

Abstract
Background and purpose:
Aminoguanidine (AG), an inhibitor of advanced glycation endproducts, has been identified as a prominent agent that prevents the fructose-induced arterial stiffening in male Wistar rats. Our aims were to examine whether AG produced benefits on the left ventricular (LV)-arterial coupling in fructose-fed (FF) animals in terms of the ventricular and arterial chamber properties.
Experimental approach:
Rats given 10% fructose in drinking water (FF) were daily treated with AG (50 mg·kg−1, i.p.) for 2 weeks and compared with the untreated FF group. In anaesthetised rats, LV pressure and ascending aortic flow signals were recorded to calculate LV end-systolic elastance (E es, an indicator of myocardial contractility) and effective arterial volume elastance (E a). The optimal afterload (Q load) determined by the ratio of E a to E es was used to measure the coupling efficiency between the left ventricle and its vasculature.
Key results:
There was a significant interaction between fructose and AG in their effects on E a. Fructose loading significantly elevated E a and AG prevented the fructose-derived deterioration in arterial chamber elastance. Both fructose and AG affected E es and Q load, and there was an interaction between fructose and AG for these two variables. Both E es and Q load exhibited a decline with fructose feeding but showed a significant rise after AG treatment in the FF rats.
Conclusions and Implications:
AG prevented not only the contractile dysfunction of the heart caused by fructose loading, but also the fructose-induced deterioration in matching left ventricular function to the arterial system.
Keywords: aminoguanidine, effective arterial volume elastance, fructose loading, end-systolic elastance, ventricular–arterial coupling
Introduction
Fructose is a major component of human diets and fructose intake has increased steadily during the past two decades. However, the reducing free carbonyl group of fructose, as in the case of glucose, can react nonenzymatically with amino groups of biological molecules to form irreversible advanced glycation endproducts (AGEs) (Schalkwijk et al., 2004). This AGE formation and the AGE-derived cross-links on collagen of the blood vessel walls account largely for the increased arterial stiffening during fructose loading (Lin et al., 2004) and diabetes (Chang et al., 2006). Diabetes was also reported to increase AGE formation on two cardiac proteins, ryanodine receptors/calcium-release channels (Bidasee et al., 2003) and sarcoplasmic reticulum Ca2+-ATPase (Bidasee et al., 2004), by which left ventricular (LV) function could be impaired. As a glycating agent, fructose should receive more attention as it is more potent in forming AGEs than glucose (McPherson et al., 1988) and accumulates in organs where the sorbitol pathway is active (Suárez et al., 1989).
Aminoguanidine (AG), a nucleophilic hydrazine compound, is a prototype of scavenging agents that inhibit AGE formation and protein–protein crosslinking in experimental diabetic rats (Brownlee et al., 1986; Huijberts et al., 1993) and in rats fed with fructose (Lin et al., 2004). An in vitro study demonstrated that the effect of AG on the prevention of fructose-induced AGE formation may be attributed in part to the trapping of the carbonyl compound derived from fructose autoxidation or protein–fructose adduct oxidation (Takagi et al., 1995). These results indicate that AG has the potential to retard the fructose-induced deterioration in the integration of LV function with the arterial system (ventricular–arterial coupling). To test this hypothesis, we examined the effects of AG on the properties of the ventricular and arterial chambers and how well they were matched in fructose-fed (FF) rats, using the end-systolic pressure-stroke volume (Pes–SV) analysis (Chang et al., 2001).
In this study, rats given 10% fructose in drinking water were treated with AG (daily peritoneal injections of 50 mg kg−1) for 2 weeks and compared with the untreated FF group. Our data indicated that AG prevents not only the contractile dysfunction of the heart caused by fructose loading, but also the fructose-induced deterioration in the efficiency of mechanical energy transferred from the left ventricle to the arterial system.
Methods
Animals and catheterization
Male Wistar rats (2 months old) were randomly divided into four groups (n=10 in each group) as follows: (1) normal diet controls (NC); (2) FF rats; (3) NC treated with AG; (4) FF rats treated with AG. The FF rats were given a 10% fructose solution, which is equivalent to a diet containing 48–57% (by calories) fructose (Dai and McNeill, 1995), for 2 weeks and compared with the age-matched NC controls. Meanwhile, the FF rats were treated for 2 weeks with AG (Sigma Chemical Co., St Louis, MO, USA) and compared with the untreated FF group. The pharmacokinetic half-life of AG was 1.4 h (Nyengaard et al., 1997). It has been shown that the most effective regime to treat diabetic rats using AG was 50 mg kg−1 by daily peritoneal injection (Degenhardt et al., 1999). Hence, we adopted this regime to treat the FF rats. All animals were allowed free access to Rodent Diet 5001 (PMI Lab, St Louis, MO, USA) and water and housed two to three per cage in a 12-h light/dark cycle animal room. There was no significant difference in water consumption between control rats and animals treated with AG (18.5±0.3 vs 20.0±0.3 ml day−1, P>0.05). Plasma glucose concentrations were determined using a SURESTEP Test Strip (Lifescan Inc., Milpitas, CA, USA). The animal experiments were conducted according to the Guide for the Care and Use of Laboratory Animals, and were approved by the Animal Care and Use Committee of the National Taiwan University.
General surgical procedures and catheterization in rats have been described (Chang et al., 2001). In brief, animals were anesthetized with sodium pentobarbital (50 mg kg−1, intraperitoneally (i.p.)), placed on a heating pad, intubated, and ventilated with a rodent respirator (Model 131, New England Medical Instruments, Medway, MA, USA). The chest was opened through the second intercostal space of the right side. An electromagnetic flow probe (model 100 series, internal circumference 8 mm, Carolina Medical Electronics, King, NC, USA) was positioned around the ascending aorta to measure the pulsatile aortic flow. A high-fidelity pressure catheter (model SPC 320, size 2F, Millar Instruments, Houston, TX, USA) was used to measure the pulsatile LV pressure via isolated right carotid artery into the left ventricle. The electrocardiogram (ECG) of lead II was recorded with a Gould ECG/Biotach amplifier (Cleveland, OH, USA). The selective LV pressure and aortic flow signals of 5–10 beats were averaged in the time domain, using the peak R wave of ECG as a fiducial point (Figure 1). A single-beat estimation technique was used to evaluate the ventricular–arterial coupling without altering LV loads (Takeuchi et al., 1991; Chang et al., 2001).
Figure 1.

Ascending aortic flow (a), LV pressure (b) and ventricular and arterial Pes–SV relationships (c). (b) The dotted line represents the isovolumic pressure curve at an end-diastolic volume, which is estimated by fitting a sinusoidal function to the isovolumic portions of the measured LV pressure (Sunagawa et al., 1980). (c) Drawing a tangential line from Pisomax to the right corner of the pressure-ejected volume loop yields a point referred to as the end-systolic equilibrium point. The dotted line connecting Pisomax to the end-systolic equilibrium point constructs the ventricular Pes–SV relationship that has the slope of Ees and the volume intercept of Veed. On the other hand, the arterial Pes–SV relationship is the dashed line connecting the end-diastolic point to the end-systolic equilibrium point, with the slope of Ea. FF, fructose-feed rats; AG, aminoguanidine.
Coupling of the left ventricle and the arterial system
To quantitate the ventricular–arterial interaction, both the left ventricle and the arterial system are considered elastic chambers with known LV end-systolic elastance (Ees) and effective arterial volume elastance (Ea), respectively; Ees represents the LV end-systolic elastance and Ea represents the effective arterial elastance (Sunagawa et al., 1983, 1984, 1985). LV pressure and ascending aortic flow signals can be recorded to construct the ventricular and arterial Pes–SV relationships to calculate Ees and Ea, respectively. Ees can be determined by the ratio of peak isovolumic pressure (Pisomax) to the effective LV end-diastolic volume (Veed) (dotted line in Figure 1c). Pisomax could be obtained by making use of a nonlinear least-squares approximation technique proposed by Sunagawa et al. (1980) (dotted line in Figure 1b). On the other hand, dividing the LV Pes by the SV yields Ea (dashed line in Figure 1c). In the steady state, Ea is independent of Ees. The efficiency of energy transmission from the left ventricle to the arterial system, that is, the optimal afterload (Qload), can be determined from the ratio of stroke work to its theoretical maximal value and can be expressed using Ea/Ees as follows (Burkhoff and Sugawa, 1986; Kubota et al., 1992):
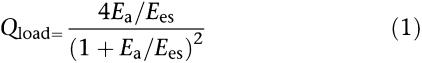 |
When Ea=Ees, Qload becomes unity and the arterial system extracts maximal energy from a given Ees and Veed.
Statistics
All data are expressed as means±s.e.m. Analysis of variance (ANOVA) was used to determine the statistical significance, whereas multiple comparisons were made for the effects of fructose and AG on the LV–arterial coupling. Significant differences were assumed at the level of P<0.05. If ANOVA for a hemodynamic variable reached the significant level, then Tukey's honestly significant difference method was used to determine the groups of rats having different mean values of the variable.
Results
Table 1 shows the effects of fructose and AG on body weight (BW), LV weight (LVW), blood glucose level, basal heart rate (HR), and cardiac output (CO). Neither fructose nor AG produced a significant difference in BW, LVW, and LVW/BW ratio, nor was there a (fructose – AG) interaction for those variables. Plasma glucose concentrations did not change significantly in FF animals, nor did they change in response to AG treatment. No interaction between the effects of fructose and AG in their effects on HR was detected in rats. By contrast, fructose loading exerted a decline in CO that was prevented by administration of AG to the FF rats.
Table 1.
Effects of fructose and aminoguanidine on body weight, LV weight, glucose level and basic hemodynamic data in male Wistar rats
| Variable | NC (n=10) | NC+AG (n=10) | FF (n=10) | FF+AG (n=10) |
|---|---|---|---|---|
| BW (g) | 325.3±9.2 | 320.0±5.1 | 312.6±6.0 | 315.7±6.4 |
| LVW (g) | 0.618±0.012 | 0.612±0.019 | 0.593±0.015 | 0.615±0.013 |
| LVW/BW | 0.189±0.003 | 0.191±0.004 | 0.190±0.002 | 0.195±0.003 |
| Glucose (mM) | 5.69±0.17 | 6.16±0.19 | 5.90±0.16 | 6.07±0.27 |
| HR (beat min−1) | 452.5±14.5 | 460.2±14.7 | 447.9±8.7 | 453.2±9.6 |
| CO (ml s−1) | 2.06±0.11 | 2.02±0.16 | 1.75±0.07* | 2.18±0.10† |
Abbreviations: AG, aminoguanidine; BW, body weight; CO, cardiac output; FF, fructose-feeding group; HR, basal heart rate; LVW, left ventricular weight; NC, normal controls.
Notes: All values are expressed as means±s.e.m.
Statistical difference (P<0.05) from the NC.
Statistical difference (P<0.05) from the FF.
Figure 2 demonstrates the effects of fructose and AG on the chamber properties of the left ventricle and the arterial system, which were derived from the LV pressure–SV plane. In comparison with the age-matched controls, the FF rats had decreased SV (Figure 2b) in the absence of any significant change in Pes (Figure 2a), which was responsible for a rise in Ea (Figure 2c). The increased Ea in rats fed with fructose was prevented by treatment with AG for 2 weeks, as evidenced by the fall of 15.2% in this variable (P<0.05). As for the chamber properties of the left ventricle, fructose loading exerted a significant decrease of 13.1% in Pisomax (Figure 2d) and an increase of 16.6% in Veed (Figure 2e), causing a marked fall in Ees (Figure 2f). After exposure to AG, the FF rats showed a significant decline in Veed and an increase in Pisomax, having a marked rise of 28.4% in Ees (P<0.01). In addition, the FF rats had increased Ea/Ees (Figure 3a) and diminished Qload (Figure 3b), when compared with the NC group. Treatment of the FF rats with AG for 2 weeks produced a significant fall of 36.1% in Ea/Ees, causing an increase of 5.7% in Qload (P<0.05).
Figure 2.

Effects of fructose and AG on the chamber properties of the arterial system and the left ventricle. Ea (c) and Ees (f) represented the elastic chamber properties of the arterial system and the left ventricle, respectively. Ea can be determined by the ratio of Pes to SV. On the other hand, Ees can be calculated from the ratio of Pisomax to Veed (see Methods). AG, aminoguanidine; FF, fructose-feeding rats; NC, normal controls.
Figure 3.

Effects of fructose and AG on the matching condition for the left ventricle coupled to the arterial system. Qload can be determined by the ratio of Ea to Ees. AG, aminoguanidine; FF, fructose-feeding rats; NC, normal controls.
Discussion and conclusions
The dietary intake of fructose in rats and humans may lead to significant elevations of the serum and tissue fructose levels (Levi and Werman, 1998; Kawasaki et al., 2002). Fructose as an effective glycating agent may play a significant role in the pathogenesis of AGE-derived abnormalities in myocardial and vascular chamber properties. Herein, we determined whether administration of AG to the rats fed with fructose produces a benefit on the coupling efficiency between the heart and the vasculature.
Effects of fructose and AG on the systolic mechanical behavior of the left ventricle
As mentioned earlier, the LV Ees could be determined by the ratio of Pisomax to Veed. Both the decreased Pisomax and the increased Veed are the major factors responsible for the diminished Ees in rats with fructose feeding. In this report, we measured LV intrinsic contractility in terms of Ees because of its independence of preload, afterload, and heart rate in a given constant contractile state of the ventricle (Suga et al., 1973; Sagawa, 1978, 1981). To our knowledge, this is the first report for assessment of the reduced contractility of the rat heart by fructose loading. In rats with experimental diabetes, Bidasee et al. (2003, 2004) showed that AGEs are formed on myocardial intracellular ryanodine receptors and sarcoplasmic reticulum Ca2+-ATPase during diabetes. The impairment of these two proteins by AGEs can diminish the amplitude of the Ca2+ transients, in that intracellular Ca2+ concentration decreases during systole and increases during diastole. These abnormalities in Ca2+ metabolism may contribute to a decrease in contractile status of the left ventricle. Thus, AGE formation by fructose loading, in myocardial cells may be responsible for the impaired chamber properties of the heart in this report. After exposure to AG, both the fructose-induced deterioration in Pisomax and Veed were markedly affected by the action of the drug, leading to a significant rise of 28.4% in Ees (Figure 2f). This suggests that AG may retard the impaired contractile status of the left ventricle that occurred in FF rats. However, the molecular basis for the prevention of fructose-derived AGEs in the myocardial proteins by AG remains to be determined.
Effects of fructose and AG on the chamber property of the arterial system
As mentioned earlier, Ea could be determined by the ratio of Pes to SV. Although there was a trend toward increasing Pes by fructose loading, no significant difference was observed between the FF rats and the NC controls. Obviously, a significant decrease in SV was the predominant factor responsible for the elevated Ea in animals with fructose feeding. The rise in Ea in the FF rats was prevented by the action of AG, as reflected in the decrease of 15.2% in this variable. This was in accord with the report from Lin et al. (2004) that AG retards the fructose-derived arterial stiffening through inhibition of AGE formation on collagens within the arterial walls. In addition, a beneficial effect of AG on resistance to blood flow in the absence of any significant changes in Pes (an approximation of mean arterial pressure) was observed when AG was administered to the FF rats (Table 1). This may result from inhibition of AGE formation by AG to preserve nitric oxide production from the endothelial cells. Thus, administration of AG to animals for 2 weeks may impart significant protection against the fructose-derived deterioration in the elastic chamber properties of the arterial system.
Effects of fructose and AG on the matching conditions for ventricular–arterial coupling
Equilibrium SV could be determined as the intersection between the ventricular and arterial Pes–SV relationships (Figure 1c). It has been shown that SV is directly proportional to Veed and is inversely related to Ea/Ees (Burkhoff and Sagawa, 1986). In the FF rats, the chamber properties of the left ventricle and the arterial system were abnormal, as evidenced by the decreased Ees and the increased Ea respectively, causing a significant rise in Ea/Ees ratio (Figure 3a). Meanwhile, ventricular dilatation developed in the FF animals, as defined by an increase in Veed. Thus, the fructose-induced fall in SV was attributed to the augmented Ea/Ees that prevails over the increased Veed.
The Qload has been used as a measure for the efficiency of energy transmission from the left ventricle to the arterial system (Sunagawa et al., 1985). As fructose loading worsened the mean value of Ea/Ees to a higher level (Figure 3a), the FF rats exhibited lower Qload than did the NC controls (Figure 3b). This suggests that the efficiency of mechanical energy transferred from the left ventricle to the vasculature may be diminished in rats with fructose feeding. To improve the matching conditions for LV–arterial coupling, administration of AG to the FF animals would be expected to diminish the discrepancy between Ea and Ees. As mentioned earlier, AG not only prevented the fructose-derived increase in Ea but also retarded the fructose-induced decrease in Ees. Thus, AG produced a significant fall of 36.1% in Ea/Ees, preventing the fructose-related decline in Qload (Figure 3b). These results suggest that treatment of the FF rats with AG for 2 weeks may optimize the matching conditions for the left ventricle to be coupled to its arterial system.
Limitations
Because Pes–SV analysis cannot be performed in conscious rats, it is difficult to evaluate the effects of pentobarbital anesthesia on the experimental animals. In this report, the results pertained only to measurements made in the open-chest rat with anesthesia.
In addition to being an AGE blocker, AG is an inhibitor of nitric oxide synthases (NOS): a potent inhibitor of the inducible isoform (iNOS); and a weaker inhibitor of endothelial isoform (eNOS) (Nilsson, 1999; Thornalley, 2003). AG may also reduce superoxide production by suppressing activation of vascular NADPH oxidase by cell-surface receptors for AGEs and by inhibition of uncoupled eNOS (Thornalley, 2003). Thus, AG might act as a protective agent in fructose-derived cardiovascular complications by both the effects mentioned to diminish peroxynitrite formation. Herein, we cannot reach any direct conclusions that AG exerts its effects only by inhibition of the AGE formation, without measuring its ability to suppress superoxide production, in FF rats.
Clinical trials of AG have been performed in diabetic patients to determine if there exists a beneficial effect on serious diabetic complications, via inhibition of AGE formation (Freedman et al., 1999; Bolton et al., 2004). Unfortunately, its applicability in the clinical setting is limited owing to rapid renal clearance and serious side effects, that is, induction of autoantibody formation, rapid progressive glomerulonephritis and anemia (Singh et al., 2001; Thornalley, 2003). However, pharmacological scavenging of dicarbonyls or stimulation of host dicarbonyl detoxification remains a worthy therapeutic strategy to prevent AGE-related disorders in the cardiovascular system (Thornalley, 2003).
Taken together, our data reveal that fructose loading diminished LV end-systolic elastance and augmented the effective Ea, causing a marked discrepancy between Ea and Ees. Thus, fructose loading worsened not only the contractile status of the left ventricle, but also the coupling efficiency between the heart and the vasculature. Treatment of the FF rats with AG for 2 weeks ameliorated the contractile status of the heart. Moreover, AG prevented the fructose-derived deterioration in the ventricular–arterial interaction with special reference to the energy transmission from the left ventricle to the arterial system.
Acknowledgments
This study was supported by grants from the National Taiwan University Hospital (NTUH 95-000 323) and from the National Science Council of Taiwan (NSC 95-2320-B-002-066).
Abbreviations
- AG
aminoguanidine
- AGEs
advanced glycation endproducts
- BW
body weight
- CO
cardiac output
- Ea
effective arterial volume elastance
- Ees
left ventricular end-systolic elastance
- HR
basal heart rate
- LVW
left ventricular weight
- Pes
end-systolic pressure of the left ventricle
- Pisomax
peak isovolumic pressure of the left ventricle
- Qload
optimal afterload
- SV
stroke volume
- Ved
end-diastolic volume of the left ventricle
- Veed
effective end-diastolic volume of the left ventricle
- V0
the zero-pressure volume
Conflict of interest
The authors state no conflict of interest.
References
- Bidasee KR, Nallani K, Yu Y, Cocklin RR, Zhang Y, Wang M, et al. Chronic diabetes increases advanced glycation end products on cardiac ryanodine receptors/calcium-release channels. Diabetes. 2003;52:1825–1836. doi: 10.2337/diabetes.52.7.1825. [DOI] [PubMed] [Google Scholar]
- Bidasee KR, Zhang Y, Shao ch, Wang M, Patel kp, Dincer ÜD, et al. Diabetes increases formation of advanced glycation end products on sarco(endo)plasmic reticulum Ca2+-ATPase. Diabetes. 2004;53:463–473. doi: 10.2337/diabetes.53.2.463. [DOI] [PubMed] [Google Scholar]
- Bolton WK, Cattran DC, Williams ME, Adler SG, Appel GB, Cartwright K, et al. Randomized trial of an inhibitor of formation of advanced glycation end products in diabetic nephropathy. Am J Nephrol. 2004;24:32–40. doi: 10.1159/000075627. [DOI] [PubMed] [Google Scholar]
- Brownlee M, Vlassara H, Kooney A, Ulrich P., Cerami A. Aminoguanidine prevents diabetes-induced arterial wall protein cross-linking. Science. 1986;232:1629–1632. doi: 10.1126/science.3487117. [DOI] [PubMed] [Google Scholar]
- Burkhoff D, Sagawa K. Ventricular efficiency predicted by an analytical model. Am J Physiol. 1986;250:R1021–R1027. doi: 10.1152/ajpregu.1986.250.6.R1021. [DOI] [PubMed] [Google Scholar]
- Chang KC, Su MJ, Peng YI, Shao CC, Wu YC, Tseng YZ. Mechanical effects of liriodenine on the left ventricular-arterial coupling in Wistar rats: pressure-stoke volume analysis. Br J Pharmacol. 2001;133:29–36. doi: 10.1038/sj.bjp.0704036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KC, Tseng CD, Wu MS, Liang JT, Tsai MS, Cho YL, et al. Aminoguanidine prevents arterial stiffening in a new rat model of type 2 diabetes. Eur J Clin Invest. 2006;36:528–535. doi: 10.1111/j.1365-2362.2006.01672.x. [DOI] [PubMed] [Google Scholar]
- Dai S, McNeill JH. Fructose-induced hypertension in rats is concentration- and duration-dependent. J Pharmacol Toxicol Methods. 1995;33:101–107. doi: 10.1016/1056-8719(94)00063-a. [DOI] [PubMed] [Google Scholar]
- Degenhardt TP, Fu MX, Voss E, Reiff K, Neidlein R, Strein K, et al. Aminoguanidine inhibits albuminuria, but not the formation of advanced glycation end-products in skin collagen of diabetic rats. Diabetes Res Clin Pract. 1999;43:81–89. doi: 10.1016/s0168-8227(98)00121-1. [DOI] [PubMed] [Google Scholar]
- Freedman BI, Wuerth JP, Cartwright K, Bain RP, Dippe S, Hershon K, et al. Design and baseline characteristics for the aminoguanidine clinical trial in overt type 2 diabetic nephropathy (ACTION II) Control Clin Trials. 1999;20:493–510. doi: 10.1016/s0197-2456(99)00024-0. [DOI] [PubMed] [Google Scholar]
- Huijberts MS, Wolffenbuttel BH, Boudier HA, Crijns FR, Kruseman AC, Poitevin P, et al. Aminoguanidine treatment increases elasticity and decreases fluid filtration of large arteries from diabetic rats. J Clin Invest. 1993;92:1407–1411. doi: 10.1172/JCI116716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki T, Akanuma H, Yamanouchi T. increased fructose concentrations in blood and urine in patients with diabetes. Diabetes Care. 2002;25:353–357. doi: 10.2337/diacare.25.2.353. [DOI] [PubMed] [Google Scholar]
- Kubota T, Alexander J, Jr, Itaya R, Todaka K, Sugimachi M, Sunagawa K, et al. Dynamic effects of carotid sinus baroreflex on ventriculoarterial coupling studied in anesthetized dogs. Circ Res. 1992;70:1044–1053. doi: 10.1161/01.res.70.5.1044. [DOI] [PubMed] [Google Scholar]
- Levi B, Werman MJ. Long-term fructose consumption accelerates glycation and several age-related variables in male rats. J Nutr. 1998;128:1442–1449. doi: 10.1093/jn/128.9.1442. [DOI] [PubMed] [Google Scholar]
- Lin YT, Tseng YZ, Chang KC. Aminoguanidine prevents fructose-induced arterial stiffening in Wistar rats: aortic impedance analysis. Exp Biol Med. 2004;229:1038–1045. doi: 10.1177/153537020422901008. [DOI] [PubMed] [Google Scholar]
- McPherson JD, Shilton BH, Walton DJ. Role of fructose in glycation and cross-linking of proteins. Biochemistry. 1988;27:1901–1907. doi: 10.1021/bi00406a016. [DOI] [PubMed] [Google Scholar]
- Nilsson BO. Biological effects of aminoguanidine: An update. Inflamm Res. 1999;48:509–515. doi: 10.1007/s000110050495. [DOI] [PubMed] [Google Scholar]
- Nyengaard JR, Chang K, Berhorst S, Reiser KM, Williamson JR, Tilton RG. Discordant effects of guanidines on renal structure and function and on regional vascular dysfunction and collagen changes in diabetic rats. Diabetes. 1997;46:94–106. doi: 10.2337/diab.46.1.94. [DOI] [PubMed] [Google Scholar]
- Sagawa K. The ventricular pressure-volume diagram revisited. Circ Res. 1978;43:677–687. doi: 10.1161/01.res.43.5.677. [DOI] [PubMed] [Google Scholar]
- Sagawa K. The end-systolic pressure-volume relation of the ventricle: definition, modifications, and clinical use. Circulation. 1981;63:1223–1227. doi: 10.1161/01.cir.63.6.1223. [DOI] [PubMed] [Google Scholar]
- Schalkwijk CG, Stehouwer CDA, van Hinsbergh vwm. Fructose-mediated non-enzymatic glycation: sweet coupling or bad modification. Diabetes Metab Res Rev. 2004;20:369–382. doi: 10.1002/dmrr.488. [DOI] [PubMed] [Google Scholar]
- Singh R, Barden A, Mori T, Beilin L. advanced glycation end-products: a review. Diabetologia. 2001;44:129–146. doi: 10.1007/s001250051591. [DOI] [PubMed] [Google Scholar]
- Suárez G, Rajaram R, Oronsky AL, Gawinowicz MA. Nonenzymatic glycation of bovine serum albumin by fructose (fructosylation): comparison with the Maillard reaction initiated by glucose. J Biol Chem. 1989;264:3674–3679. [PubMed] [Google Scholar]
- Suga H, Sagawa K, Shoukas AA. Load independence of the instantaneous pressure-volume ratio of the canine left ventricle and effects of epinephrine and heart rate on the ratio. Circ Res. 1973;32:314–322. doi: 10.1161/01.res.32.3.314. [DOI] [PubMed] [Google Scholar]
- Sunagawa K, Maughan WL, Burkhoff D, Sagawa K. Left ventricular interaction with arterial load studied in isolated canine ventricle. Am J Physiol. 1983;245:H773–H780. doi: 10.1152/ajpheart.1983.245.5.H773. [DOI] [PubMed] [Google Scholar]
- Sunagawa K, Maughan WL, Sagawa K. Optimal arterial resistance for the maximal stroke work studied in isolated canine left ventricle. Circ Res. 1985;56:586–595. doi: 10.1161/01.res.56.4.586. [DOI] [PubMed] [Google Scholar]
- Sunagawa K, Sagawa K, Maughan WL. Ventricular interaction with the loading system. Ann Biomed Eng. 1984;12:163–189. doi: 10.1007/BF02584229. [DOI] [PubMed] [Google Scholar]
- Sunagawa K, Yamada A, Senda Y, Kikuchi Y, Nakamura M, Shibahara T, et al. Estimation of the hydromotive source pressure from ejection beats of the left ventricle. IEEE Trans Biomed Eng. 1980;27:299–305. doi: 10.1109/TBME.1980.326737. [DOI] [PubMed] [Google Scholar]
- Takagi Y, Kashiwagi A, Tanaka Y, Asahina T, Kikkawa R, Shigeta Y. Significance of fructose-induced protein oxidation and formation of advanced glycation end product. J Diabetes Complicat. 1995;9:87–91. doi: 10.1016/1056-8727(94)00022-g. [DOI] [PubMed] [Google Scholar]
- Takeuchi M, Igarashi Y, Tomimoto S, Odake M, Hayashi T, Tsukamoto T, et al. Single-beat estimation of the slope of the end-systolic pressure-volume relation in the human left ventricle. Circulation. 1991;83:202–212. doi: 10.1161/01.cir.83.1.202. [DOI] [PubMed] [Google Scholar]
- Thornalley PJ. Use of aminoguanidine(Pimagedine) to prevent the formation of advanced glycation endproducts. Arch Biochem Biophys. 2003;419:31–40. doi: 10.1016/j.abb.2003.08.013. [DOI] [PubMed] [Google Scholar]