

Abstract
Background and purpose:
Using the neonatal rat spinal cord in vitro, we investigated the action of glibenclamide, a drug possessing dual pharmacological effects, namely block of KATP channels and of the cystic fibrosis transmembrane conductance regulator (CFTR).
Experimental approach:
Intra- and extracellular recordings were performed on motoneurons and interneurons. RT-PCR and western immunoblotting were used to determine gene and protein expression.
Key results:
Glibenclamide (50 μM) facilitated mono- and polysynaptic reflexes, hyperpolarized motoneuron resting potential, increased action potential amplitude, decreased Renshaw cell-mediated recurrent inhibition, and increased network excitability by depressing GABA- and glycine-mediated transmission. The action of glibenclamide was mimicked by tolbutamide (500 μM) or the CFTR blocker diphenylamine-2,2-dicarboxylic acid (500 μM). The action of glibenclamide was independent from pharmacological inhibition of the Na+–K+ pump with strophanthidin (4 μM) and was associated with a negative shift in the extrapolated reversal potential for CI- dependent synaptic inhibition. On interneurons, intracellularly-applied 8-bromo-cAMP elicited an inward current and resistance decrease; effects antagonized by the selective CFTR antagonist, CFTRinh-172 (5 μM). RT-PCR and western immunoblotting indicated strong expression of the CFTR in neonatal rat spinal cord.
Conclusions and implications:
These data suggest the CFTR expressed in motoneurons and interneurons of the neonatal spinal cord is involved in the control of Cl- homeostasis and neuronal excitability. CFTR appeared to contribute to the relatively depolarized equilibrium potential for synaptic inhibition, an important process to control hyperexcitability and seizure-predisposition in neonates.
Keywords: synaptic inhibition, CFTR, GABA, glycine, chloride transporter, motoneuron
Introduction
Sulphonylurea drugs have been used to investigate the role of adenosine triphosphate (ATP)-sensitive K+ channels (KATP) in controlling the excitability of central neurons (Crepel et al., 1993; Mironov et al., 1998). In certain brain areas that possess intrinsic electrical rhythmicity, like the brainstem respiratory centres and associated nuclei, KATP channels pace the frequency of bursting and the duration of single bursts (Pierrefiche et al., 1996; Sharifullina et al., 2005). Because this mechanism relies on the cyclic intracellular consumption and neosynthesis of ATP, it represents a powerful process for relating neuronal electrical discharges to metabolic activity.
In the spinal cord, inherent rhythmicity can be readily observed in locomotor networks that express a stable pattern of regular electrical discharges (Kiehn and Butt, 2003). As we have previously shown that the ATP-dependent Na+–K+ pump is a major controller of spinal network bursting (Rozzo et al., 2002), we wondered whether periodic changes in intracellular ATP might control the activity of KATP conductances and thus limit neuronal excitability. One simple functional test for this possibility is to apply a KATP channel blocker, like glibenclamide (Bryan et al., 2004), and to monitor the resultant changes in network responses. Using this approach, we obtained unexpected results, which raised the issue of a novel mechanism for the control of spinal network excitability. Furthermore, our data contributed to the expansion of the current understanding of the mechanisms controlling gamma amino-butyric acid (GABA) and glycine inhibitory transmission.
Methods
Electrophysiology
Four- to eight-day-old (P4–P8) Wistar rats were killed by decapitation under urethane anaesthesia (0.2 ml intraperitoneally of a 10% wt vol−1 solution) and the lumbar region was isolated. This procedure is in accordance with the regulation of the Italian Animal Welfare Act and is approved by the local authority veterinary service. For motoneuron studies, the spinal cord was hemisected sagitally and superfused (7.5 ml min−1) with Krebs solution of the following composition (in mM): 113 NaCl, 4.5 KCl, 1 MgCl2·7H2O, 2 CaCl2, 1 NaH2PO4, 25 NaHCO3 and 11 glucose, gassed with 95% O2–5% CO2; pH 7.4 at room temperature.
Experimental design and analysis
To produce antidromic or orthodromic responses, ventral (VRs) or dorsal roots (DRs) were stimulated with suction electrodes (stimulus intensity=0.3–10 V; 0.05–0.2 ms). Orthodromic responses were recorded from the VR ipsilateral to the stimulated DR. As described previously (Marchetti et al., 2001), functionally identified L3–L5 motoneurons were recorded, under current clamp, with sharp electrodes filled with 2 M KMeSO4, whereas direct current (DC)-coupled VR responses were obtained with suction electrodes. We also compared motoneuronal depolarizations recorded as DC responses from VRs following bath-applied (25 s) N-methyl-D-aspartate (NMDA) (20 μM), α-amino-3-hydroxy-5-methyl-isoxazole-4-propionic acid (AMPA) (20 μM), GABA (500 μM) or glycine (500 μM) in control solution and in the presence of glibenclamide (50 μM). Recurrent (Renshaw cell-dependent) inhibitory postsynaptic potentials (ipsps) were recorded from motoneurons in control or in 3 mM kynurenic acid solution without shape and latency change (Marchetti et al., 2002). For single-electrode voltage-clamp (switching frequency=3–5 kHz; gain ⩾5 nA mV−1), electrodes were filled with 2 M Cs2SO4 plus 20 mM QX-314 (to minimize Na+-dependent spikes). To explore the Cl− reversal potential ( ) of motoneurons, inhibitory postsynaptic currents (ipscs) were evoked by antidromic stimulation (0.05 Hz) at various holding potentials in the presence of kynurenic acid and averaged.
) of motoneurons, inhibitory postsynaptic currents (ipscs) were evoked by antidromic stimulation (0.05 Hz) at various holding potentials in the presence of kynurenic acid and averaged.  was calculated by extrapolation from plots of synaptic charge versus holding potential. Although rectification of GABA-evoked responses from mouse spinal neurons in culture has been demonstrated at values positive to −50 mV (Bormann et al., 1987), other studies indicate it to be virtually absent from rat slice preparations (Gao and Ziskind-Conhaim, 1995). As our recordings were obtained with Cs+- and QX-314-filled microelectrodes, the contribution by voltage-activated conductances to the measured responses should have been minimized. Furthermore, to reduce to a minimum the artefacts due to the limited current passing ability of the sharp electrodes, we excluded from the analysis data obtained at membrane potentials more positive than −60 mV, thus making it feasible to extrapolate the
was calculated by extrapolation from plots of synaptic charge versus holding potential. Although rectification of GABA-evoked responses from mouse spinal neurons in culture has been demonstrated at values positive to −50 mV (Bormann et al., 1987), other studies indicate it to be virtually absent from rat slice preparations (Gao and Ziskind-Conhaim, 1995). As our recordings were obtained with Cs+- and QX-314-filled microelectrodes, the contribution by voltage-activated conductances to the measured responses should have been minimized. Furthermore, to reduce to a minimum the artefacts due to the limited current passing ability of the sharp electrodes, we excluded from the analysis data obtained at membrane potentials more positive than −60 mV, thus making it feasible to extrapolate the  value from current/voltage plots. DR stimulus intensity was graded to produce either monosynaptic responses (i.e., threshold (Th) intensity just enough to induce a detectable response; Marchetti et al., 2001) or polysynaptic responses when the stimulus was >1 × Th value. The monosynaptic nature of 1 × Th-evoked VR responses or of excitatory postsynaptic potentials (epsps) was confirmed by their short latency and stability to a 10 Hz stimulus train (Fulton and Walton, 1986).
value from current/voltage plots. DR stimulus intensity was graded to produce either monosynaptic responses (i.e., threshold (Th) intensity just enough to induce a detectable response; Marchetti et al., 2001) or polysynaptic responses when the stimulus was >1 × Th value. The monosynaptic nature of 1 × Th-evoked VR responses or of excitatory postsynaptic potentials (epsps) was confirmed by their short latency and stability to a 10 Hz stimulus train (Fulton and Walton, 1986).
Lamina X interneurons were recorded in coronal spinal slices (250 μm) superfused with Krebs solution. Visually identified interneurons (⩽15 μm soma diameter) were whole-cell patch clamped with a solution containing (mM) KCl 130, NaCl 5, MgCl2 2, CaCl2 0.1, 4-(2-hydroxyethyl)-1-piperazine-ethanesulphonic acid 10, ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′,-tetraacetic acid 5, ATP-Mg 2, GTP-Na 1 (pH 7.2 with KOH 280–300 mosmol l–1). Patch electrodes had 4–5 MΩ resistance. Cells were chosen for analysis if series resistance (RS) increases did not exceed 10% (no decrease was routinely observed). The junction potential was not taken into account in the data presented. Voltage and current pulse generation and data acquisition were performed with a PC, using pClamp 9.0 software. All recorded currents were filtered at 3 kHz and sampled at 10 kHz. Data were quantified as mean±s.e., with statistical significance assessed by use of Student's t-test.
Western blot
The protocol used for immunoblotting of lumbar spinal cord and lung samples was as described in Supplementary Figure 1. The anti-cystic fibrosis transmembrane conductance regulator (CFTR) antibody (Ab; 1:300) against amino-acid residues 1468–1480 of the human CFTR (accession no. P13569) was used with horseradish peroxidase-conjugated goat anti-rabbit IgG (1:2000; 1 h).
Reverse transcriptase–polymerase chain reaction (RT–PCR)
Total RNA was isolated from rat lumbar spinal cord and lung following the Invitrogen protocol based on Trizol extraction as indicated in Supplementary Figure 1.
Drugs and reagents
For electrophysiological experiments, drugs were applied via the superfusion system. We used bicuculline methiodide, diphenylamine-2,2′-dicarboxylic acid (DPC), strychnine hydrochloride, 4-hydroxyquinoline-2-carboxylic acid (kynurenic acid), GABA, cesium sulphate, lidocaine N-ethyl bromide (QX-314) and tolbutamide purchased from Sigma (Sigma-Aldrich, Milan, Italy). Potassium methyl sulphate was purchased from Fluka (Buchs, Switzerland). Glibenclamide was purchased from Tocris (Tocris Bioscience, Ellisville, MO, USA). CFTRinh-172 (3-((3-trifluoromethyl)phenyl)-5-((4-carboxyphenyl)methylene)-2-thioxo-4-thiazolidinone) was purchased from Calbiochem (Darmstadt, Germany). For Western blotting experiments, the protease inhibitors cocktail was purchased from Roche (F Hoffmann-La Roche Ltd, Basel, Switzerland). Bicinchoninic acid assay was purchased from Sigma-Aldrich. Nu-PAGE Novex (3–8%) was purchased from Invitrogen (Invitrogen SRL, San Giuliano Milanese, Italy). Anti-CFTR Ab was purchased from Alomone (Alomone Labs Ltd, Jerusalem, Israel).
Results
Network electrophysiology
We first examined the effects of glibenclamide on standard synaptic transmission evoked by electrical stimulation of a single DR. Although concentrations of glibenclamide in the submicromolar range had no effect (data not shown), the averaged monosynaptic and polysynaptic responses evoked by weak (1 × Th) or strong (>1 × Th) DR stimulation (Marchetti et al., 2001) were enhanced by 50 μM glibenclamide, as exemplified in Figure 1a (monosynaptic responses are depicted in the inset). The drug was applied for 20 min to ensure steady-state effects after tissue equilibration, as shown in Figure 1b in which changes in the polysynaptic response area and peak are plotted versus time (glibenclamide application indicated by horizontal bar). Figure 1c shows the facilitating effects of 50 μM glibenclamide on the size of the mono- and polysynaptic responses. Although the largest increment was observed for responses to weak stimuli, all responses were significantly increased, regardless of the stimulus intensity. Lower concentrations (1–10 μM) of glibenclamide were ineffective (5±10% change in response amplitude; P>0.05; n=4). A higher concentration of glibenclamide (100 μM) did not produce effects significantly larger than 50 μM on the mono- or polysynaptic responses (n=3; data not shown).
Figure 1.

Glibenclamide enhances spinal circuit excitability. (a) Monosynaptic (inset) and polysynaptic responses evoked by weak (1 × Th) or strong (4 × Th) DR stimulation, respectively, and recorded from a lumbar VR are increased after 20 min application of 50 μM glibenclamide. Responses are averages of 5–50 events. (b) Time course of glibenclamide evoked increase in area and peak amplitude of polysynaptic reflexes. Note that 20 min exposure was adequate for observing steady-state effects. (c) Histograms plotting VR reflex area (% of control) for different DR stimulus intensities in the presence of glibenclamide (50 μM). For each one of the stimulation intensities (n=8 preparations), a significant (P<0.05) increase in response area was observed. (d) VR depolarization amplitude (as % of control) evoked by 25 s bath application of NMDA (20 μM) or AMPA (20 μM) was significantly (*P<0.05) enhanced by glibenclamide, whereas GABA (500 μM)- or glycine (500 μM)-evoked depolarizations were significantly depressed (*P<0.05; n=5).
As the effects of glibenclamide implied changes in network activity, we next investigated spinal network output following changes in excitability owing to application of neurotransmitter agents (Kerkut and Bagust, 1995). Thus, we recorded VR DC depolarizations evoked by NMDA (20 μM), AMPA (20 μM), GABA (500 μM) and glycine (500 μM) on the same preparations. Figure 1d shows that depolarizations to NMDA or AMPA were significantly increased, whereas depolarizations to GABA or glycine were significantly depressed.
Motoneuron electrophysiology
The glibenclamide-dependent changes in synaptic transmission and the differential alterations in responses to excitatory and inhibitory amino-acid agonists suggested a complex modulation of transmitter receptor function. This was further explored by obtaining intracellular recordings from single motoneurons. In particular, while monitoring effects from the same cell, it was possible to compare changes in the epsp induced by DR stimulation with changes in the recurrent ipsp evoked by antidromic stimulation of the corresponding VR (Marchetti et al., 2002, 2005). This protocol enabled detailed information to be obtained on the properties of excitatory and inhibitory neurotransmission.
Figure 2a shows that, when applied to a single motoneuron, glibenclamide depressed the recurrent ipsp, whereas it enhanced the monosynaptic epsp elicited by low-Th DR stimulation. This contrasting condition is quantified in Figure 2b in which the large potentiation of the epsp was opposed by the significant fall in the ipsp amplitude.
Figure 2.

Contrasting effect of glibenclamide on motoneuron ipsp and epsp. (a) Intracellular recording from a single motoneuron showed opposite changes in recurrent ipsp (evoked by VR stimulation; first response) and monosynaptic epsp (second response). Value at the start of trace indicates membrane potential, whereas fast upward deflections are the stimulus artefacts. The epsp had a longer latency because of the longer distance between stimulation site and recorded cell. (b) Histograms depict average fall in recurrent ipsp amplitude with rise in mono epsp amplitude (*P<0.05; n=8).
In addition to such changes in synaptic physiology, we also investigated the ability of glibenclamide to alter some basic electrophysiological properties of motoneurons. To this end, we eliminated the contributions made by network-distributed glibenclamide-sensitive conductances, by inhibiting synaptic transmission with the pharmacological antagonists kynurenic acid (3 mM), bicuculline (20 μM) and strychnine (1 μM). Under these conditions, as shown in Figure 3a, glibenclamide slowly hyperpolarized the motoneuron membrane potential (on average by 3.0±0.9 mV; P<0.007; n=11) with increased input resistance (29±10%; P<0.003; n=11), as exemplified by the larger amplitude of the hyperpolarizing electrotonic potentials evoked by intracellular current pulses (Figure 3b). Analogous results (25±5% rise in input resistance) were obtained with tolbutamide (500 μM; n=13), a KATP channel blocker weaker than glibenclamide. The effects of either sulphonylurea could not be reversed after 30 min washout. Together with membrane hyperpolarization and resistance increase, glibenclamide intermittently inhibited action potential generation owing to membrane hyperpolarization, as indicated in Figure 3a.
Figure 3.

Motoneuron hyperpolarization together with increase in antidromic spike overshoot and input resistance following glibenclamide application. (a) Intracellular recording from single motoneuron (initial resting potential=−81 mV; large deflections are antidromic spikes) to which glibenclamide was applied as indicated by the horizontal bar. Note gradual membrane hyperpolarization to −83 mV plus increase in spike amplitude and occasional spike failure. (b) Faster speed traces show electrotonic potentials induced by −0.2 nA pulses to measure input resistance followed by VR stimuli to evoke antidromic spikes (truncated). Note the absence of spontaneous synaptic events owing to the use of Krebs solution containing synaptic receptor blockers (3 mM kynurenic acid, 2 μM strychnine, 20 μM bicuculline). Voltage calibration as in (a).
Figure 4a shows that glibenclamide increased the slope of the current/voltage curve throughout the range of membrane potential values tested. This effect of glibenclamide was associated with a potentiation of the peak amplitude of the spike (3.6±1.1 mV; P<0.009; n=11) even when the cell membrane potential was repolarized to the control level (Figure 4b). When plotted from the same cell, the spike overshoot showed a good correlation with the resistance increase (r=0.89), as depicted in Figure 4c. Although Table 1 shows that other spike parameters were unchanged, motoneuron firing was facilitated by glibenclamide as indicated by the greater number of spikes fired for the same current pulse (Figure 4d). These results are quantified in Figure 4e (n=6).
Figure 4.

Changes in motoneuron properties after application of glibenclamide. (a) Current/voltage plot shows increased slope, indicating rise in membrane resistance produced by glibenclamide. (b) Larger spike overshoot in the presence of glibenclamide persisted even after cell repolarization to control membrane potential, although the other spike parameters were unchanged. Value indicates membrane potential. (c) Strong correlation between rise in overshoot and resistance increase (n=8 cells). Data are expresses as % increment over control. (d) Superimposed records from the same motoneuron in control or glibenclamide solution. At the same level of membrane potential (−70 mV; 0.2 nA current injected during application of glibenclamide), in control solution the cell fired two action potentials in response to intracellular current injection, whereas it generated nine spikes in the presence of glibenclamide. (e) Plots showing that glibenclamide increased the firing rate (ordinate) of motoneurons during injection of 1 s current pulses of varying amplitude (abscissae). Each data point shows similar % increment versus control (P<0.05; n=6).
Table 1.
Effects of glibenclamide (50 μM) on electrical properties of motoneurons
| Control | Glibenclamide | P-value | |
|---|---|---|---|
| Vrest, mV | −72±1.4 | −75±1.3 | 0.007a |
| Rin, MΩ | 21±1.8 | 25±2.4 | 0.03a |
| Overshoot, mV | 15±1.2 | 19±1.1 | 0.009a |
| Spike rise time, ms | 1.3±0.1 | 1.3±0.1 | 0.98 |
| Inflection point, mV | −47±1.6 | −46±1.5 | 0.6 |
| Time to peak, ms | 3.3±0.2 | 3.5±0.2 | 0.1 |
| Stimulus threshold, V | 0.6±0.1 | 0.6±0.1 | 0.09 |
Abbreviations: Vrest=resting membrane potential; Rin=input resistance.
Data presented are means±s.e.; n=11.
Statistically significant difference. Inflection point is measured at the break between the initial segment and somatodendritic spike.
Collectively, these data show that glibenclamide evoked a gradual rise in motoneuron excitability and this was associated with membrane hyperpolarization and spike potentiation, which were not due to a block of spike-dependent K+ currents. These results prompted us to consider a constitutively active mechanism regulating background conductance.
The excitability of rat spinal neurons is powerfully controlled by the ATP-consuming electrogenic Na+–K+ pump (Ballerini et al., 1997; Rozzo et al., 2002). Therefore, the effects of glibenclamide might have been secondary to the Na+–K+ pump operation driven by larger intracellular availability of ATP. To test this possibility, the action of glibenclamide was studied after applying the potent Na+–K+ pump blocker, strophanthidin. Figure 5a shows that strophanthidin evoked motoneuron depolarization (Ballerini et al., 1997; Rozzo et al., 2002) with antidromic spike failure at −51 mV (see inset). This effect was reverted to hyperpolarization to −75 mV (with return of spike generation and emergence of synaptic activity) by subsequent application of glibenclamide. Figure 5b demonstrates that glibenclamide significantly hyperpolarized the membrane potential despite the block of the Na+–K+ pump.
Figure 5.

Glibenclamide affected motoneuron excitability even when the Na+–K+ pump was pharmacologically blocked. (a) Top trace shows intracellular recording from motoneuron (resting potential of −78 mV with full antidromic spike; see inset indicated with arrow) slowly depolarized by strophanthidin to −51 mV with associated spike failure (see inset) because of membrane depolarization. Application of glibenclamide (open bar) repolarized the membrane potential to −75 mV with return of antidromic spike and emergence of spontaneous synaptic events (see inset). (b) Histograms showing average change in membrane potential obtained after 20 min application of strophanthidin (with respect to resting membrane potential taken as 0 mV), and 40 min strophanthidin plus 20 min glibenclamide application (with respect to potential attained in the presence of strophanthidin). Both values significantly (P<0.05; n=6) differ from control. The larger effect of glibenclamide in the presence of strophanthidin might be explained by recovery of voltage-dependent channels from deactivation occurring at −50 mV.
One important effect of glibenclamide is to block CFTR (Schultz et al., 1999), a membrane protein involved in Cl− transport in a variety of epithelia cells, particularly in the lungs (Sheppard and Welsh, 1999; Nilius and Droogmans, 2003), whereas it is supposed to be minimally expressed in the central nervous system (Mulberg et al., 1994; Hincke et al., 1995). We considered the possibility that glibenclamide might have produced its electrophysiological actions by blocking CFTR rather than KATP channels. Thus, the effect of the CFTR inhibitor DPC (500 μM; Schultz et al., 1999) was investigated on motoneurons to see if it mimicked the actions of glibenclamide. Figure 6a shows that bath-applied DPC gradually hyperpolarized the motoneuron membrane potential, an effect accompanied by enhanced height of the antidromic spike (Figure 6b). On average, DPC significantly increased the membrane potential and resistance of motoneurons as indicated in Figure 6c (n=5). These data therefore demonstrate that DPC had an identical action to glibenclamide on spinal motoneurons.
Figure 6.

DPC changes resting and spike properties of motoneurons. (a) Continuous intracellular record from motoneuron shows gradual hyperpolarization of membrane potential from −69 to −73 mV following application of DPC. (b) Antidromic spike of the same motoneuron in control conditions and after applying DPC (each response is an average of >20): for the same level of membrane potential (obtained by injecting 0.3 nA depolarizing current) there is a large increment in the spike peak. (c) Histograms indicating the hyperpolarization in resting membrane potential (left) and the input resistance increase (right) observed after applying DPC (n=5). *P<0.05.
Cl−-dependent inhibition of motoneurons
The reduction in Renshaw cell-mediated ipsp by glibenclamide suggested that a change in Cl− permeability may contribute to its action. On immature neurons, experimental and theoretical studies show that GABAA receptors mediate membrane depolarization via increased Cl− permeability, with minimal HCO3 involvement (Cupello, 2003). Hence, it is possible to use responses evoked by synaptic inhibition to determine whether glibenclamide changes the  and, consequently, neuronal excitability. For this purpose, we recorded ipscs from motoneurons impaled with a sharp, voltage-clamping electrode to minimize perturbation of the intracellular milieu (Fisher and Nistri, 1993), even though sharp electrodes have inherently limited current passing ability and constrain the range of membrane potentials to be explored under voltage clamp conditions. Figure 7a shows an example of the change in the Renshaw cell-mediated ipsc of a motoneuron in which the synaptic response was depressed by glibenclamide together with a significant shift (on average −25±4 mV, n=5) in the extrapolated ipsc reversal potential (see Figure 7b and c) providing 48% reduction in the amplitude of the inhibitory synaptic Cl− current at −70 mV membrane potential. This value does not take into account the small hyperpolarization (on average −3 mV) owing to block by glibenclamide of the resting Cl− permeability. More direct tests of
and, consequently, neuronal excitability. For this purpose, we recorded ipscs from motoneurons impaled with a sharp, voltage-clamping electrode to minimize perturbation of the intracellular milieu (Fisher and Nistri, 1993), even though sharp electrodes have inherently limited current passing ability and constrain the range of membrane potentials to be explored under voltage clamp conditions. Figure 7a shows an example of the change in the Renshaw cell-mediated ipsc of a motoneuron in which the synaptic response was depressed by glibenclamide together with a significant shift (on average −25±4 mV, n=5) in the extrapolated ipsc reversal potential (see Figure 7b and c) providing 48% reduction in the amplitude of the inhibitory synaptic Cl− current at −70 mV membrane potential. This value does not take into account the small hyperpolarization (on average −3 mV) owing to block by glibenclamide of the resting Cl− permeability. More direct tests of 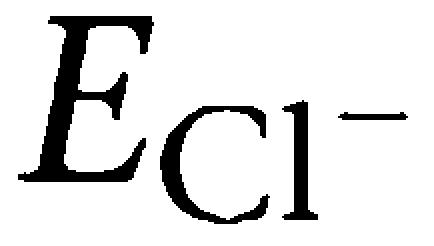 were complicated by the fact that blind patching of motoneurons in the isolated spinal cord was difficult because of rapid electrode block. On the other hand, the viability of motoneurons in thin slices was usually poor owing to damage of these large cells.
were complicated by the fact that blind patching of motoneurons in the isolated spinal cord was difficult because of rapid electrode block. On the other hand, the viability of motoneurons in thin slices was usually poor owing to damage of these large cells.
Figure 7.

Glibenclamide-evoked change in reversal potential of recurrent ipscs of motoneurons. (a) Examples of recurrent ipscs (average from 150 responses) at three levels of holding potential under voltage clamp conditions. Note depression of ipsc with glibenclamide application (25 min). (b and c) Plots show charge transfer/voltage relations for control (open circles) or glibenclamide (filled circles) responses with histograms indicating the negative shift of the extrapolated  value (*P<0.05; n=6).
value (*P<0.05; n=6).
The effects of glibenclamide on interneurons
The next question that arose was whether the observed changes induced by glibenclamide are cell specific and thus restricted to motoneurons. Lamina X interneurons also make monosynaptic contacts on motoneurons and are mainly responsible for the synaptic inhibition that produces the rhythmic alternation of motor discharges at the intrasegmental level (Birinyi et al., 2003; Kiehn, 2006). On such interneurons (patch-clamped in thin slice preparations at −60 mV), we activated CFTR by applying, via the intracellular solution, the stable cyclic adenosine monophosphate (cAMP) analogue 8-bromo-cAMP (400 μM; Schultz et al., 1999). As shown by the example in Figure 8, shortly after patch breakthrough, the interneuron generated a slow inward current, which stabilized at −127 pA. On average, the 8-bromo-cAMP-induced inward current was −126±25 pA (n=6) and was associated with a decrease in the input resistance, to 57±12% of the value recorded immediately after breakthrough. The recently discovered inhibitor of CFTR, the thiazolidinone CFTRinh-172 (5 μM; Thiagarajah et al., 2004), was bath-applied and reversed the effect of 8-bromo-cAMP to generate an outward current (87 pA). On average, with respect to the effects observed in the presence of 8-bromo-cAMP, the outward current evoked by CFTRinh-172 was 66±7 pA (n=5) with a resistance increase (125±11%); both effects were reversed after a 10 min washout. In the absence of 8-bromo-cAMP, neither glibenclamide nor CFTRinh-172 changed the baseline current or input resistance (n=13).
Figure 8.

The CFTR inhibitor CFTRinh-172 reversed the action of 8-bromo-cAMP on interneurons. Voltage clamp trace (under whole-cell patch clamp configuration at −60 mV) of spinal interneuron recorded with an electrode containing 8-bromo-cAMP. Note that shortly after establishing the whole-cell configuration (arrow), there is a slowly developing inward current that is antagonized by CFTRinh-172.
CFTR in the neonatal rat spinal cord
We next investigated the presence of CFTR in the neonatal rat spinal cord. In these experiments, mRNA was extracted from rat lung (a tissue enriched in CFTR expression; Jentsch et al., 2002) and lumbar spinal cord. The lumbar spinal cord contained a 170 nucleotide-long band (lanes 1 and 2) corresponding to the expected size of the CFTR amplified sequence (Figure 9a). As a control for the detection of DNA, total mRNA was incubated with actin-specific oligonucleotides (lanes 3 and 4 for spinal cord and lung, respectively). The relative amount of CFTR mRNA in the spinal cord compared to that in the lung was estimated (after normalization with actin) to be 77±3% (n=3).
Figure 9.

Presence of CFTR in spinal cord tissue. (a) RT–PCR analysis for the presence of CFTR mRNA in rat lumbar spinal cord (lane 1) and lung (lane 2). The predicted fragment of 171 bp for CFTR mRNA is present in both lanes. Lanes 3 and 4 show mRNA signals for actin from spinal cord and lung, respectively. bp=base pair. (b) Expression of CFTR protein in membrane extracts from rat lumbar spinal cord (lane 2) and lung (lane 3); NIH 3T3 membranes were used as negative control (lane 1). Peptide-inhibited samples are shown in lanes 4 and 5 for spinal and lung membranes, respectively.
Immunoblots of membrane fractions of rat lumbar spinal cord and lungs showed the presence of CFTR (Figure 9b, lanes 2 and 3) with bands at 180 and 200 kDa, corresponding to unglycosilated and glycosilated CFTR (Mulberg et al., 1994; Hincke et al., 1995). Preincubation of the CFTR Ab with its immunogenic antigen eliminated the expression of CFTR, by Western blot, from both the lung and spinal membrane fractions (Figure 9b, lanes 4 and 5). Likewise, no signal was observed with NIH 3T3 cell samples (Figure 9b, lane 1). Densitometric estimates of CFTR expression indicated that the heavier band from lungs was 41±5% (n=11) of that of spinal tissue, whereas the lighter band was less in the spinal cord (10±2%) than in the lungs.
Immunocytochemical staining of rat spinal cord sections with the fluorescent-conjugated anti-CFTR Ab revealed diffuse positivity, particularly intense in the nucleus of neurons (Supplementary Figure 2), although the nucleus of lung epithelial cells was surprisingly unstained. A BLAST search indicated that the rat epitope recognized by the CFTR Ab had 80% sequence homology with the nuclear receptor corepressor 2 (accession no. NP035554), a nuclear factor (molecular weight of 270 kDa; van der Laan et al., 2005) that regulates transcription of development-related genes in the rat brain (Martinez de Arrieta et al., 2000). This observation precluded the use of this Ab to map CFTR immunoreactivity in the spinal cord, although it did not invalidate the Western blot data, as confirmed by the largely different molecular weight and the tests with cell membranes that do not contain this corepressor (Figure 9b).
Discussion
The principal finding of the present report is the demonstration of CFTR expression in the neonatal rat spinal cord. Pharmacological block of CFTR led to profound changes in network excitability chiefly owing to alterations in the  , and thus in the ability of GABA (and glycine) to operate as an inhibitory neurotransmitter.
, and thus in the ability of GABA (and glycine) to operate as an inhibitory neurotransmitter.
Changes in network activity evoked by glibenclamide
Although increased network excitability and larger epsps in the presence of glibenclamide might have been compatible with pharmacological block of KATP channels, the observed motoneuron hyperpolarization together with resistance increase was clearly inconsistent with this interpretation and were probably the cause of the larger spike amplitude (without further changes in spike width or decay). Furthermore, the fall in recurrent ipsp amplitude indicated that it was unlikely that the overall increase in synaptic transmission was merely caused by larger neuronal input resistance. It was also difficult to understand why K+ channel inhibition was involved in the differential effects of glutamate, GABA or glycine receptor activation. Hence, it seemed useful to determine whether a unifying hypothesis, that was not dependent on inhibition of the K+ conductance, could account for the complex effects of glibenclamide. An increase in the mono- (and poly-) synaptic reflexes, as well as in AMPA- and NMDA-mediated responses, together with the decreased effects of GABA or glycine (and ipsps) could all be explained by assuming a reduction in a neuronal conductance operating at resting potential and also controlling the action of GABA and glycine. A Cl− conductance appeared to be a suitable candidate for this role and this led us to consider the involvement of CFTR, because the effects of glibenclamide were observed at concentrations that typically block this transporter rather than KATP channels (Schultz et al., 1999). The observed effects of glibenclamide reached a maximum at 50 μM; doubling the drug concentration did not intensify the effect. Because 10 μM concentrations were sub-threshold, these observations indicate a very steep dose–response relationship for glibenclamide.
CFTR as a Cl− regulator
The CFTR, an integral membrane protein controlling Cl− secretion, is expressed in epithelial cells of intestine, airways, secretory glands, bile ducts and epididymis (Sheppard and Welsh, 1999; Nilius and Droogmans, 2003), and is involved in the regulation of fluid flow and ion concentrations (Sheppard and Welsh, 1999; Nilius and Droogmans, 2003). Genetic mutations of CFTR underlie cystic fibrosis, a disease characterized by disrupted epithelial Cl− transport (Lewis et al., 2003). Glibenclamide is an effective inhibitor of CFTR function (Schultz et al., 1999). Nevertheless, because CFTR is thought to be virtually absent from the central nervous system (Hincke et al., 1995), glibenclamide has been widely used to study the role of KATP channels in the brain (Mironov et al., 1998; Yamada and Inagaki, 2002). The present study is, therefore, the first to suggest, based on electrophysiological and molecular biology data, that CFTR is present in the spinal cord of the newborn rat and that its activity contributes to the resting membrane potential and Cl−-dependent inhibition mediated by GABA and glycine.
How could CFTR contribute to  in newborn spinal neurons?
in newborn spinal neurons?
In the adult brain, Cl− is continuously extruded by the membrane transporter KCC2 to generate a rather negative equilibrium potential for this anion (Ben Ari, 2002; Payne et al., 2003). In the neonatal brain, transporter immaturity is responsible for the impaired Cl−-dependent inhibition mediated by GABA and, thus, for the lower convulsion threshold (Ben Ari, 2002; Rivera et al., 2005). Conversely, in the newborn spinal cord, the expression of KCC2 is large enough (although not as high as in the adult) to make unclear the origin of intracellular Cl− accumulation underlying the GABA or glycine-evoked depolarization (Hubner et al., 2001; Ueno et al., 2002; Stein et al., 2004). Because only Cl− has been shown to be responsible for the effects of GABA or glycine on spinal motoneurons (Hamill et al., 1983; Wu et al., 1992; Cupello, 2003), and the  value for passively distributed Cl− would be very negative (−87 mV), other mechanisms for the depolarized
value for passively distributed Cl− would be very negative (−87 mV), other mechanisms for the depolarized  should be sought.
should be sought.
On motoneurons recorded with sharp electrodes, glibenclamide significantly attenuated inhibitory synaptic currents generated by Renshaw cells. The limited current passing ability of sharp electrodes did not allow us to observe the value of  directly. It was therefore obtained by extrapolation, on the assumption that the inhibitory synaptic current displayed minimal rectification (Gao and Ziskind-Conhaim, 1995). Even if the precise value of
directly. It was therefore obtained by extrapolation, on the assumption that the inhibitory synaptic current displayed minimal rectification (Gao and Ziskind-Conhaim, 1995). Even if the precise value of 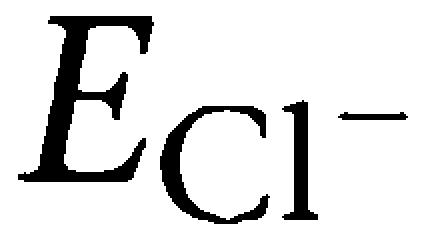 could not be obtained, the negative shift in the current/voltage plot plus the increased membrane resistance suggested that glibenclamide-mediated modulation of a Cl− conductance contributed to the leak conductance of motoneurons. As glibenclamide is able to block CFTR, we suggest that this membrane protein is responsible for the observed changes induced by glibenclamide. Our proposal is supported by the analogous results obtained on motoneurons with tolbutamide and DPC, both considered to be CFTR inhibitors (Schultz et al., 1999), and, on interneurons, with CFTRinh-172 (Thiagarajah et al., 2004). On interneurons, the use of the whole-cell patch clamp technique precluded the normal operation of CFTR, as previously reported, and made it necessary to use 8-bromo-cAMP as a stable activator of CFTR (Sheppard and Welsh, 1992; Bachmann et al., 2000; Sheppard et al., 2004; Wright et al., 2004). In the presence of 8-bromo-cAMP, the inward current and resistance decrease recorded from interneurons were readily antagonized by CFTRinh-172, indicating that CFTR expression and function were not confined to motoneurons.
could not be obtained, the negative shift in the current/voltage plot plus the increased membrane resistance suggested that glibenclamide-mediated modulation of a Cl− conductance contributed to the leak conductance of motoneurons. As glibenclamide is able to block CFTR, we suggest that this membrane protein is responsible for the observed changes induced by glibenclamide. Our proposal is supported by the analogous results obtained on motoneurons with tolbutamide and DPC, both considered to be CFTR inhibitors (Schultz et al., 1999), and, on interneurons, with CFTRinh-172 (Thiagarajah et al., 2004). On interneurons, the use of the whole-cell patch clamp technique precluded the normal operation of CFTR, as previously reported, and made it necessary to use 8-bromo-cAMP as a stable activator of CFTR (Sheppard and Welsh, 1992; Bachmann et al., 2000; Sheppard et al., 2004; Wright et al., 2004). In the presence of 8-bromo-cAMP, the inward current and resistance decrease recorded from interneurons were readily antagonized by CFTRinh-172, indicating that CFTR expression and function were not confined to motoneurons.
The basic properties of CFTR differ from those of other Cl− channels or transporters (Sheppard and Welsh, 1999). In fact, although CFTR can form Cl− channels when expressed in heterologous cells, it has recently been suggested that, in physiological conditions, it mainly acts as a regulator of other membrane channels (Nilius and Droogmans, 2003); this process is also expected to occur in the neonatal rat spinal cord.
The most obvious target for this effect of CFTR is the NKCC1 transporter; this accumulates Cl− in neonatal brain neurons to produce a value of  responsible for the depolarization induced by GABA or glycine (Dzhala et al., 2005). Our hypothesis is that CFTR stimulated NKCCl to increase intracellular Cl−, as previously observed in cell culture (Adam et al., 2005). This accords with the notion that CFTR enhances the functional expression of NKCC1 (Shumaker and Soleimani, 1999). It should be emphasized that the present study is the first to apply such a hypothesis to the neonatal spinal cord: future studies are needed to demonstrate conclusively that this process occurs in the newborn rat spinal cord and to identify its precise cellular location, once more suitable Abs become available.
responsible for the depolarization induced by GABA or glycine (Dzhala et al., 2005). Our hypothesis is that CFTR stimulated NKCCl to increase intracellular Cl−, as previously observed in cell culture (Adam et al., 2005). This accords with the notion that CFTR enhances the functional expression of NKCC1 (Shumaker and Soleimani, 1999). It should be emphasized that the present study is the first to apply such a hypothesis to the neonatal spinal cord: future studies are needed to demonstrate conclusively that this process occurs in the newborn rat spinal cord and to identify its precise cellular location, once more suitable Abs become available.
Despite the preliminary nature of the present study, our electrophysiological results following application of glibenclamide enabled us to outline a functional role of CFTR in controlling  and excitability of neonatal spinal neurons.
and excitability of neonatal spinal neurons.
CFTR transcripts and membrane expression in spinal tissue
The presence of CFTR in the brain is purportedly limited to the hypothalamus (Mulberg et al., 1994; Weyler et al., 1999). Nevertheless, our demonstration of the presence of CFTR mRNA and protein expression in neonatal spinal cord tissue provides a compelling reason to consider that glibenclamide (and tolbutamide, DPC or CFTRinh-172) induces its effects by inhibiting CFTR. As the mature glycosylated form of CFTR (Cheng et al., 1990) was strongly expressed in membrane extracts, it could be an important regulator of Cl− transport in the neonatal spinal cord. The immunocytochemical identification of rat CFTR-positive neurons was fraught with technical problems owing to Ab crossreactivity and did not allowed us to identify the cell types immunoreactive for CFTR.
Could the effects of glibenclamide be attributed to KATP channel block?
The two prime candidates for the formation of functional KATP channels in the brain are the Kir6.2 subunit of the K+ channel family and the SUR1 element of the glibenclamide-sensitive sulphonylurea receptor (Aguilar-Bryan and Bryan, 1999). Although Kir6.2 subunits are moderately expressed in the adult rat spinal cord (Thomzig et al., 2005), their expression in the developing spinal cord has not been measured. Nevertheless, Kir6.2−/− mice do not exhibit abnormal behavioural phenotypes (Yamada et al., 2001), indicating a minimal contribution of this subunit to motor or sensory functions. This accords with the finding that most brain KATP channels operate only in ATP-depleted metabolic states such as hypoxia and are usually closed during resting conditions (Yamada and Inagaki, 2002). For these reasons, it is unlikely that the effects of glibenclamide on neonatal neurons at rest or during synaptic transmission involved inhibition of KATP channels, because all the electrophysiological parameters (spike amplitude, input resistance) indicated adequate cell oxygenation and made it unlikely that KATP activation was involved. The present data thus suggest that care is needed when interpreting effects of sulphonylurea drugs applied to neurons in the central nervous system, as they are unlikely to be simply due to block of KATP channels.
Functional implications
In the neonatal rat spinal cord, although GABA and glycine depolarize neurons because their  is positive to resting potential, these transmitters exert strong synaptic inhibition (Marchetti et al., 2002). The present data suggest that the CFTR contributed to the depolarized
is positive to resting potential, these transmitters exert strong synaptic inhibition (Marchetti et al., 2002). The present data suggest that the CFTR contributed to the depolarized  by promoting Cl− intracellular transport. Membrane depolarization evoked by GABA (or glycine)-opened Cl− channels may inhibit neurons through voltage-dependent inactivation of the Na+ (and Ca2+) conductances needed for firing action potentials, in analogy to GABA-mediated presynaptic inhibition in the adult spinal cord (Rudomin, 2002). Blocking these events with CFTR inhibitors would then hyperpolarize neurons and increase their excitability via voltage-dependent reactivation of Na+ (and Ca2+) channels plus enhanced input resistance.
by promoting Cl− intracellular transport. Membrane depolarization evoked by GABA (or glycine)-opened Cl− channels may inhibit neurons through voltage-dependent inactivation of the Na+ (and Ca2+) conductances needed for firing action potentials, in analogy to GABA-mediated presynaptic inhibition in the adult spinal cord (Rudomin, 2002). Blocking these events with CFTR inhibitors would then hyperpolarize neurons and increase their excitability via voltage-dependent reactivation of Na+ (and Ca2+) channels plus enhanced input resistance.
External data objects
Acknowledgments
This work was supported by grants from MIUR (FIRB) and FVG. We thank Manuela Simonetti for her help with the PCR experiments and to Dr Elsa Fabbretti and Marianna D'Arco for their support with Western blotting.
Abbreviations
- Ab
antibody
- DR
dorsal root
- epsp
excitatory postsynaptic potential
- ipsc
inhibitory postsynaptic current
- ipsp
inhibitory postsynaptic potential
- Th
threshold
- VR
ventral root
Conflict of interest
The authors state no conflict of interest.
Supplementary Information accompanies the paper on British Journal of Pharmacology website (http://www.nature.com/bjp)
References
- Adam G, Ousingsawat J, Schreiber R, Kunzelmann K. Increase in intracellular Cl− concentration by cAMP- and Ca2+-dependent stimulation of M1 collecting duct cells. Pflugers Arch. 2005;449:470–478. doi: 10.1007/s00424-004-1356-4. [DOI] [PubMed] [Google Scholar]
- Aguilar-Bryan L, Bryan J. Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocr Rev. 1999;20:101–135. doi: 10.1210/edrv.20.2.0361. [DOI] [PubMed] [Google Scholar]
- Bachmann A, Russ U, Waldegger S, Quast U. Potent stimulation and inhibition of the CFTR Cl− current by phloxine B. Br J Pharmacol. 2000;131:433–440. doi: 10.1038/sj.bjp.0703600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballerini L, Bracci E, Nistri A. Pharmacological block of the electrogenic sodium pump disrupts rhythmic bursting induced by strychnine and bicuculline in the neonatal rat spinal cord. J Neurophysiol. 1997;77:17–23. doi: 10.1152/jn.1997.77.1.17. [DOI] [PubMed] [Google Scholar]
- Ben Ari Y. Excitatory actions of GABA during development: the nature of the nurture. Nat Rev Neurosci. 2002;3:728–739. doi: 10.1038/nrn920. [DOI] [PubMed] [Google Scholar]
- Birinyi A, Viszokay K, Weber I, Kiehn O, Antal M. Synaptic targets of commissural interneurons in the lumbar spinal cord of neonatal rats. J Comp Neurol. 2003;461:429–440. doi: 10.1002/cne.10696. [DOI] [PubMed] [Google Scholar]
- Bormann J, Hamill OP, Sakmann B. Mechanism of anion permeation through channels gated by glycine and gamma-aminobutyric acid in mouse cultured spinal neurones. J Physiol. 1987;385:243–286. doi: 10.1113/jphysiol.1987.sp016493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan J, Vila-Carriles WH, Zhao G, Babenko AP, Aguilar-Bryan L. Toward linking structure with function in ATP-sensitive K+ channels. Diabetes. 2004;53:S104–S112. doi: 10.2337/diabetes.53.suppl_3.s104. [DOI] [PubMed] [Google Scholar]
- Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, et al. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990;63:827–834. doi: 10.1016/0092-8674(90)90148-8. [DOI] [PubMed] [Google Scholar]
- Crepel V, Rovira C, Ben-Ari Y. The K+ channel opener diazoxide enhances glutamatergic currents and reduces GABAergic currents in hippocampal neurons. J Neurophysiol. 1993;69:494–503. doi: 10.1152/jn.1993.69.2.494. [DOI] [PubMed] [Google Scholar]
- Cupello A. Neuronal transmembrane chloride electrochemical gradient: a key player in GABAA receptor activation physiological effect. Amino Acids. 2003;24:335–346. doi: 10.1007/s00726-002-0350-4. [DOI] [PubMed] [Google Scholar]
- Dzhala VI, Talos DM, Sdrulla DA, Brumback AC, Mathews GC, Benke TA, et al. NKCC1 transporter facilitates seizures in the developing brain. Nat Med. 2005;11:1205–1213. doi: 10.1038/nm1301. [DOI] [PubMed] [Google Scholar]
- Fisher ND, Nistri A. A study of the barium-sensitive and -insensitive components of the action of thyrotropin-releasing hormone on lumbar motoneurons of the rat isolated spinal cord. Eur J Neurosci. 1993;5:1360–1369. doi: 10.1111/j.1460-9568.1993.tb00922.x. [DOI] [PubMed] [Google Scholar]
- Fulton BP, Walton K. Electrophysiological properties of neonatal rat motoneurones studied in vitro. J Physiol. 1986;370:651–678. doi: 10.1113/jphysiol.1986.sp015956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao B, Ziskind-Conhaim L. Development of glycine- and GABA-gated currents in rat spinal cord. J Neurophysiol. 1995;74:113–121. doi: 10.1152/jn.1995.74.1.113. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Bormann J, Sakmann B. Activation of multiple-conductance state chloride channels in spinal neurones by glycine and GABA. Nature. 1983;305:805–808. doi: 10.1038/305805a0. [DOI] [PubMed] [Google Scholar]
- Hincke MT, Nairn AC, Staines WA. Cystic fibrosis transmembrane conductance regulator is found within brain ventricular epithelium and choroid plexus. J Neurochem. 1995;64:1662–1668. doi: 10.1046/j.1471-4159.1995.64041662.x. [DOI] [PubMed] [Google Scholar]
- Hubner CA, Stein V, Hermans-Borgmeyer I, Meyer T, Ballanyi K, Jentsch TJ. Disruption of KCC2 reveals an essential role of K+–Cl− cotransport already in early synaptic inhibition. Neuron. 2001;30:515–524. doi: 10.1016/s0896-6273(01)00297-5. [DOI] [PubMed] [Google Scholar]
- Jentsch TJ, Stein V, Weinreich F, Zdebik AA. Molecular structure and physiological function of chloride channels. Physiol Rev. 2002;82:503–568. doi: 10.1152/physrev.00029.2001. [DOI] [PubMed] [Google Scholar]
- Kerkut GA, Bagust J. The isolated mammalian spinal cord. Prog Neurobiol. 1995;46:1–48. doi: 10.1016/0301-0082(94)00055-m. [DOI] [PubMed] [Google Scholar]
- Kiehn O. Locomotor circuits in the mammalian spinal cord. Annu Rev Neurosci. 2006;29:279–306. doi: 10.1146/annurev.neuro.29.051605.112910. [DOI] [PubMed] [Google Scholar]
- Kiehn O, Butt SJ. Physiological, anatomical and genetic identification of CPG neurons in the developing mammalian spinal cord. Prog Neurobiol. 2003;70:347–361. doi: 10.1016/s0301-0082(03)00091-1. [DOI] [PubMed] [Google Scholar]
- Lewis MJ, Lewis EH, III, Amos JA, Tsongalis GJ. Cystic fibrosis. Am J Clin Pathol. 2003;120:S3–S13. doi: 10.1309/DUUVFR11W595V7FE. [DOI] [PubMed] [Google Scholar]
- Marchetti C, Beato M, Nistri A. Evidence for increased extracellular K+ as an important mechanism for dorsal root induced alternating rhythmic activity in the neonatal rat spinal cord in vitro. Neurosci Lett. 2001;304:77–80. doi: 10.1016/s0304-3940(01)01777-3. [DOI] [PubMed] [Google Scholar]
- Marchetti C, Pagnotta S, Donato R, Nistri A. Inhibition of spinal or hypoglossal motoneurons of the newborn rat by glycine or GABA. Eur J Neurosci. 2002;15:975–983. doi: 10.1046/j.1460-9568.2002.01927.x. [DOI] [PubMed] [Google Scholar]
- Marchetti C, Taccola G, Nistri A. Activation of group I metabotropic glutamate receptors depresses recurrent inhibition of motoneurons in the neonatal rat spinal cord in vitro. Exp Brain Res. 2005;164:406–410. doi: 10.1007/s00221-005-2368-9. [DOI] [PubMed] [Google Scholar]
- Martinez de Arrieta C, Koibuchi N, Chin WW. Coactivator and corepressor gene expression in rat cerebellum during postnatal development and the effect of altered thyroid status. Endocrinology. 2000;141:1693–1698. doi: 10.1210/endo.141.5.7467. [DOI] [PubMed] [Google Scholar]
- Mironov SL, Langohr K, Haller M, Richter DW. Hypoxia activates ATP-dependent potassium channels in inspiratory neurones of neonatal mice. J Physiol. 1998;509:755–766. doi: 10.1111/j.1469-7793.1998.755bm.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulberg AE, Wiedner EB, Bao X, Marshall J, Jefferson DM, Altschuler SM. Cystic fibrosis transmembrane conductance regulator protein expression in brain. Neuroreport. 1994;5:1684–1688. doi: 10.1097/00001756-199408150-00035. [DOI] [PubMed] [Google Scholar]
- Nilius B, Droogmans G. Amazing chloride channels: an overview. Acta Physiol Scand. 2003;177:119–147. doi: 10.1046/j.1365-201X.2003.01060.x. [DOI] [PubMed] [Google Scholar]
- Payne JA, Rivera C, Voipio J, Kaila K. Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 2003;26:199–206. doi: 10.1016/S0166-2236(03)00068-7. [DOI] [PubMed] [Google Scholar]
- Pierrefiche O, Bischoff AM, Richter DW. ATP-sensitive K+ channels are functional in expiratory neurones of normoxic cats. J Physiol. 1996;494:399–409. doi: 10.1113/jphysiol.1996.sp021501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Kaila K. Two developmental switches in GABAergic signalling: the K+–Cl− cotransporter KCC2 and carbonic anhydrase CAVII. J Physiol. 2005;562:27–36. doi: 10.1113/jphysiol.2004.077495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozzo A, Ballerini L, Abbate G, Nistri A. Experimental and modeling studies of novel bursts induced by blocking Na+ pump and synaptic inhibition in the rat spinal cord. J Neurophysiol. 2002;88:676–691. doi: 10.1152/jn.2002.88.2.676. [DOI] [PubMed] [Google Scholar]
- Rudomin P. Selectivity of the central control of sensory information in the mammalian spinal cord. Adv Exp Med Biol. 2002;508:157–170. doi: 10.1007/978-1-4615-0713-0_19. [DOI] [PubMed] [Google Scholar]
- Schultz BD, Singh AK, Devor DC, Bridges RJ. Pharmacology of CFTR chloride channel activity. Physiol Rev. 1999;79:S109–S144. doi: 10.1152/physrev.1999.79.1.S109. [DOI] [PubMed] [Google Scholar]
- Sharifullina E, Ostroumov K, Nistri A. Metabotropic glutamate receptor activity induces a novel oscillatory pattern in neonatal rat hypoglossal motoneurones. J Physiol. 2005;563:139–159. doi: 10.1113/jphysiol.2004.079509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard DN, Gray MA, Gong X, Sohma Y, Kogan I, Benos DJ, et al. The patch-clamp and planar lipid bilayer techniques: powerful and versatile tools to investigate the CFTR Cl− channel. J Cyst Fibros Suppl. 2004;2:101–108. doi: 10.1016/j.jcf.2004.05.046. [DOI] [PubMed] [Google Scholar]
- Sheppard DN, Welsh MJ. Effect of ATP-sensitive K+ channel regulators on cystic fibrosis transmembrane conductance regulator chloride currents. J Gen Physiol. 1992;100:573–591. doi: 10.1085/jgp.100.4.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard DN, Welsh MJ. Structure and function of the CFTR chloride channel. Physiol Rev. 1999;79:S23–S45. doi: 10.1152/physrev.1999.79.1.S23. [DOI] [PubMed] [Google Scholar]
- Shumaker H, Soleimani M. CFTR upregulates the expression of the basolateral Na+–K+–2Cl− cotransporter in cultured pancreatic duct cells. Am J Physiol. 1999;277:C1100–C1110. doi: 10.1152/ajpcell.1999.277.6.C1100. [DOI] [PubMed] [Google Scholar]
- Stein V, Hermans-Borgmeyer I, Jentsch TJ, Hubner CA. Expression of the K+Cl− cotransporter KCC2 parallels neuronal maturation and the emergence of low intracellular chloride. J Comp Neurol. 2004;468:57–64. doi: 10.1002/cne.10983. [DOI] [PubMed] [Google Scholar]
- Thiagarajah JR, Song Y, Haggie PM, Verkman AS. A small molecule CFTR inhibitor produces cystic fibrosis-like submucosal gland fluid secretions in normal airways. FASEB J. 2004;18:875–877. doi: 10.1096/fj.03-1248fje. [DOI] [PubMed] [Google Scholar]
- Thomzig A, Laube G, Pruss H, Veh RW. Pore-forming subunits of K-ATP channels, Kir6.1 and Kir6.2, display prominent differences in regional and cellular distribution in the rat brain. J Comp Neurol. 2005;484:313–330. doi: 10.1002/cne.20469. [DOI] [PubMed] [Google Scholar]
- Ueno T, Okabe A, Akaike N, Fukuda A, Nabekura J. Diversity of neuron-specific K+–Cl− cotransporter expression and inhibitory postsynaptic potential depression in rat motoneurons. J Biol Chem. 2002;277:4945–4950. doi: 10.1074/jbc.M109439200. [DOI] [PubMed] [Google Scholar]
- van der Laan S, Lachize SB, Schouten TG, Vreugdenhil E, de Kloet ER, Meijer OC. Neuroanatomical distribution and colocalisation of nuclear receptor corepressor (N-CoR) and silencing mediator of retinoid and thyroid receptors (SMRT) in rat brain. Brain Res. 2005;1059:113–121. doi: 10.1016/j.brainres.2005.08.011. [DOI] [PubMed] [Google Scholar]
- Weyler RT, Yurko-Mauro KA, Rubenstein R, Kollen WJ, Reenstra W, Altschuler SM, et al. CFTR is functionally active in GnRH-expressing GT1–7 hypothalamic neurons. Am J Physiol. 1999;277:C563–C571. doi: 10.1152/ajpcell.1999.277.3.C563. [DOI] [PubMed] [Google Scholar]
- Wright AM, Gong X, Verdon B, Linsdell P, Mehta A, Riordan JR, et al. Novel regulation of cystic fibrosis transmembrane conductance regulator (CFTR) channel gating by external chloride. J Biol Chem. 2004;279:41658–41663. doi: 10.1074/jbc.M405517200. [DOI] [PubMed] [Google Scholar]
- Wu WL, Ziskind-Conhaim L, Sweet MA. Early development of glycine- and GABA-mediated synapses in rat spinal cord. J Neurosci. 1992;12:3935–3945. doi: 10.1523/JNEUROSCI.12-10-03935.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Inagaki N. ATP-sensitive K+ channels in the brain: sensors of hypoxic conditions. News Physiol Sci. 2002;17:127–130. doi: 10.1152/nips.01384.2001. [DOI] [PubMed] [Google Scholar]
- Yamada K, Ji JJ, Yuan H, Miki T, Sato S, Horimoto N, et al. Protective role of ATP-sensitive potassium channels in hypoxia-induced generalized seizure. Science. 2001;292:1543–1546. doi: 10.1126/science.1059829. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.