

Abstract
Background and purpose:
We report the development of a very efficient cell-based high throughput screening (HTS) method, which utilizes a novel bio-sensor that selectively detects apoptosis based on the fluorescence resonance energy transfer (FRET) technique.
Experimental approach:
We generated a stable HeLa cell line expressing a FRET-based bio-sensor protein. When cells undergo apoptosis, they activate a protease called ‘caspase-3'. Activation of this enzyme will cleave our sensor protein and cause its fluorescence emission to shift from a wavelength of 535 nm (green) to 486 nm (blue). A decrease in the green/blue emission ratio thus gives a direct indication of apoptosis. The sensor cells are grown in 96-well plates. After addition of different chemical compounds to each well, a fluorescence profile can be measured at various time-points using a fluorescent plate reader. Compounds that can trigger apoptosis are potential candidates as anti-cancer drugs.
Key results:
This novel cell-based HTS method is highly effective in identifying anti-cancer compounds. It was very sensitive in detecting apoptosis induced by various known anti-cancer drugs. Further, this system detects apoptosis, but not necrosis, and is thus more useful than the conventional cell viability assays, such as those using MTT. Finally, we used this system to screen compounds, isolated from two plants used in Chinese medicine, and identified several effective compounds for inducing apoptosis.
Conclusions and Implications:
This FRET-based HTS method is a powerful tool for identifying anti-cancer compounds and can serve as a highly efficient platform for drug discovery.
Keywords: drug discovery, HTS, FRET, green fluorescent protein, apoptosis, caspase, anticancer drugs, herbal medicine, podophyllotoxin, tanshinone
Introduction
Currently, one of the major cancer treatments is chemotherapy. Most chemotherapy drugs such as vincristine, paclitaxel, and etoposide (ET), exert their anticancer effects by arresting cells at different stages of the cell cycle and then promoting them into apoptotic cell death (Gidding et al., 1999; Sleiman and Stewart, 2000; Das et al., 2001). As these anticancer drugs cannot discriminate between cancer cells and noncancer cells, many normal cells are also killed during the process of chemotherapy. This nonspecific cytotoxicity damages the patient's immune system and generates many side effects including neutropenia, vomiting, hair loss, peripheral neurotoxicity, etc. (Perry et al., 1976; Einzig et al., 1991). Developing new anticancer drugs with a higher potency and specificity against cancer cells has therefore become an important goal in biomedical research.
The first step to drug discovery is to screen for drug leads from a large pool of compounds. At present, there is a lack of efficient cell-based methods that can rapidly screen compounds by detecting apoptotic cell death (Watanabe et al., 2002). For example, the commonly used in vitro caspase activity assay utilizes cell extracts from a large population of cells. Furthermore, although the TdT-mediated dUTP nick end labeling method is only applicable to fixed cells but not living cells, assays involving Annexin V require the use of a fluorescent microscope or fluorescence-activated cell sorting analysis. These methods are time-, labor- and cost-consuming and therefore, will be difficult for a high throughput drug screening. In this paper, we report the development of a high throughput screening method for detecting caspase-3 activation during the process of apoptosis in living cells grown in a 96-well plate. Caspase-3 is a cysteine protease that is specifically activated by apoptotic inducers. When caspase-3 is activated, it can cleave a number of cellular proteins at specific cleavage sites (Nicholson, 1999; Wolf and Green, 1999), inactivating these vital proteins and consequently leading to an inevitable cell death. As activation of caspase-3 is the most critical step in apoptosis, it serves a dual purpose – as a decision point for cells committing to apoptotic cell death and as a landmark event for the detection of apoptosis (Samali et al., 1999).
Our high throughput screen uses a stable cell line (HeLa-C3) expressing a caspase biosensor, which is an engineered protein consisting of three parts: a donor cyan fluorescent protein (CFP), a 16-amino-acid peptide linker containing the caspase-3 cleavage site and an acceptor yellow fluorescent protein (YFP) (Luo et al., 2001). The method of detection is based on the effects of fluorescence resonance energy transfer (FRET) (Cubitt et al., 1995; Heim and Tsien, 1996). The basic principle of this method is that in the absence of activated caspase-3 (i.e. under normal growth conditions), the fluorescent emission energy of the excited donor fluorescent protein CFP can be transferred to the acceptor molecule YFP, leading to the emission of yellow fluorescent light. When caspase-3 is activated during apoptosis, the activated protease will cleave the biosensor (CFP-linker-YFP) at the linker peptide containing the recognition sequence for caspase-3, DEVD. Cleavage of the sensor protein abolishes the effect of FRET, resulting in a change in the emission profile of the sensor protein because fluorescent light is emitted from CFP instead of YFP. Thus, by measuring the fluorescence emission ratio between YFP and CFP, we can detect the amount of activation of caspase-3 in living cells during the process of apoptosis (Luo et al., 2001).
This paper reports the detailed design of this drug screening system. To demonstrate its capabilities, we have tested this system using a number of known anticancer drugs, including camptothecin, ET, hydroxyurea, paclitaxel and vincristine. Furthermore, we have used this screening system to detect the potential anticancer activity of a group of purified herbal compounds, including six compounds from Salvia miltiorrhiza Bunge (Danshen) and six compounds from Podophyllum emodi Wall.
Methods
Generation of stable HeLa-C3 cell line
The mammalian construct comprised the CFP-DEVD-YFP fusion gene encoding the sensor C3 protein was previously generated and characterized by our group (Luo et al., 2001). The plasmid DNA of sensor C3 was introduced into HeLa cells by electroporation (Luo and Chang, 2004). Transfected cells were seeded in a 90 mm culture dish, and 48 h after electroporation (cell confluence reached 60%), cells were trypsinized and seeded into two six-well plates in a selection medium containing 400 μg ml−1 geneticin (Invitrogen, Carlsbad, CA, USA). The concentration of geneticin was increased to 500 μg ml−1 5 days later and the culture medium was changed every 3–4 days with a fresh medium containing 500 μg ml−1of geneticin. About 2 weeks after the transfection, GFP-positive clones were first selected under a fluorescence microscope, then individually trypsinized and seeded into 90 mm plates. GFP-positive clones with high purity and strong fluorescence were further selected by this ‘local trypsinization' and ‘subculture' method. Finally, three clones were chosen for further examination of cell morphology and apoptotic assay. Clone no. 7 displayed a normal cell morphology and cell division pattern, similar to that of HeLa cells, and could undergo apoptotic cell death when treated with standard apoptotic inducers such as ultraviolet (UV)-irradiation and tumor necrosis factor-α (TNF-α). We have compared the kinetics of caspase-3 activation between HeLa and HeLa-C3 cells during UV-induced apoptosis using an in vitro caspase-3 activity assay. Results show that the two cell lines have a similar kinetics, which suggests that introduction of sensor C3 did not induce significant changes in caspase-3 activation in HeLa cells (data not shown). This stable cell line was designated as HeLa-C3 and used for developing the caspase-sensor-based high throughput screening platform in this paper.
Cell culture and drug addition
HeLa-C3 cells were cultured in minimum essential medium (MEM) (Invitrogen) containing 10% fetal bovine serum (Invitrogen), 100 U ml−1 penicillin, 100 μg ml−1 streptomycin (Invitrogen) and 500 μg ml−1 geneticin (Invitrogen), in a 5% CO2 humidity incubator at 37°C. In order to test the effects of different compounds or drugs, 10 000 HeLa-C3 cells were seeded into each well of a 96-well plate. After an overnight culture, medium was removed and 100 μl of fresh medium containing compounds to be tested were added. At various time points, the plate was read by a Perkin–Elmer Victor3 plate reader with excitation at 440±10 nm (Perkin–Elmer, Wellesley, MA, USA). The emission was measured at 486+8 nm for CFP and 535+8 nm for YFP.
Caspase-3 activity assay
Cells treated with different stimuli were collected and lysed in lysis buffer (20 mM Tris-HCl, pH 7.5, 10 mM NaH2PO4, 10 mM Na2HPO4, 130 mM NaCl, 10 mM sodium pyrophosphate, and 1% Triton X-100) containing protease inhibitors of aprotinin (10 μg ml−1), leupeptin (10 μg ml−1), pepstatin A (10 μg ml−1), phenylmethylsulfonyl fluoride (10 μg ml−1) and ethylenediaminetetraacetic acid (EDTA) (2 mM). The cell lysates were centrifuged at 16 000 g for 15 min at 4°C. Caspase-3 activity assay was performed in a 96-well plate. For each assay, 5 μl of the cell lysates with about 30–40 μg of proteins was first mixed with 93 μl of the assay buffer (20 mM (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), 10 mM KCl, 2.5 mM MgCl2, 1 mM EDTA, 1 mM ethylene glycol bis(β-aminoethylether)-N,N,N',N',-tetraacetic acid, and 1 mM dithiothreitol, pH 7.5), plus 2 μl of the caspase-3 substrate (Ac-DEVD-AMC) (1 μg μl−1) (BD Biosciences, San Diego, CA, USA). Then the mixture was incubated at 37°C in darkness for 1 h. The fluorescent intensity was measured using a Perkin–Elmer Victor3 plate reader with excitation wavelength at 355 nm and emission wavelength at 460 nm. Caspase-3 activity was presented in units of ‘activity per μg protein'. The protein concentration was measured by the Bradford method employing a Bio-Rad protein assay (Bio-Rad, Hercules, CA, USA).
DNA laddering assay
DNA laddering assay was performed according to Herrmann's method with some modifications (Herrmann et al., 1994). A cell pellet collected from a 90 mm Petri dish was resuspended in 100 μl of lysis buffer (1% NP-40, 50 mM Tris-HCl, 20 mM EDTA, pH 7.5) containing 1 mg ml−1 protease K and 1 mg ml−1 RNase A and incubated at 37oC for 1 h. Proteins in the cell lysate were removed by phenol–chloroform (USB, Cleveland, OH, USA) extraction. Genomic DNA was precipitated by adding ethanol to a final concentration of 70% to the supernatant and was then dissolved in Tris–EDTA buffer and resolved on a 2% agarose gel.
Cell viability measurement using (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (MTT) assay
The cell proliferation Kit I (MTT) from Roche (Basel, Switzerland) was used to measure cell viability. Cells were grown in a 96-well plate overnight and treated with different conditions. At each designated time point, 10 μl of the labeling solution was added into each well of the 96-well plate. After 2 h incubation in the CO2 incubator, 100 μl of the solubilization solution was added to dissolve purple crystals which are the products of MTT substrates. After 22 h incubation, the absorbance was measured by a Perkin–Elmer Victor3 plate reader at 595 nm.
Western blot analysis
Fifty micrograms (μg) of protein from the same cell lysate as used for caspase-3 activity assay was separated by 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred onto a Hybond ECL nitrocellulose membrane (Amersham, Piscataway, NJ, USA). After blocking with 5% non-fat milk, the membranes were incubated with different antibodies at the following conditions: overnight at 4°C with either rabbit-anti-caspase-3 (#9662) (Cell Signaling, Danvers, MA, USA) at 1:2000 dilution, or mouse-anti-β-tubulin antibody (Calbiochem) at 1:1000 dilution, and 1 h incubation at room temperature for mouse-anti-GFP (sc-9996) (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) at 1:2000 dilution. The membranes were then incubated with a horseradish peroxidase-conjugated mouse (Bio-Rad, 170–6516) or rabbit (Bio-Rad, 170–6515) secondary antibody at 1:5000 dilutions for 1 h and developed using the ECLWestern-blotting analysis system (Amersham).
Imaging techniques
HeLa-C3 cells were grown overnight on a 25-mm round coverslip mounted onto a 35-mm Petri dish with an observation window and treated with various compounds for different amounts of time. The fluorescent images of living cells were obtained using an inverted fluorescence microscope (Zeiss Axiovert 35) equipped with a FRET filter set. The cells were excited using an excitation filter (440±10 nm) plus a diachronic mirror of 455 nm. The emission images of YFP (535±12.5 nm) and CFP (480±15 nm) were recorded using a computer-controlled SPOT CCD camera (Diagnostic Instruments, Sterling Heights, MI, USA). The digital fluorescent images were then processed using MetaMorph software (Universal Imaging Co., Downingtown, PA, USA). The phase images of the cells were obtained using the same inverted fluorescence microscope.
Y/C emission ratio measurement and calculation
Cells cultured in 96-well plates were treated with various compounds. At the indicated time points, the emission signals of YFP (535±8 nm) and CFP (486±8 nm) were respectively measured by a Perkin–Elmer Victor3 plate reader using an excitation light of 440±10 nm. The background fluorescence was then measured from the wells containing only medium. After subtracting the background fluorescence from the recorded signal, net YFP and CFP readings were obtained. In this paper, Y/C emission ratio is used to represent the effect of FRET, which is equal to the net YFP reading divided by the net CFP reading from the same well.
Data analysis
Results are shown either as representative examples or as mean values (with s.d.) from the number of experiments shown.
Materials
Camptothecin (Calbiochem, San Diego, CA, USA) and ET (Calbiochem) were dissolved in dimethylsulfoxide (DMSO) (Sigma-Aldrich, St Louis, MO, USA) at a concentration of 100 mM. Paclitaxel (Sigma-Aldrich) and vincristine sulfate (Sigma-Aldrich) were dissolved in DMSO at 50 μM. The stock concentrations of TNF-α (10 μg ml−1), cycloheximide (10 mg ml−1) and nocodazole (100 μg ml−1) were prepared in water. Thirty percent H2O2 (8.826 M) was diluted to 5 M with double-distilled H2O as a stock solution and then added to the cells at 1:1000 dilution. The pan-caspase inhibitor Z-VAD-FMK (G7232) was purchased from Promega (Madison, WI, USA). Compounds prepared from Danshen and Podophyllum emodi Wall. were kindly provided by Dr Houwei Luo from China Pharmaceutical University, Nanjing, China (Luo et al., 1985; Xie et al., 2000). Tanshinone compounds were dissolved in DMSO at 5 mM and Podophyllum compounds were first dissolved in DMSO at 50 mg ml−1 or 100 mg ml−1and then further diluted to make 40 μM stock concentrations with DMSO. All stock solutions were sterilized by filtration using filters with a pore size of 0.22 μm. Hoechst 33342 in a stock solution of 10 mg ml−1 in water was purchased from Invitrogen.
Results
Activation of caspase-3 during apoptosis can be detected from HeLa-C3 cells using a fluorescent plate reader
We generated a stable cell line (HeLa-C3) which consistently expresses the gene encoding the fusion protein of CFP-DEVD-YFP (sensor C3) (Luo et al., 2001). Under normal growth conditions as shown in the control panel of Figure 1a, most HeLa-C3 cells emit green fluorescent light owing to the energy transfer from CFP to YFP. When these HeLa-C3 cells were treated with different apoptotic stimuli (including paclitaxel, TNF-α and UV-irradiation) for various periods of time, the fluorescent light emitted changed from a green to a blue color, indicating that the fluorescent energy can no longer be transferred from CFP to YFP (Figure 1a). When we measured the changes in fluorescent intensity emitted from HeLa-C3 cells seeded in a 96-well plate using a fluorescent plate reader (Perkin–Elmer Victor3) after 9 h of UV treatment, the Y/C emission ratio reduced from 6.20 to 3.14, with a net reduction value of 3.06. An even greater reduction in Y/C emission ratio of 3.80 was detected 36 h after treatment with paclitaxel (Figure 1b). Moreover, the reductions in the Y/C emission ratio after UV irradiation can be prevented by pre-treatment with the pan-caspase inhibitor (20 μM) for 2 h. This result suggests that these fluorescent changes were caused by caspase-3 activation. Consistent with this finding, we also found that the reduction of Y/C emission ratio was correlated with the increase in caspase-3 activity. Results in Figure 1c show that significant increases of caspase-3 activity were detected in both UV- and paclitaxel-treated cells using an in vitro caspase-3 activity assay. Meanwhile, very little caspase-3 activity increase was detected from cells pretreated with pan-caspase inhibitor (Figure 1c). These results suggest that we can use HeLa-C3 cells to monitor the activation of caspase-3 in a population of cells treated with different types of apoptotic stimuli.
Figure 1.

The fluorescence emitted from HeLa-C3 cells changes from green to blue in response to induction of apoptosis. (a) HeLa-C3 cells grown on a cover-slip-based observation chamber were treated with three apoptotic inducers: 100 nM paclitaxel for 24 h, 10 ng ml−1 TNF-α plus 10 μg ml−1cycloheximide for 12 h and 12 h after UV-irradiation for 5 min. The phase images and fluorescent images of YFP and CFP were recorded from the same observation field in the green and blue channels, respectively. Merged color images of YFP (green) and CFP (blue) of HeLa-C3 cells show a significant increase in blue color exclusively in cells with cell shrinkage morphologies. The phase images show cell shrinkage and apoptotic bodies, which are significant indicators of apoptosis. Bar, 25 μm. (b) HeLa-C3 cells grown in a 96-well plate were treated with UV-irradiation (UV) for 5 min in the absence or presence of the pan caspase inhibitor Z-VAD-FMK (20 μM), or treated with 100 nM of paclitaxel (Tax) for 18 and 36 h. At the indicated time points, the fluorescence intensity was measured using a fluorescent plate reader. Data shown are means±s.d. from three experiments. (c) Caspase-3 activity was measured from HeLa-C3 cells using an in vitro assay described in Methods. The decrease in the Y/C emission ratio of the HeLa-C3 cells correlates well with the increase of caspase-3 activity during apoptosis. Data shown are means±s.d. from three experiments.
HeLa-C3 cells are specific for detecting apoptosis but not necrosis
As discussed in the Introduction, the biosensor in the HeLa-C3 cells was designed to detect caspase-3 activation during apoptosis in living cells (Luo et al., 2001). In principle, this detection should be specific only to apoptotic cell death during which caspase-3 is activated and insensitive to necrotic cell death when caspase-3 is not activated. In order to verify this, we compared differential responses of HeLa-C3 cells to apoptotic stimulus (UV-irradiation for 5 min) and necrotic stimulus (continued treatment with 5 mM H2O2). Results of Western blotting in Figure 2a show that caspase-3 was cleaved into its active form 3 h after UV-irradiation. At the same time, the fusion protein of sensor C3 was also cleaved into two polypeptides: YFP-DEVD and CFP, both of which were recognized by the GFP antibody (Figure 2a). For cells treated with a necrotic inducer (5 mM H2O2) for 2–12 h, however, no cleavage of caspase-3 or sensor protein was observed (Figure 2a) even though over 95% of cells were undergoing cell death (Figure 2c). Fragmented DNA, a clear indication of apoptotic cell death, was clearly observable in HeLa-C3 cells treated with UV-irradiation. Intact DNA was observable in cells continually treated with 5 mM H2O2, indicating that apoptotic cell death did not occur (Figure 2b).
Figure 2.

The CFP-DEVD-YFP fusion protein in HeLa-C3 cells is specifically cleaved during apoptosis but not necrosis. HeLa-C3 cells were treated with either an apoptotic inducer (5 min UV-irradiation) or a necrotic stimulus (5 mM H2O2) for various times and analyzed by Western blotting (a) and agarose gel electrophoresis (b). The cleavage of sensor protein was observed only during UV-induced apoptosis as indicated by caspase-3 activation (a) and DNA fragmentation (b). (c) HeLa-C3 cells grown in a 96-well plate were irradiated with 5 min UV light and then cultured for 9 h or treated with 5 mM H2O2 for 8 h. After detecting the fluorescence intensity of the cells using a fluorescent plate reader, cell viability was determined from the same sample well by MTT assay. Data shown are means±s.d. from three experiments.
To further examine the specificity of this HeLa-C3-based screening method, we performed the following experiment. HeLa-C3 cells were treated with 5 mM H2O2 for 8 h, and fluorescence emitted from YFP and CFP were measured separately using a fluorescent plate reader. Afterwards, cell viability from the same group of cells was determined by MTT assay. The results in Figure 2c show that although most HeLa-C3 cells were killed in the treatment with the inducer of necrosis, H2O2, no significant changes in the Y/C emission ratio were detected. In contrast, when cells were induced into apoptosis by UV-irradiation, a significant reduction in the Y/C emission ratio was detected (Figure 2c). These results suggest that HeLa-C3 cells can selectively detect apoptotic, but not necrotic, cell deaths.
HeLa-C3 cells can detect caspase-3 activation during apoptosis induced by multiple anticancer drugs
To evaluate whether HeLa-C3 cells can be used as a high throughput screening platform for selecting novel anticancer drug candidates, we tested this system with a variety of known anticancer compounds, including paclitaxel, vincristine, ET, hydroxyurea and camptothecin. Paclitaxel is a well-known anticancer drug. It can arrest cells in mitosis by preventing microtubule disassembly and induce these arrested mitotic cells into apoptosis (Bhalla et al., 1993; Mollinedo and Gajate, 2003). Figure 3a shows the changes in the Y/C emission ratio from HeLa-C3 cells during the course of paclitaxel treatment at three different concentrations. About 50% of the total reduction in the Y/C emission ratio was seen from cells treated with 10 nM paclitaxel for ∼36 h, and a more significant decrease of nearly 75% in the Y/C ratio was seen from cells treated with 50 nM paclitaxel for over 30 h. No fluorescent change was seen at a lower concentration of paclitaxel (1 nM) and the control cells.
Figure 3.

A significant reduction in the Y/C emission ratio can be seen from HeLa-C3 cells treated with different anticancer drugs: paclitaxel (a), vincristine (b), ET (c) and hydroxyurea (d). A summary of FRET effects in HeLa-C3 cells in response to multiple anticancer drugs at their optimal concentrations (e). The error bars represent the s.d. from three independent experiments.
We further tested this high throughput drug screening system with another microtubule interfering agent, vincristine, which can block cells in mitosis and then induce them into apoptosis (Takano et al., 1993; Mollinedo and Gajate, 2003). In this study, three concentrations of vincristine (1, 10 and 50 nM) were added to HeLa-C3 cells. Screening results showed that 10 nM of vincristine reduced the Y/C emission ratio to about 4, and a higher concentration of vincristine at 50 nM further reduced the Y/C ratio to less than 3 (as indicated by the dotted line) within 48 h (Figure 3b). We further compared the cytotoxic effect of vincristine at concentrations of 10 and 50 nM based on cell morphology. We found that about 30–40% of the cells exhibited a clear cell shrinkage morphology after treatment with drug at 10 nM for 48 h and even more cells were dead (80–90%) with a higher concentration of vincristine (50 nM) (data not shown).
The third drug we tested was ET, an inhibitor of DNA topoisomerase II. ET can block DNA synthesis during S-phase of the cell cycle, and then induce cells into apoptotic cell death (Fearnhead et al., 1994). From the screening results in Figure 3c, we can see that the effective concentration of ET for activating apoptosis is between 1 and 100 μM. Also, significant reductions in the Y/C emission ratio were seen 60 h after addition of the drug, suggesting that ET may need more time to activate caspase-3 and induce apoptotic cell death in these HeLa-C3 cells than paclitaxel or vincristine.
The last drug used to test our system was hydroxyurea, which belongs to the category of antimetabolites. It prevents cell division by inhibiting ribonucleotide reductase, leading to the depletion of adenine required for DNA synthesis. Hydroxyurea failed to induce cell death even at 10 mM. At 40 mM, hydroxyurea finally brought the emission ratio of Y/C down to almost 2 at 48 h (Figure 3d). Figure 3e summarizes the screening results obtained from all five anticancer drugs at their most effective concentrations. The graphs show that about three- to four-fold reductions in the emission ratio of Y/C can be detected by a fluorescent plate reader after cells were induced into apoptosis. This demonstrates that HeLa-C3 cells are very sensitive in detecting caspase-3 activation induced by multiple anticancer drugs.
Reduction in the Y/C emission ratio reflects the decrease of cell viability owing to apoptosis but not necrosis
The emission ratio of Y/C starts to decline in HeLa-C3 cells when intracellular caspase-3 is activated during apoptosis. This reduction of Y/C emission ratio is the readout of our drug screening system. It only indicates the level of caspase-3 activation from a population of apoptotic cells. However, it does not directly represent the percentage of cell death. Because the purpose of drug screening is to identify compounds that can effectively kill cancer cells, it is therefore necessary to determine the correlation between Y/C emission ratio changes and cell viability in our HeLa-C3-based screening system.
To achieve this goal, we measured the effects of four distinctive apoptotic stimuli: paclitaxel, nocodazole (Bumbasirevic et al., 1995), ET and UV-irradiation, along with four different necrotic stimuli: H2O2, NP-40, Triton X-100 and treatment with pure water. All inducers were individually applied to cells grown in a 96-well plate. Fluorescent intensities of YFP and CFP (under CFP excitation) were recorded, using a fluorescent plate reader. Cell viability from the same group of cells was then determined by MTT assay and the Y/C emission ratio was plotted against cell viability for each treatment. Results were summarized in Figure 4, which shows that the reduction in Y/C emission ratio correlates very well with the decrease in cell viability when cells were treated with various apoptotic stimuli. In most cases, the Y/C emission ratio was reduced from 6 to 2 in two phases. In the first phase, the Y/C ratio reduced from 6 to 3, whereas the cell viability only decreased about 20%. In the second phase, the Y/C ratio changed from 3 to 2, but cell viability decreased more significantly, from 80% to close to 0%. This two-phase phenomenon may be caused by the time delay between caspase-3 activation and completion of cell death as assessed by the MTT method, which measures the activity of a metabolic enzyme inside a cell. The necrotic stimuli, on the other hand, caused very different effects in cells. Although necrotic stimuli significantly reduced cell viability, very little change in the Y/C emission ratio was detected. Consequently, all the data corresponding to the necrotic stimuli appear in the lower right corner of the graph, indicating there is little correlation between necrotic cell death and reduction of the Y/C emission ratio. These results thus clearly demonstrate that, the drug screening method based on HeLa-C3 cells can identify apoptotic cell death over necrotic cell death in a wide range of death inducers. Based on the positive correlation between the reduction of Y/C emission ratio and the decrease of cell viability established in Figure 4, the screening threshold for Y/C emission ratio was determined to be 3. We found that when the Y/C emission ratio is reduced to a level below 3, most cells will die via apoptosis. If, however, the Y/C value is above 3, the cells will either die by necrosis or only a minority of cells will undergo apoptosis.
Figure 4.

A clear correlation between a reduction in Y/C emission ratios and a decrease in cell viability is exhibited in cells treated with apoptotic stimuli but not in cells treated with necrotic inducers. HeLa-C3 cells grown in 96-well plates were treated with four different apoptotic inducers: 50 nM paclitaxel for 12–48 h, 50 ng ml−1 nocodazole for 12–48 h, 100 μM ET for 12–48 h and 3–12 h after UV-irradiation for 5 min, and with four different necrotic inducers: H2O2 at 3, 5 and 7 mM for 2–8 h, 0.01 and 0.1% NP40 for 0.5 h, 0.1% Triton X-100 for 0.5 h and double-distilled H2O for 0.5 h. The fluorescence intensity and cell viability of HeLa-C3 cells from different treatments were obtained in similar ways as described in Figure 2c. Data shown are from a single representative experiment.
Application of HeLa-C3 cells in screening tanshinone family compounds for apoptotic effects
After establishing that the HeLa-C3-based high throughput screening method is sensitive and reliable for detecting compounds capable of inducing cancer cells to enter apoptosis, we used this system to screen compounds purified from two herbal plants: Salvia miltiorrhiza Bunge and Podophyllum emodi Wall.
S. miltiorrhiza Bunge is a well-known traditional Chinese medicinal plant and the dried root of this plant is called Danshen. To evaluate the apoptotic effects of Danshen compounds, we purified six tanshinone family compounds, including tanshinone I (TI), tanshinone IIA (TIIA), tanshinone IIB (TIIB), cryptotanshinone (Crypto), methylenetanshinquinone (MTQ) and sodium tanshinone IIA sulfonate (TanNa) (Luo et al., 1985), and further tested these compounds using HeLa-C3-based screening method. The chemical structures and molecular weights of these compounds are shown in Figure 5.
Figure 5.

Molecular structures of tanshinone family compounds.
To conduct the drug screening assay, all compounds were dissolved in DMSO except sodium TanNa, which was dissolved in double-distilled H2O. All compounds were added to cells in concentrations of 5, 10 and 20 μM, respectively. The fluorescent emissions of YFP and CFP (under the excitation of CFP) were measured using a fluorescent plate reader at different time points. The Y/C emission ratio was plotted against time for each tanshinone compound and the graphs for all six compounds are shown in Figure 6. In comparison to the control conditions, all compounds except TanNa were capable of inducing apoptosis, as indicated by the significant reductions in the Y/C emission ratio. Among these compounds, Crypto and MTQ showed the strongest ability to activate caspase-3 at 20 μM (Figure 6a and b). Forty-eight hours after addition of the compounds, the Y/C emission ratio dropped to a value around 3, the threshold previously set for determining the capability of a compound to induce apoptosis.
Figure 6.

Screening results of tanshinone family compounds. HeLa-C3 cells grown in 96-well plates were treated with six structurally related compounds of the tanshinone family at three concentrations (5, 10, 20 μM) for up to 48 h. The fluorescence intensities of YFP and CFP were detected separately using a fluorescent plate reader. The graphs show the efficacy of each compound in activating caspase-3 in HeLa-C3 cells. (a) Crypto, (b) MTQ, (c) TanNa, (d)TI, (e) TIIA, (f) TIIB. (g) Results of Y/C emission ratio levels at all three concentrations after HeLa-C3 cells were treated with six tanshinone family compounds for 48 h. Error bars in (a–f) represent s.d. from three independent experiments and those in (g) represent the s.e.m. from seven independent experiments.
TI appeared to be equally effective at both medium (10 μM) and high (20 μM) concentrations in reducing the Y/C emission ratio to less than 3 within 40 h of treatment (Figure 6d). Microscopic analysis showed that some crystals were formed in the cell culture medium containing TI, suggesting that this compound has a very low solubility in water. Both TIIA and TIIB induced a significant reduction in the Y/C emission ratio at the concentration of 20 μM in around 40 h (Figure 6e and f). No crystals were observed from these two compounds at this concentration. TanNa was chemically modified from tanshinone IIA by adding a sulfonate group to the furan ring of the compound. This modification greatly increased its solubility in water. However, very little apoptotic effect was detected at the three concentrations tested (Figure 6c). This negative apoptotic effect of TanNa may be caused by its chemical modification, which prevented this compound from crossing the cell membrane (Liu et al., 1991).
To objectively compare the apoptotic effects of all six compounds on HeLa-C3 cells, we have plotted the average Y/C emission ratio detected at 48 h from three independent experiments for all six compounds, each at the same three concentrations (Figure 6g). The results show that among all the tanshinone compounds tested, TIIA at 20 μM has the lowest Y/C emission ratio, indicating that this compound has the best apoptotic effect among this group of herbal compounds. TI and TIIB at concentrations of 10 and 20 μM also induced a dramatic reduction in the Y/C emission ratio. Thus, we can conclude from this drug screening assay that TI, TIIA, and TIIB are the most potent apoptotic inducers among the six tanshinone family compounds tested in this study.
Using HeLa-C3 cells to screen podophyllotoxin (S1) family compounds for apoptotic effects
We were interested in compounds of the S1 family because a well-known anticancer drug, ET was developed from S1 (Felix and Senn, 1975; Walker et al., 1991). S1 is present in various plant species and is particularly abundant in the genus Podophyllum. In the present study, six S1-related compounds (Figure 7), including podophyllotoxin (S1), 4′-demethylpodophyllotoxin (S4), deoxypodophyllotoxin (DP1), dehydropodophyllotoxin (UP), podophyllotoxone (P8), and 1β, 2β, 3β, 4′-demethylpicropodophyllone (DP3), were extracted and purified from rhizomes of a Chinese herbal plant, P. emodi Wall., and their potencies in inducing apoptosis were examined using HeLa-C3 cells.
Figure 7.

Molecular structures of podophyllotoxin family compounds.
Based on the primary screening results of S1, we have selected three drug concentrations (10, 20 and 40 nM) for testing all six compounds plus ET. Although the structure of S1 is very similar to that of ET, their apoptotic potencies are very different. Results in Figure 8a–c show that ET demonstrated a negligible effect on HeLa-C3 cells at all three concentrations tested. In contrast, S1 consistently showed higher potency at both 20 and 40 nM concentrations and greatly reduced the Y/C emission ratio in less than 36 h of drug treatment (Figure 8b and c). These results of drug screening using HeLa-C3 cells also agreed with morphological observations of HeLa cells. Cells treated with 20 nM of S1 started to round up at 12 h, and reached a maximum state of mitotic arrest at 24 h. Cell death as indicated by cell shrinkage started at 24 h, and most cells were dead after 48 h of drug treatment (Figure 8e). Almost no cell death and reduction in the Y/C ratio were detected from cells treated with 10 nM of S1 for up to 48 h of treatment (Figure 8a and e). After comparing the potencies between podophyllotoxin and ET, we found that podophyllotoxin induced significant apoptosis at a much lower concentration of 20 nM, although a 5000-fold higher concentration of ET (100 μM) was required to produce a similar effect (Figure 3c).
Figure 8.

Screening results of S1 family compounds. HeLa-C3 cells grown in 96-well plates were treated with six podophyllotoxin family compounds plus ET at 10 nM (a), 20 nM (b) and 40 nM (c) for up to 48 h. Changes in the Y/C emission ratio were measured and used to compare the efficacy of each compound in inducing caspase-3 dependent apoptosis. (d) Apoptotic effects of DP1 and S1 were directly compared with the microtubule interfering agents nocodazole and vincristine, each at a concentration of 10 nM. (e) HeLa cells were treated with 10 and 20 nM S1 and 10 nM DP1 for up to 48 h. Phase images showing mitotic arrested cells with rounded cell morphology and apoptotic cells with cell shrinkage. Cells treated with 10 nM DP1 were also stained with Hoechst 33342, a DNA dye. The fluorescent images in the enlarged pictures show that 10 nM DP1 arrested cells in prometaphase at 12 h and caused DNA fragmentation after 24 h of drug treatment. The scale bars in the pictures of phase contrast and Hoechst staining are 50 and 25 μm, respectively. The error bars represent s.d. from three independent experiments.
Among all six compounds tested in this study, DP1, the parental compound of podophyllotoxin, showed the highest potency against HeLa-C3 cells. At 10 nM, DP1 was the only compound capable of reducing the Y/C emission ratio to near 3 in 48 h, indicating that DP1 can effectively induce apoptosis in HeLa-C3 cells (Figure 8a). We also observed cell morphology changes during the course of drug treatment. After adding 10 nM of DP1 to HeLa cells for 12 h, many cells appeared to be arrested in mitosis with a rounded-up morphology. More rounded-up cells were observed at 24 h (Figure 8e). Cell death as indicated by cell shrinkage was first seen at 24 h and more dead cells appeared at 48 h (Figure 8e). We further examined at which stage of mitosis cells were arrested by DP1 using nuclear DNA staining. HeLa cells were treated with DP1 for various lengths of time and stained with a cell-permeable DNA dye, Hoechst 33342 (100 ng ml−1), for 30 min. As shown in Figure 8e, DP1 arrested cells in prometaphase at 12 h with mitotic cell morphology and condensed chromosomes. After prolonged treatment with 10 nM DP1 for 24, 36 and 48 h, fragmented chromosomal DNA could be clearly observed, demonstrating that the cells were undergoing apoptotic cell death (Figure 8e).
P8 and 4′-demethylpodophyllotoxin (S4) showed an interesting trend in which the compounds demonstrated some apoptotic effect only at the highest concentration tested, 40 nM (Figure 8c). Very little apoptotic effect was detected using HeLa-C3 cells from UP and another novel compound 1β, 2β, 3β, 4′-demethylpicro-podophyllone (DP3) (Xie et al., 2000).
It has been reported that both podophyllotoxin and DP1 can inhibit microtubule assembly and arrest cells in mitosis (Damayanthi and Lown, 1998). In order to compare the potency of S1 and DP1 with other microtubule-interfering agents, S1 and DP1 were tested against two commonly known microtubule-interfering agents, vincristine and nocodazole. All four compounds were added to HeLa-C3 cells at the same concentration, 10 nM, and incubated for up to 48 h. Results in Figure 8d show that nocodazole and podophyllotoxin had little effect in activating caspase-3 at 10 nM. In contrast, DP1 showed the strongest effect in reducing the Y/C ratio to lower than 3 in less than 48 h. It should be noted that at a higher concentration such as 100 nM, apoptotic effect of nocodazole can be clearly detected using HeLa-C3 cells.
Discussion
A cell-based, high throughput screen method for anticancer activity was established in this study. The cornerstone of this method was the stable cell line derived from HeLa cells that produces a caspase sensor protein. We have shown that these sensor proteins can be cleaved by caspase-3 when HeLa-C3 cells undergo apoptotic cell death. As a result of this cleavage, the fluorescence emission changes from a yellowish green color to a cyan/blue color. This color change can be easily detected by a fluorescent plate reader. We have also demonstrated that HeLa-C3 cells are only sensitive in detecting caspase-dependent apoptotic cell death but not necrosis. Therefore, it can be used as a specific sensor for selecting compounds with the ability to induce cancer cells into apoptosis. We further tested this drug screening system using various known inducers of apoptosis, with positive results. Furthermore, a clear correlation between caspase activation detected by HeLa-C3 cells and cell death measured by the standard MTT assay was also established in this study.
Zhang et al. (1999) introduced a simple statistical parameter (Z′ factor) for comparing qualities between different high throughput screening assays . In order to evaluate the quality of HeLa-C3-based drug screening system, we have calculated the Z′ factor for our screening platform. In Zhang's paper, the Z′ factor was defined as
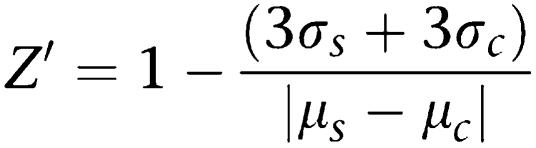 |
In our calculation, μs is the mean value of Y/C emission ratio produced by the tested anticancer drugs and μc is the mean value of Y/C emission ratio of the negative control. σs and σc are the corresponding standard deviations for the tested drugs and the negative control, respectively. We have calculated the Z′ value from every tested anticancer drug at the effective concentration shown in Figure 3e. The final average Z′ value from these five anticancer drugs is 0.87±0.02. According to Zhang's paper, a high throughput screening method with a Z′ value larger than 0.5 is considered an excellent assay. Thus, in terms of screening quality, our screening method is a good system for detecting apoptotic cell death.
The first GFP-FRET based in vivo assay for drug screening was reported by Jay Jones et al. (2000). They demonstrated a good correlation between FRET changes and caspase-3 activation. In this work, we have made several improvements on the FRET assay. First, our HeLa-C3 cells have stronger fluorescent signals as they were derived from a single clone. Second, our sensor protein is more sensitive to apoptotic stimuli because it gives a greater reduction in the Y/C emission ratio when cells undergo apoptotic cell death. Third, our system allowed us to set a threshold value of 3 in the Y/C emission ratio to indicate a compound with significant apoptotic activity. We have illustrated that setting this single threshold is sufficient to identify correctly several anticancer drugs tested in this study. An important feature of this screening threshold is that it represents an absolute value, as unlike readings of fluorescence signals; the Y/C emission ratio generated by this system is independent of the number of cells involved. This is why a single threshold can be used to determine the outcome of the screening results. As shown in Figure 3e, most anticancer drugs tested in this experiment did reach this threshold within 48 h of drug treatment. Thus, any compound that can reduce the value of the Y/C ratio to a value below 3 can be considered a ‘hit' in this pre-clinical drug screening. We consider that setting a single threshold of Y/C ratio provides a simple and consistent method for high throughput drug screening and allows faster computerization of data processing and data analysis. For compounds having a significant reduction in the Y/C emission ratio, one should further examine the slope of Y/C emission ratio changes. Generally speaking, a curve of Y/C emission ratio with a steep slope usually indicates a fast activation of caspase-3, whereas a slope of gentle decline represents slow caspase-3 activation. Finally, we confirmed that the Y/C emission ratio measured by our HeLa-C3 cells only responds to apoptotic inducers but not necrotic stimuli.
In comparison with the conventional apoptotic and cell viability assays, our method has the following advantages:
HeLa-C3 cells can detect caspase-3 activation in living cells during the process of apoptosis.
It is specific only to apoptosis but not necrosis.
It allows the assessment of the kinetics of drug action in activating caspases from the same population of cells during the course of drug treatment.
It can rapidly measure the concentration–dependent effects of a drug.
The HeLa-C3 cells can be grown on 96-well plates and fluorescent light emitted from these cells can be detected using a fluorescent plate reader. Thus, HeLa-C3-based method can be used for a high throughput screen for compounds with apoptotic effects.
It is easy to operate and provides rapid results. More importantly, it is highly cost-effective.
We are in the process of generating more stable cell lines containing the sensor protein C3, using various cancer cells including HL-60, so that we can screen compounds that are effective on certain types of cancer cells.
Herbal medicine has been used to treat various diseases, including cancer, for many years in Asian countries. These medicines have therefore been considered a rich source for new drug discoveries. In this study, we have focused on two herbal plants, S. miltiorrhiza Bunge and P. emodi Wall., screening compounds isolated from these plants for pro-apoptotic activity using our HeLa-C3 cell system.
S. miltiorrhiza has been used to treat hematological abnormalities, heart diseases (Xia et al., 2003), hepatitis and cancer. Many of the constituents of Danshen have been isolated and can be classified into two groups: polyphenolic acids and diterpenoid quinone pigments such as the tanshinones. Over 40 tanshinone compounds have been isolated and TIIA, Crypto, TI and dihydrotanshinone are the major constituents (Wu et al., 1991). It has been reported that TI and IIA have growth inhibitory effects on some human cancer cell lines and TIIA may inhibit cell growth in some human leukemia cells by inducing them into apoptosis (Yoon et al., 1999). In vivo studies have also shown that TIIA can inhibit the growth of a human breast infiltrating duct carcinoma orthotopically implanted in nude mice (Wang et al., 2005). In this study, we have purified six tanshinone family compounds and evaluated their apoptotic effects using our newly developed HeLa-C3-based drug screening system. The results show that TIIA was the most potent compound among the six tanshinone family compounds. The screening results also indicated that TI and TIIB may be good candidates for further anticancer drug development. One major concern for compounds in the tanshinone family is that they have very low solubility in water. Chemical modification on the structure of tanshinone compounds or physically packaging them into water-soluble micelles may increase their solubility in water. Nonetheless, the mechanism by which tanshinone compounds induce apoptosis is not yet well understood.
The second herbal plant used for the drug screening was P. emodi Wall., one of the Podophyllaceae family which includes the American species, P. peltatum, also known as May Apple, the Indian or Chinese species, P. emodi or P. hexandrum, and another Chinese Taiwan species P. pleiantum (Imbert, 1998; Gordaliza et al., 2000). The major ingredient of the species we have studied is podophyllotoxin. Both podophyllotoxin and its derivatives have been used to treat various diseases including viral infections (Sudo et al., 1998), venereal warts (Wantke et al., 1993) and rheumatoid arthritis (Berglund et al., 1978) for many years. Some derivatives of podophyllotoxin have been found to be very effective against cancer cell lines (Subrahmanyam et al., 1998), and also solid human tumor xenografts (i.e. lung and colon cancers) (Utsugi et al., 1996). Podophyllotoxin exerts its cytotoxicity effect by inhibiting microtubule assembly and thus promoting cells to die via apoptosis (Damayanthi and Lown, 1998). Owing to its high toxicity, podophyllotoxin has not been used as an anticancer drug for chemotherapy. However, chemical modification of podophyllotoxin by adding a glucose group to its tubulin-binding site has produced the chemotherapy drug, ET (Felix and Senn, 1975). The mechanism of eradicating cancer cells by ET, however, is completely different from that of podophyllotoxin. ET is an inhibitor of DNA topoisomerase II, which can arrest cells in S phase and promote them into apoptosis (Walker et al., 1991). In contrast, podophyllotoxin arrests cells in M phase and induces M-phase arrested cells into apoptosis (Damayanthi and Lown, 1998; Imbert, 1998; Gordaliza et al., 2000).
The screening results of the podophyllotoxin family compounds show that podophyllotoxin (S1) and DP1 have the highest anticancer efficacy among the seven podophyllotoxin compounds including ET. Both S1 and DP1 can arrest cells in mitosis and then induce them into apoptosis. S1 and DP1 have long been known for their toxic effects against both cancer and normal cells. Unfortunately, S1 can only be applied topically to treat venereal warts and intake of S1 causes major health complications such as gastrointestinal irritation. However, in this study we showed that DP1, in concentrations as low as 10 nM, can effectively activate caspase-3 and induce HeLa-C3 cells into apoptosis in just 48 h. It is clear that at 10 nM, DP1 has the most potent apoptotic activity of the podophyllotoxin family compounds, with even greater efficiency than the anticancer drug vincristine. The fact that DP1 outperformed vincristine, a well-known anticancer drug, suggests that DP1 may be a good candidate for the development of a more effective anticancer drug. In the past, S1 has been the major focus for chemical modification as it is the parent compound of the anticancer drug ET. With the new understanding that DP1 is more potent than S1, DP1 can become the new focus for developing a more effective anticancer drug. Hence, further research on chemical modifications of ET's parent compounds should continue, in the hopes of discovering a drug with greater potency than ET and fewer side effects than podophyllotoxin.
Acknowledgments
This work was supported by the Research Grants Council of Hong Kong (HKUST6466/05 M and AoE/B-15/01) and the Emerging High Impact Areas project of Hong Kong University of Science and Technology.
Abbreviations
- Ac-DEVD-AMC (7-amino-4-methylcoumarin
N-acetyl-L-aspartyl-L-glutamyl-L-valyl-L-aspartic acid amide
- CFP
cyan fluorescent protein
- Crypto
cryptotanshinone
- DP1
deoxypodophyllotoxin
- DP3, 1β, 2β, 3β
4′-demethlypicropodophyllone
- DTT
1,4-dithiothreitol
- FRET
fluorescence resonance energy transfer
- GFP
green fluorescent protein
- HTS
high throughput screening
- MIAs
microtubule-interfering agents
- MTQ
methylenetanshinquinone
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NP-40
Nonidet P-40
- P8
podophyllotoxone
- PMSF
phenylmethylsulfonyl fluoride
- S1
podophyllotoxin
- S4
4′-demethylpodophyllotoxin
- TanNa
sodium tanshinone IIA sulfonate
- TE buffer
Tris-EDTA buffer
- TI
tanshinone I
- TIIA
tanshinone IIA
- TIIB
tanshinone IIB
- TNF-α
tumor necrosis factor-α
- UP
dehydropodophyllotoxin
- YFP
yellow fluorescent protein
- Z-VAD-FMK
carbobenzoxy-valyl-aspartyl-[O-methyl]fluoromethylketone
Conflict of interest
The authors state no conflict of interest.
References
- Berglund K, Johansson BG, Laurell AB, Sjoholm A, Sturfelt G. Inflammatory parameters in rheumatoid arthritis during and after administration of an anti-mitotic agent (Podophyllum lignan derivatives) Scand J Rheumatol. 1978;7:61–63. [PubMed] [Google Scholar]
- Bhalla K, Ibrado AM, Tourkina E, Tang C, Mahoney ME, Huang Y. Taxol induces internucleosomal DNA fragmentation associated with programmed cell death in human myeloid leukemia cells. Leukemia. 1993;7:563–568. [PubMed] [Google Scholar]
- Bumbasirevic V, Skaro-Milic A, Mircic A, Djuricic B.Apoptosis induced by microtubule disrupting drugs in normal murine thymocytes in vitro Scanning Microsc 19959509–516.discussion 516–518 [PubMed] [Google Scholar]
- Cubitt AB, Heim R, Adams SR, Boyd AE, Gross LA, Tsien RY. Understanding, improving and using green fluorescent proteins. Trends Biochem Sci. 1995;20:448–455. doi: 10.1016/s0968-0004(00)89099-4. [DOI] [PubMed] [Google Scholar]
- Damayanthi Y, Lown JW. Podophyllotoxins: current status and recent developments. Curr Med Chem. 1998;5:205–252. [PubMed] [Google Scholar]
- Das GC, Holiday D, Gallardo R, Haas C. Taxol-induced cell cycle arrest and apoptosis: dose-response relationship in lung cancer cells of different wild-type p53 status and under isogenic condition. Cancer Lett. 2001;165:147–153. doi: 10.1016/s0304-3835(01)00404-9. [DOI] [PubMed] [Google Scholar]
- Einzig AI, Hochster H, Wiernik PH, Trump DL, Dutcher JP, Garowski E, et al. A phase II study of taxol in patients with malignant melanoma. Invest New Drugs. 1991;9:59–64. doi: 10.1007/BF00194546. [DOI] [PubMed] [Google Scholar]
- Fearnhead HO, Chwalinski M, Snowden RT, Ormerod MG, Cohen GM. Dexamethasone and etoposide induce apoptosis in rat thymocytes from different phases of the cell cycle. Biochem Pharmacol. 1994;48:1073–1079. doi: 10.1016/0006-2952(94)90142-2. [DOI] [PubMed] [Google Scholar]
- Felix W, Senn HJ. Clinical study of the new podophyllotoxin derivative, 4′-demethylepipodophyllotoxin 9-(4,6-o-ethylidene- beta-D-glucopyranoside) (NSC-141540; VP-16-213), in solid tumors. Cancer Chemother Rep. 1975;59:737–742. [PubMed] [Google Scholar]
- Gidding CE, Kellie SJ, Kamps WA, de Graaf SS. Vincristine revisited. Crit Rev Oncol Hematol. 1999;29:267–287. doi: 10.1016/s1040-8428(98)00023-7. [DOI] [PubMed] [Google Scholar]
- Gordaliza M, Castro MA, del Corral JM, Feliciano AS. Antitumor properties of podophyllotoxin and related compounds. Curr Pharm Des. 2000;6:1811–1839. doi: 10.2174/1381612003398582. [DOI] [PubMed] [Google Scholar]
- Heim R, Tsien RY. Engineering green fluorescent protein for improved brightness, longer wavelengths and fluorescence resonance energy transfer. Curr Biol. 1996;6:178–182. doi: 10.1016/s0960-9822(02)00450-5. [DOI] [PubMed] [Google Scholar]
- Herrmann M, Lorenz HM, Voll R, Grunke M, Woith W, Kalden JR. A rapid and simple method for the isolation of apoptotic DNA fragments. Nucleic Acids Res. 1994;22:5506–5507. doi: 10.1093/nar/22.24.5506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbert TF. Discovery of podophyllotoxins. Biochimie. 1998;80:207–222. doi: 10.1016/s0300-9084(98)80004-7. [DOI] [PubMed] [Google Scholar]
- Jones J, Heim R, Hare E, Stack J, Pollok BA. Development and application of a GFP-FRET intracellular caspase assay for drug screening. J Biomol Screen. 2000;5:307–318. doi: 10.1177/108705710000500502. [DOI] [PubMed] [Google Scholar]
- Liu MZ, Huang YS, Xiao WQ. No promoting effects of sodium tanshinone II-A sulfonate on growth and metastasis of Lewis carcinoma. Zhongguo Yao Li Xue Bao. 1991;12:534–537. [PubMed] [Google Scholar]
- Luo HW, Wu BJ, Yong ZG, Niwa M, Hirata Y. Pigments from Salvia miltiorrhiza. Phytochemistry. 1985;24:815–817. [Google Scholar]
- Luo KQ, Chang DC. The gene-silencing efficiency of siRNA is strongly dependent on the local structure of mRNA at the targeted region. Biochem Biophys Res Commun. 2004;318:303–310. doi: 10.1016/j.bbrc.2004.04.027. [DOI] [PubMed] [Google Scholar]
- Luo KQ, Yu VC, Pu Y, Chang DC. Application of the fluorescence resonance energy transfer. Biochem Biophys Res Commun. 2001;283:1054–1060. doi: 10.1006/bbrc.2001.4896. [DOI] [PubMed] [Google Scholar]
- Mollinedo F, Gajate C. Microtubules, microtubule-interfering agents and apoptosis. Apoptosis. 2003;8:412–450. doi: 10.1023/a:1025513106330. [DOI] [PubMed] [Google Scholar]
- Nicholson DW. Caspase structure, proteolytic substrates, and function during apoptotic cell death. Cell Death Differ. 1999;6:1028–1042. doi: 10.1038/sj.cdd.4400598. [DOI] [PubMed] [Google Scholar]
- Perry MC, Moertel CG, Schutt AJ, Reitemeier RJ, Hahn RG. Phase II studies of dianhydrogalactitol and VP-16-213 in colorectal cancer. Cancer Treat Rep. 1976;60:1247–1250. [PubMed] [Google Scholar]
- Samali A, Zhivotovsky B, Jones D, Nagata S, Orrenius S. Apoptosis: cell death defined by caspase activation. Cell Death Differ. 1999;6:495–496. doi: 10.1038/sj.cdd.4400520. [DOI] [PubMed] [Google Scholar]
- Sleiman RJ, Stewart BW. Early caspase activation in leukemic cells subject to etoposide-induced G2-M arrest: evidence of commitment to apoptosis rather than mitotic cell death. Clin Cancer Res. 2000;6:3756–3765. [PubMed] [Google Scholar]
- Subrahmanyam D, Renuka B, Rao CV, Sagar PS, Deevi DS, Babu JM, et al. Novel D-ring analogues of podophyllotoxin as potent anti-cancer agents. Bioorg Med Chem Lett. 1998;8:1391–1396. doi: 10.1016/s0960-894x(98)00232-7. [DOI] [PubMed] [Google Scholar]
- Sudo K, Konno K, Shigeta S, Yokota T. Inhibitory effects of podophyllotoxin derivatives on herpes simplex virus replication. Antivir Chem Chemother. 1998;9:263–267. doi: 10.1177/095632029800900307. [DOI] [PubMed] [Google Scholar]
- Takano Y, Okudaira M, Harmon BV. Apoptosis induced by microtubule disrupting drugs in cultured human lymphoma cells. Inhibitory effects of phorbol ester and zinc sulphate. Pathol Res Pract. 1993;189:197–203. doi: 10.1016/S0344-0338(11)80092-0. [DOI] [PubMed] [Google Scholar]
- Utsugi T, Shibata J, Sugimoto Y, Aoyagi K, Wierzba K, Kobunai T, et al. Antitumor activity of a novel podophyllotoxin derivative (TOP-53) against lung cancer and lung metastatic cancer. Cancer Res. 1996;56:2809–2814. [PubMed] [Google Scholar]
- Walker PR, Smith C, Youdale T, Leblanc J, Whitfield JF, Sikorska M. Topoisomerase II-reactive chemotherapeutic drugs induce apoptosis in thymocytes. Cancer Res. 1991;51:1078–1085. [PubMed] [Google Scholar]
- Wang X, Wei Y, Yuan S, Liu G, Lu Y, Zhang J, et al. Potential anticancer activity of tanshinone IIA against human breast cancer. Int J Cancer. 2005;116:799–807. doi: 10.1002/ijc.20880. [DOI] [PubMed] [Google Scholar]
- Wantke F, Demmer CM, Gotz M, Jarisch R. Podophyllum: an irritant on patch testing. Contact Dermatitis. 1993;29:274–275. doi: 10.1111/j.1600-0536.1993.tb03567.x. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Hitomi M, van der Wee K, Rothenberg F, Fisher SA, Zucker R, et al. The pros and cons of apoptosis assays for use in the study of cells, tissues, and organs. Microsc Microanal. 2002;8:375–391. doi: 10.1017/S1431927602010346. [DOI] [PubMed] [Google Scholar]
- Wolf BB, Green DR. Suicidal tendencies: apoptotic cell death by caspase family proteinases. J Biol Chem. 1999;274:20049–20052. doi: 10.1074/jbc.274.29.20049. [DOI] [PubMed] [Google Scholar]
- Wu WL, Chang WL, Chen CF. Cytotoxic activities of tanshinones against human carcinoma cell lines. Am J Chin Med. 1991;19:207–216. doi: 10.1142/S0192415X91000284. [DOI] [PubMed] [Google Scholar]
- Xia Z, Gu J, Ansley DM, Xia F, Yu J. Antioxidant therapy with Salvia miltiorrhiza decreases plasma endothelin-1 and thromboxane B2 after cardiopulmonary bypass in patients with congenital heart disease. J Thorac Cardiovasc Surg. 2003;126:1404–1410. doi: 10.1016/s0022-5223(03)00970-x. [DOI] [PubMed] [Google Scholar]
- Xie XM, Luo HW, Niwa M. Stereochemistry studies of 1 beta, 2 beta, 3 beta, 4′-demethylpicropodophyllone. J China Pharmaceutical University. 2000;31:174–176. [Google Scholar]
- Yoon Y, Kim YO, Jeon WK, Park HJ, Sung HJ. Tanshinone IIA isolated from Salvia miltiorrhiza BUNGE induced apoptosis in HL60 human premyelocytic leukemia cell line. J Ethnopharmacol. 1999;68:121–127. doi: 10.1016/s0378-8741(99)00059-8. [DOI] [PubMed] [Google Scholar]
- Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]