corrects their glucose intolerance and enhances insulin secretion. When considered collectively these observations suggest that osteocalcin is a bone-derived hormone involved in the regulation of energy metabolism. This does not exclude the possibility that osteoblasts may secrete additional hormones regulating energy metabolism. Other cell types, such as adipocytes, also secrete multiple hormones regulating energy metabolism (Spiegelman and Flier, 2001).
The evidence provided suggests that uncarboxylated osteocalcin is mediating the metabolic functions of this hormone. The mechanisms whereby OST-PTP affects this post-translational modification remain unknown. It should be emphasized that most circulating hormones associate with a regulatory protein and are inactive. Thus, that only 10% of osteocalcin is bioactive is in line with this general rule in endocrinology (DeGroot and Jameson, 2001). Furthermore, at least one other γ-carboxylated protein, prothrombin, becomes active, as thrombin, when the carboxylated residues are removed (Furie and Furie, 1988).
Skeleton and regulation of energy metabolism
Our results add further credence to the concept that bone and energy metabolisms exert reciprocal regulations. Indeed, the resistance of Esp −/ − mice to obesity, glucose intolerance and insulin resistance together with the osteoblast-specific expression of osteocalcin identify the skeleton as a rheostat regulating glucose metabolism. They also raise the possibility that skeleton may contribute to the development of the metabolic syndrome since Esp −/ − mice do not develop obesity or diabetes. Clinical observations showing that serum osteocalcin levels are significantly lower in type 2 diabetic patients, and become normal following improvement of glycemic control, are also consistent with this idea (Rosato et al., 1998).
Lastly, our results raise teleological questions. For instance, why would a bone-specific hormone regulate energy metabolism, and what is the need for a hormone favoring βcell proliferation and insulin secretion? In both cases we can only speculate. For the first question, given the large surface it covered skeleton is an excellent site of hormone synthesis. Along this line it is possible that other hormones remain to be identified in osteoblasts. Alternatively, osteocalcin and possibly other hormones may have been recruited to skeleton through tinkering during evolution. As for the second question, it is conceivable that the pro-proliferation function of osteoblast-secreted hormones may have been required during evolution to maintain the size of the islets constant in periods of food deprivation.
EXPERIMENTAL PROCEDURES
Mice generation
Generation of Esp-nLacZ and Osteocalcin −/ − mice was previously reported (Dacquin et al., 2004; Ducy et al., 1996). To generate osteoblast-specific Esp-deficient (Esposb −/ −) mice a targeting vector harboring LoxP sites within introns 23 and 35 as well as a floxed neomycin resistance cassette was electroporated into ES cells. Targeted ES cells were injected in 129Sv/EV blastocysts to generate chimeric mice harboring the floxed allele (Espflox). Espflox/+ mice were crossed with α1(I)collagen-Cre mice to generate Esposb −/+ mice and their progeny was intercrossed to obtain Esposb −/ − mice. Adiponectin+/ − mice were generated according to a previously described strategy (Maeda et al., 2002). Transgenic α1(I)-Esp mice were generated by pronuclei injection of a construct fusing full length Esp cDNA with the osteoblast-specific fragment of the mouse type I collagen promoter (Dacquin et al., 2002). All procedures involving animals were approved by the IACUC and conform to the relevant regulatory standards.
Metabolic studies
For glucose tolerance test (GTT) glucose (2g/kg body weight (BW)) was injected intraperitoneally (IP) after an overnight fast and blood glucose was monitored using blood glucose strips and the Accu-Check glucometer (Roche) at indicated times. For glucose stimulated insulin secretion test (GSIS) glucose (3g/kg BW) was injected IP after an overnight fast; sera were collected from tails and insulin measured as described (Mauvais-Jarvis et al., 2000). For insulin tolerance test (ITT) mice were fasted for 6 hours, injected IP with insulin (0.2U/kg BW) and blood glucose levels were measured at indicated times as described (Mauvais-Jarvis et al., 2002). ITT data are presented as percentage of initial blood glucose concentration. Hyperinsulinemic-euglycemic clamps were performed at Penn State Mouse Metabolic Phenotyping Center. Briefly, Esp −/ −, Ocn −/ −, and WT littermates (n=4~8 for each group) were fasted overnight, and a 2-hr hyperinsulinemic (2.5mU/kg/min)-euglycemic clamps were performed following intravenous administration of [3-3H] glucose and 2-deoxy-D-[1-14C] glucose as previously described (Kim et al., 2004). Gold thioglucose (600mg/kg BW, USP) was injected IP, mice were sacrificed 3 months later for analysis. High fat diet (HFD) studies were performed as described (Elefteriou et al., 2006). Body weight was measured every 3 weeks after HFD; GTT and ITT were performed in mice fed a HFD for 6 weeks. Streptozotocin (150mg/kg single injection) was injected IP and blood glucose measured as described above every 2 days thereafter. After 8 days pancreas were isolated to measure insulin content as previously described (Mauvais-Jarvis et al., 2000). Food intake was measured using metabolic cages as the daily change of food weight. Energy expenditure was measured using metabolic cages connected to a calorimeter. Heat values (Kcal/Hr) were recorded over 2 days and reported to each mouse BW.
Laboratory measurements
Blood was collected by heart puncture of isoflurane-anesthetized mice in the fed and fasted states. Colorimetric assays were used to measure serum levels of free fatty acids and of triglycerides (Sigma). Serum levels of insulin and leptin (Crystal Chem Inc. kit), adiponectin, Resistin, Amylin, and PYY (Linco kit), C-peptide (Gentaur kit), IGF-1 (DSL kit), were quantified by ELISA. Human and mouse osteocalcin levels by IRMA (Immunotopics kit).
Mouse islets and primary cells isolation and culture
Islets were isolated on Histopaque gradient. In brief, after clamping the common bile duct at its entrance to the duodenum, 1mg/ml collagenaseP in M199 medium was injected into the duct. The swollen pancreas was surgically removed and incubated at 37°C for 17 min. Digested pancreata were dispersed by pipetting, rinsed twice, filtered through a Spectra-mesh (400 μm), resuspended in Histopaque and overlaid with M199 medium. Islets were collected following centrifugation at 1,700g for 20 min, washed twice with cold M199 medium, resuspended in M199/1%NCS or αMEM/1%FBS medium and cultured at 37°C in 5% CO2.
Primary adipocytes were isolated from epididymal fat pads by collagenase digestion. Briefly, minced adipose tissue was digested by 1mg/ml collagenaseP in KRP Buffer (20mM HEPES, 120mM NaCl, 6mM KCl, 1.2mM MgSO4, 1mM CaCl2, 0.6mM Na2HPO4, 0.4mM NaH2PO4, 2.5mM D-glucose, 2%BSA, pH 7.4) for 1 h at 37°C. The isolated cells were washed twice with KRP Buffer before being cultured in αMEM/1%FBS at 37°C in 5%CO2.
Primary osteoblasts were prepared from calvaria of 5 day-old pups as previously described (Ducy et al., 2000a) and were cultured in αMEM/10% FBS in the presence of 100μg/ml ascorbic acid and 5mM β-glycerophosphate for 5 days. Skin fibroblasts were isolated by collagenase digestion (0.5mg/ml) and were cultured in αMEM/10% FBS. Twenty-four hours before addition of primary islets (or adipocytes) osteoblasts (or fibroblasts) were placed in αMEM/1%FBS. For warfarin treatment ROS17/2.8 osteoblastic cells were maintained in DMEM/F12/10%FBS until being supplemented with 50μM warfarin or vehicle in DMEM/F12/1%FBS for 48h prior to co-culture with adipocytes. After 4h of co-culture either in the presence or absence of (1μm) culture inserts (Falcon) islets (or adipocytes) were collected for RNA isolation using TRIZOL.
Gene expression analyses
Real-time PCR were performed on DNAseI-treated total RNA converted to cDNA using primers were from SuperArray and the Taq SYBR Green Supermix with ROX on an MX3000 instrument; β-actin amplification was used as an internal reference for each sample except in figure 7L were Glucagon expression was used. X-gal staining was carried out as described (Dacquin et al., 2004).
Histology
Livers were cryoembedded, sectioned at 5μm and stained with Oil red O. Fat and pancreatic tissues were fixed in 10% neutral formalin, embedded in paraffin, sectioned at 5μm and sections were stained with hematoxylin and eosin (H&E). Immunohistochemistry was performed using rabbit anti-insulin (SantaCruz, 1:100) and mouse anti-Ki67 (Vector, 1:100) antibodies and ABC Elite kits (Vector). To evaluate cell sizes or numbers 5 to 10 sections (each 50μm apart) were analyzed using a 40x objective on a Leica microscope outfitted with a CCD camera (SONY) and using the Osteomeasure software. β-cell area represents the surface positive for insulin immunostaining divided by the total pancreatic surface. β-cell mass was calculated as β-cell area multiplied by pancreatic weight. At least 3 mice were analyzed per condition. Tibias anterior muscles were fixed in 4% PFA/2% glutaraldehyde/0.1 M sodium cacodylate pH 7.3, post-fixed in 1% osmium tetraoxide and embedded in epoxy resin (Epon). Ultrathin sections were stained in 4% aqueous Uranyl Acetate and 2 min in Reynolds’ Lead Citrate and examined with a JEOL 2000FX. Ten electron micrographs per mouse were digitized and the area of each clearly distinguishable mitochondrial was analyzed using ImageJ software. Fifteen to 25 individual mitochondria were measured in 4 mice of each genotype.
Osteocalcin production and hydroxyapatite (HA) binding assay
GST-osteocalcin fusion protein was bacterially produced and purified on glutathione beads according to standard procedures. Osteocalcin was then cleaved out from the GST subunit using thrombin and its purity was assessed by SDS-PAGE. Sera from 1 month-old mice or supernatant from warfarin-treated osteoblast cultures were added to HA slurry to achieve a final concentration of 25mg slurry/ml. After 15min HA beads were pelleted by centrifugation and HA-bound osteocalcin was eluted with 0.5M sodium phosphate buffer, pH 8.0. Osteocalcin present in eluates and initial samples was measured by IRMA. Values represent percentage of HA-bound osteocalcin over initial osteocalcin content.
Statistics
Results are given as means±standard deviations except in Figures 2B and 5F where means± standard errors of the mean are shown. Statistical analyses were performed using unpaired, two-tailed Student’s t or ANOVA tests followed by post hoc tests.
Supplementary Material
Acknowledgments
We thank X. Liu, S. Houlihan, L. Malynowsky and R. Wilson for their assistance, Drs. D. Accili, T. Kitamura and R. Leibel for suggestions and advices and Dr. R. Axel for critical reading of the manuscript. MDM is a scholar of the Fonds de la recherche en santé du Québec. This work was supported by a fellowship of JSPS (EH) and grants from NIH (GK), the American Diabetes Association and the Pennsylvania Department of health (JK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Berkner KL. The vitamin K-dependent carboxylase. Annu Rev Nutr. 2005;25:127–149. doi: 10.1146/annurev.nutr.25.050304.092713. [DOI] [PubMed] [Google Scholar]
- Brecher G, Laqueur GL, Cronkite EP, Edelman PM, Schwartz IL. The Brain Lesion of Goldthioglucose Obesity. J Exp Med. 1965;121:395–401. doi: 10.1084/jem.121.3.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousin W, Courseaux A, Ladoux A, Dani C, Peraldi P. Cloning of hOST-PTP: the only example of a protein-tyrosine-phosphatase the function of which has been lost between rodent and human. Biochem Biophys Res Commun. 2004;321:259–265. doi: 10.1016/j.bbrc.2004.06.137. [DOI] [PubMed] [Google Scholar]
- Dacquin R, Mee PJ, Kawaguchi J, Olmsted-Davis EA, Gallagher JA, Nichols J, Lee K, Karsenty G, Smith A. Knock-in of nuclear localised beta-galactosidase reveals that the tyrosine phosphatase Ptprv is specifically expressed in cells of the bone collar. Dev Dyn. 2004;229:826–834. doi: 10.1002/dvdy.20003. [DOI] [PubMed] [Google Scholar]
- Dacquin R, Starbuck M, Schinke T, Karsenty G. Mouse alpha1(I)-collagen promoter is the best known promoter to drive efficient Cre recombinase expression in osteoblast. Dev Dyn. 2002;224:245–251. doi: 10.1002/dvdy.10100. [DOI] [PubMed] [Google Scholar]
- DeGroot L, Jameson JL. Endocrinology. Philadelphia: Elsevier; 2001. [Google Scholar]
- Ducy P, Amling M, Takeda S, Priemel M, Schilling AF, Beil FT, Shen J, Vinson C, Rueger JM, Karsenty G. Leptin inhibits bone formation through a hypothalamic relay: a central control of bone mass. Cell. 2000a;100:197–207. doi: 10.1016/s0092-8674(00)81558-5. [DOI] [PubMed] [Google Scholar]
- Ducy P, Desbois C, Boyce B, Pinero G, Story B, Dunstan C, Smith E, Bonadio J, Goldstein S, Gundberg C, et al. Increased bone formation in osteocalcin-deficient mice. Nature. 1996;382:448–452. doi: 10.1038/382448a0. [DOI] [PubMed] [Google Scholar]
- Ducy P, Schinke T, karsenty G. The osteoblast: A sophisticated fibroblast under central surveillance. Science. 2000b;289:1501–1504. doi: 10.1126/science.289.5484.1501. [DOI] [PubMed] [Google Scholar]
- Elefteriou F, Benson MD, Sowa H, Starbuck M, Liu X, Ron D, Parada LF, Karsenty G. ATF4 mediation of NF1 functions in osteoblast reveals a nutritional basis for congenital skeletal dysplasiae. Cell Metab. 2006;4:441–451. doi: 10.1016/j.cmet.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. 1998;395:763–770. doi: 10.1038/27376. [DOI] [PubMed] [Google Scholar]
- Furie B, Furie BC. The molecular basis of blood coagulation. Cell. 1988;53:505–518. doi: 10.1016/0092-8674(88)90567-3. [DOI] [PubMed] [Google Scholar]
- Harada S, Rodan GA. Control of osteoblast function and regulation of bone mass. Nature. 2003;423:349–355. doi: 10.1038/nature01660. [DOI] [PubMed] [Google Scholar]
- Hauschka PV, Lian JB, Cole DE, Gundberg CM. Osteocalcin and matrix Gla protein: vitamin K-dependent proteins in bone. Physiol Rev. 1989;69:990–1047. doi: 10.1152/physrev.1989.69.3.990. [DOI] [PubMed] [Google Scholar]
- Kadowaki T, Yamauchi T. Adiponectin and adiponectin receptors. Endocr Rev. 2005;26:439–451. doi: 10.1210/er.2005-0005. [DOI] [PubMed] [Google Scholar]
- Karsenty G. Convergence between bone and energy homeostases: Leptin regulation of bone mass. Cell Metab. 2006;4:341–348. doi: 10.1016/j.cmet.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Higashimori T, Park SY, Choi H, Dong J, Kim YJ, Noh HL, Cho YR, Cline G, Kim YB, Kim JK. Differential effects of interleukin-6 and -10 on skeletal muscle and liver insulin action in vivo. Diabetes. 2004;53:1060–1067. doi: 10.2337/diabetes.53.4.1060. [DOI] [PubMed] [Google Scholar]
- Kushner JA, Ciemerych MA, Sicinska E, Wartschow LM, Teta M, Long SY, Sicinski P, White MF. Cyclins D2 and D1 are essential for postnatal pancreatic beta-cell growth. Mol Cell Biol. 2005;25:3752–3762. doi: 10.1128/MCB.25.9.3752-3762.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le May C, Chu K, Hu M, Ortega CS, Simpson ER, Korach KS, Tsai MJ, Mauvais-Jarvis F. Estrogens protect pancreatic beta-cells from apoptosis and prevent insulin-deficient diabetes mellitus in mice. Proc Natl Acad Sci U S A. 2006;103:9232–9237. doi: 10.1073/pnas.0602956103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda N, Shimomura I, Kishida K, Nishizawa H, Matsuda M, Nagaretani H, Furuyama N, Kondo H, Takahashi M, Arita Y, et al. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat Med. 2002;8:731–737. doi: 10.1038/nm724. [DOI] [PubMed] [Google Scholar]
- Maruyama H, Hisatomi A, Orci L, Grodsky GM, Unger RH. Insulin within islets is a physiologic glucagon release inhibitor. J Clin Invest. 1984;74:2296–2299. doi: 10.1172/JCI111658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauro LJ, Olmsted EA, Skrobacz BM, Mourey RJ, Davis AR, Dixon JE. Identification of a hormonally regulated protein tyrosine phosphatase associated with bone and testicular differentiation. J Biol Chem. 1994;269:30659–30667. [PubMed] [Google Scholar]
- Mauvais-Jarvis F, Ueki K, Fruman DA, Hirshman MF, Sakamoto K, Goodyear LJ, Iannacone M, Accili D, Cantley LC, Kahn CR. Reduced expression of the murine p85alpha subunit of phosphoinositide 3-kinase improves insulin signaling and ameliorates diabetes. J Clin Invest. 2002;109:141–149. doi: 10.1172/JCI13305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauvais-Jarvis F, Virkamaki A, Michael MD, Winnay JN, Zisman A, Kulkarni RN, Kahn CR. A model to explore the interaction between muscle insulin resistance and beta-cell dysfunction in the development of type 2 diabetes. Diabetes. 2000;49:2126–2134. doi: 10.2337/diabetes.49.12.2126. [DOI] [PubMed] [Google Scholar]
- Murshed M, Schinke T, McKee MD, Karsenty G. Extracellular matrix mineralization is regulated locally; different roles of two gla-containing proteins. J Cell Biol. 2004;165:625–630. doi: 10.1083/jcb.200402046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otabe S, Yuan X, Fukutani T, Wada N, Hashinaga T, Nakayama H, Hirota N, Kojima M, Yamada K. Overexpression of Human Adiponectin in Transgenic Mice Results in Suppression of Fat Accumulation and Prevention of Premature Death by High-Calorie Diet. Am J Physiol Endocrinol Metab. 2007 doi: 10.1152/ajpendo.00645.2006. [DOI] [PubMed] [Google Scholar]
- Price PA. Gla-containing proteins of bone. Connect Tissue Res. 1989;21:51–57. doi: 10.3109/03008208909049995. discussion 57–60. [DOI] [PubMed] [Google Scholar]
- Raju B, Cryer PE. Loss of the decrement in intraislet insulin plausibly explains loss of the glucagon response to hypoglycemia in insulin-deficient diabetes: documentation of the intraislet insulin hypothesis in humans. Diabetes. 2005;54:757–764. doi: 10.2337/diabetes.54.3.757. [DOI] [PubMed] [Google Scholar]
- Rosato MT, Schneider SH, Shapses SA. Bone turnover and insulin-like growth factor I levels increase after improved glycemic control in noninsulin-dependent diabetes mellitus. Calcif Tissue Int. 1998;63:107–111. doi: 10.1007/s002239900498. [DOI] [PubMed] [Google Scholar]
- Spiegelman BM, Flier JS. Obesity and the regulation of energy balance. Cell. 2001;104:531–543. doi: 10.1016/s0092-8674(01)00240-9. [DOI] [PubMed] [Google Scholar]
- Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, Patel HR, Ahima RS, Lazar MA. The hormone resistin links obesity to diabetes. Nature. 2001;409:307–312. doi: 10.1038/35053000. [DOI] [PubMed] [Google Scholar]
- Teitelbaum SL, Ross FP. Genetic regulation of osteoclast development and function. Nat Rev Genet. 2003;4:638–649. doi: 10.1038/nrg1122. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, Messaddeq N, Harney JW, Ezaki O, Kodama T, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439:484–489. doi: 10.1038/nature04330. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama-Kasaoka N, et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med. 2001;7:941–946. doi: 10.1038/90984. [DOI] [PubMed] [Google Scholar]
- Zacchigna L, Vecchione C, Notte A, Cordenonsi M, Dupont S, Maretto S, Cifelli G, Ferrari A, Maffei A, Fabbro C, et al. Emilin1 links TGF-beta maturation to blood pressure homeostasis. Cell. 2006;124:929–942. doi: 10.1016/j.cell.2005.12.035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.