

Abstract
Therapeutic drug monitoring is not routinely used for cytotoxic agents. There are several reasons, but one major drawback is the lack of established therapeutic concentration ranges. Combination chemotherapy makes the establishment of therapeutic ranges for individual drugs difficult, the concentration-effect relationship for a single drug may not be the same as that when the drug is used in a drug combination. Pharmacokinetic optimization protocols for many classes of cytotoxic compounds exist in specialized centres, and some of these protocols are now part of large multicentre trials. Nonetheless, methotrexate is the only agent which is routinely monitored in most treatment centres. An additional factor, especially in antimetabolite therapy, is the existence of pharmacogenetic enzymes which play a major role in drug metabolism. Monitoring of therapy could include assay of phenotypic enzyme activities or genotype in addition to, or instead of, the more traditional measurement of parent drug or drug metabolites. The cytotoxic activities of mercaptopurine and fluorouracil are regulated by thiopurine methyltransferase (TPMT) and dihydropyrimidine dehydrogenase (DPD), respectively. Lack of TPMT functional activity produces life-threatening mercaptopurine myelotoxicity. Very low DPD activity reduces fluorouracil breakdown producing severe cytotoxicity. These pharmacogenetic enzymes can influence the bioavailability, pharmacokinetics, toxicity and efficacy of their substrate drugs.
Keywords: antimetabolites, cytosine arabinoside, cytotoxic drugs, fluorouracil, mercaptopurine, methotrexate, therapeutic drug monitoring
Introduction
Drugs used to treat cancer inhibit the mechanisms of cell proliferation. Alkylating agents (e.g. cyclophosphamide, busulfan, carboplatin) readily form covalent bonds with the bases in DNA and so prevent DNA replication by the introduction of crosslinks in the double helix. Anticancer antibiotics (e.g. daunorubicin) intercalate between the DNA base pairs. Intercalation stabilizes the DNA-topoisomerase II complex and stops the reversible ‘swivelling’ at the DNA replication fork which is required for effective replication of the DNA template. The epipodophyllotoxins (e.g. etoposide, teniposide) also act on DNA, topoisomerase II inducing DNA strand breaks. The steroid hormones (e.g. prednisolone), via receptor binding, interfere with DNA synthesis and alter intracellular metabolism. The vinca alkaloids (e.g. vincristine) prevent the formation of the mitotic spindle, whilst the antimetabolites (e.g. methotrexate, 6-mercaptopurine) directly interfere with DNA formation by inhibiting purine and pyrimidine biosynthesis [1].
Cancer chemotherapy is in the main empirical with the majority of cytotoxics given at a fixed dosage based either on body surface area or weight. These compounds have a narrow therapeutic index, and in general they lack a simple index of pharmacological effect with which to monitor treatment. Cytotoxic drugs are therefore ideal candidates for the development of therapeutic drug monitoring (TDM) programmes, but many difficulties have hindered the development of such programmes for anticancer drugs. These include not only the lack of a clear relationship between drug dose and therapeutic effect but also extensive drug metabolism and arduous assay techniques. In addition, any one centre treats a relatively small number of patients in what are usually combination chemotherapy regimens. There are many treatment variables which may confound the interpretation of data and make it difficult to establish therapeutic ranges for any individual compound. For an effective TDM programme an established therapeutic range is required.
In cancer chemotherapy there can be a significant time lag between the measurement of drug in biological tissues and the ultimate end point of efficacy, whether that be cure or recurrence of active disease. Several studies have established a relationship between plasma drug concentrations and dose-limiting toxicities for a number of cytotoxic drugs, e.g. busulfan [2, 3], carboplatin [4, 5] and etoposide [6, 7]. Although relationships between systemic exposure and overall therapeutic effect have been reported [2, 8] there are very few prospective randomised trials designed to determine whether therapeutic intervention based on a derived therapeutic range results in a better clinical outcome [9].
Maximal efficacy of anticancer treatment is of prime importance because of the potential repercussions in cancer patients, particularly those with drug responsive cancers. With modern treatment 95% of children with acute lymphoblastic leukaemia (ALL) will achieve disease remission and more than 70% can look forward to long-term leukaemia-free survival. The vast majority of children who are cured are those in whom a first remission is achieved and maintained for 6–8 years [10]. If first remission drug treatment fails the chances of long-term disease-free survival are poor [10, 11].
The importance of antimetabolite therapy to the ALL chemotherapeutic regimens is illustrated by the results of the UK nationwide trial ALL VIII [12]. Before 1980 the 4 year disease-free survival for ALL in Britain was less than 50%. A more detailed and rigid nationwide protocol was introduced (UKALL VIII) for the existing antimetabolite therapy which resulted in an improvement in long-term survival by 15–20% [12]. The existence of such a nationwide protocol aided the development of a TDM programme within these trials [8, 13]. This review will focus on antimetabolite chemotherapeutic agents, drugs for which a number of studies have established relationships between the drug, or its active metabolite, and response to therapy.
The drugs
The antimetabolite drugs mercaptopurine (MP), methotrexate (MTX) and cytosine arabinoside (Ara-C) play an important role in all major therapeutic protocols for the treatment of leukaemia. Fluorouracil (FU) is used extensively in the treatment of a number of solid tumours including colorectal, breast and head and neck cancers. Antimetabolites are structurally similar to endogenous compounds, MTX is a folate analogue, FU and Ara-C are pyrimidines, and MP a purine. Antimetabolites are metabolized like their normal analogues [14] and so interfere with the synthesis of nucleic acids or are incorporated into DNA as false bases. All antimetabolites are metabolized to intracellular active metabolites which are retained within the cell. MTX is polyglutamated whilst the purine and pyrimidine analogues are anabolized to nucleotides (Table 1). MTX is the only antimetabolite whose dosage is regularly individualized based on TDM, but pharmacokinetic optimization protocols for the purine and pyrimidine antimetabolites exist in specialized centres and such protocols are now part of large muticentre trials. The identification of slow and fast metabolic phenotypes, which have been described for MP,FU and Ara-C, has aided the development of TDM programmes, and these nucleotide antimetabolites are now potential candidates for routine TDM.
Table 1.
Antimetabolite drugs: mode of action.

Mercaptopurine (MP)
What to measure
MP is an extensively metabolized prodrug. There are three major metabolic routes; oxidation, phosphoribosylation and S-methylation (Figure 1) [15]. Oxidation of the thiopurine ring, catalysed by xanthine oxidase, is a major catabolic route producing the 6-thio analogue of uric acid [14]. This enzyme activity varies from tissue to tissue, it is highest in the intestine and liver but absent from circulating blood cells [16]. Population studies have shown a four-to 10-fold interindividual variation in xanthine oxidase activity [17, 18]. The influence of interindividual variations in xanthine oxidase activity on the outcome of thiopurine therapy has yet to be clearly defined, but we do know that decreased amounts of xanthine oxidase potentiate the cytotoxic effects of MP, as illustrated by the coadministration of the xanthine oxidase inhibitor allopurinol [15, 19].
Figure 1.
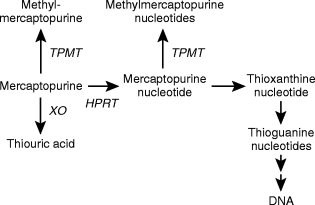
The intracellular metabolism of mercaptopurine. The principle metabolic routes are catalysed by thiopurine methyltransferase (TPMT), xanthine oxidase (XO) and hypoxanthine phosphoribosyltransferaase (HPRT).
The purine ‘salvage’ enzyme hypoxanthine phosphoribosyltransferase (HPRT) catalyses the initial step in the biological activation of MP. The formation of intracellular thionucleotide metabolites is essential for thiopurine cytotoxicity [20]. MP eventually forms 6-thioguanine nucleotides (TGNs), but a variety of intermediary thionucleotides are formed en-route to the TGNs (Figure 1). These fraudulent nucleotides can potentially produce their cytotoxic and immunosuppressive actions by many different mechanisms. Cytotoxicity has been directly linked to the incorporation of drug derived TGNs into DNA [21, 22], but in addition some thionucleotide metabolites can inhibit de novo purine synthesis [13, 23].
S-methylation of the thiopurine is catalysed by thiopurine methyltransferase (TPMT) an enzyme which exhibits a wide interindividual variation. The genetic polymorphism that controls red blood cell (RBC) TPMT activity controls the enzyme activity in all other cells and tissues, including the lymphoblast [24–26]. In a Caucasian population 89% of individuals inherit high activity (two wild type alleles), 11%‘intermediate’ (one wild type and one variant allele) and 1 in 300 have no functional TPMT activity (two variant alleles) [24, 27]. MP and its initial nucleotide are both methylated by TPMT and the resultant methylated compounds are formed at the expense of TGNs. Because a number of centres have reported correlations between TPMT activity and the response to both MP and the immunosuppressive agent azathioprine (a MP pro-drug) [28–30], and a lack of functional TPMT activity is associated with profound, life threatening myelosuppression [28, 30–32], a knowledge of basal TPMT activity has been suggested prior to drug therapy [33, 34]. Both TPMT phenotypic activity and TGN metabolite concentrations can be conveniently measured using the RBC as a surrogate tissue [35–37], (Figure 2).
Figure 2.
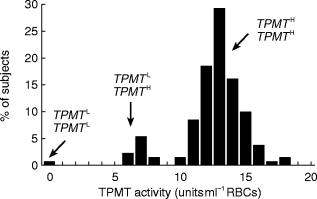
Thiopurine methyltransferase (TPMT) genetic polymorphism. Frequency distribution histogram of TPMT activity in 130 unrelated control children. Presumed genotypes at the locus TPMT are shown. Leukaemic children who lack functional TPMT activity (extreme left of histogram) are acutely sensitive to MP ]30, 31[, whilst children with very high TPMT activities (extreme right of histogram) do not respond to standard dosages of MP ]28, 54[. Redrawn from ]28[.
Methods of analysis
The available assays for the quantification of parent drug or intracellular thionucleotide metabolites are based on h.p.l.c. technology, and the biological tissues sampled include plasma [38, 39], urine [40], red blood cells [35], neutrophil granulocytes and lymphocytes [41]. The lower limits of detection are 30 pmol ml−1 biological fluid (30 nm), and 5–30 pmol 8×10−8 cells. The major intracellular metabolites monitored by reverse-phase h.p.l.c. with u.v. detection and isocratic gradient elution are the TGNs and methyl-MP nucleotides [35, 41]. A common feature of many published methods is the hydrolysis of the thionucleotide back to the parent thiopurine prior to h.p.l.c. quantification. The specific thionucleotide metabolites can be measured with gradient elution from an anion-exchange column [42]. Simple reverse-phase h.p.l.c. methodology is used for the measurement of plasma MP concentrations and these assays have been used in routine analysis [43, 44]. The major catabolite 6-thiouric acid can also be measured in plasma [45], but the lack of commercially available 6-thiouric acid has perhaps hindered the development of routine assays. The clinical use of 6-thiouric acid monitoring remains unproven, but in theory urine concentrations could be used as a compliance indicator.
TPMT phenotype is measured in the RBC using radiochemical [37, 46] or h.p.l.c. assays [36, 47], Measurement of TPMT activity has become a standard clinical test in some centres [48]. The assay for drug metabolites or enzyme activities requires 100 μl packed RBCs or RBC lysate, respectively. TPMT genotype can also be defined for some individuals, and a number of variant alleles for low TPMT activity have been defined [33, 49, 50]. But the most common variant allele in 283 clinical laboratory specimens from the USA (TPMT3 A, 55% of variant alleles) was absent in a Korean population [33].
Therapeutic drug monitoring
In the continuing chemotherapy of childhood ALL MP plasma concentrations have been suggested as a method of monitoring systemic exposure to MP [43, 44]. MP urine concentrations have been correlated with MP plasma AUC, and have been suggested as a possible alternative to repeat blood sampling [40]. The MP plasma profile differs in the same child when studied on repeated occasions with the same dose of drug (75 mg m−2), nonetheless MP plasma concentrations show a relationship to the response to treatment [38, 43]. In children sampled on repeated occasions throughout continuing chemotherapy, disease relapse was associated with mean peak plasma levels below 135 ng ml−1 and a mean AUC value below 251 ng ml−1 h, whilst myelotoxicity was associated with mean AUC values above 363 ng ml−1 h and mean peak plasma levels above 166 ng ml−1 [43]. There was no overlap in mean MP pharmacokinetic parameters between those children who suffered disease relapse and those who experienced severe myelotoxicity.
Pharmacokinetic-pharmacodynamic correlations have been made measuring RBC TGNs [8], and RBC drug metabolites can be used to monitor the efficacy of therapy and compliance with oral chemotherapy in childhood ALL [51]. RBC TGNs can be used as a measure of the systemic exposure to MP derived active metabolites, and the degree of drug induced myelosuppression can be related to TGN concentrations [28, 41, 52]. In continuing chemotherapy protocols for childhood ALL, which consist of prolonged periods of oral antimetabolite therapy, leukaemia-free survival is significantly associated with RBC TGN concentrations independently of other prognostic factors [8]. In a group of children with ALL taking the protocol standard dose of MP (75 mg m−2), the population median TGN concentration was 275 pmol 8×10−8 RBCs. The recurrence of active disease was associated with the formation of TGN concentrations below 275 pmol, and this subgroup of children had significantly higher activities of the pharmacogenetic enzyme TPMT [28]. In such antimetabolite based protocols the product of RBC TGNs and MTX polyglutamate concentrations has also been linked to treatment failure [53]. Those children who accumulated mean RBC MTX.TGN concentrations <813 (nmol/mmol haemoglobin)2 had a significantly poorer event-free survival [53]. Unfortunately, TGN concentrations per number of RBCs [8, 28] and the product of TGN and MTX concentrations per mmol of haemoglobin [53] cannot be directly compared.
Monitoring benefits
In MP chemotherapy efficacy and toxicity are interrelated; myelosuppression is required for a successful anticancer effect [28]. The aim of treatment with thiopurine drugs in the UK ALL protocols is controlled myelosuppression, and during continuing chemotherapy myelosuppression is used as a surrogate measure of response to thiopurine therapy. TPMT activities measured at disease diagnosis show a significant negative relationship to the duration of cytopenia driven MP withdrawal during continuing therapy [54]. Those children with very high TPMT activities fail to respond to standard MP dosages, and do not experience cytotoxicity [28, 54]. The TPMT deficient individual produces grossly elevated RBC TGN concentrations and experiences profound myelosuppression at standard MP dosages (Figure 2) [29–32]. Enzyme deficiency can be detected by phenotypic TPMT activity [36, 37] or genotype [33, 50]. Alternatively, the continued accumulation of elevated TGNs could be detected after a test dose of MP [55].
Approximately 400 children per year enter UK ALL protocols which contain 2 years continuing therapy, one or two will be TPMT deficient. Such children can be managed on MP by the use of attenuated dosage regimens, e.g. 7.5 mg m−2 daily or on alternate days [30, 31, 55]. If thiopurine therapy is to be individualized then routine measurement of TPMT activity must be added to the list of agents that show a concentration-effect relationship. A knowledge of pretreatment TPMT phenotype would identify both those individuals who would be unduly sensitive and those ‘resistant’ to thiopurine standard dosages. For the latter group, awareness of drug tolerance would encourage titration to toxicity, and for the former the use of attenuated dosages.
Drug interactions
Inhibition of MP oxidation or S-methylation by concurrent drug therapy will potentiate thiopurine cytotoxicity. Allopurinol is a well documented inhibitor of xanthine oxidase catalysed MP oxidation [19]. With the 6MP pro-drug azathioprine it is recommended that, during concurrent allopurinol therapy the azathioprine dose is reduced by two-thirds. This decreases, but does not abolish, the risk of myelotoxicity [56]. However, no reduction in the dosage of the MP sister compound thioguanine (2-amino MP) is required with allopurinol coadministration [19]. Thioguanine is not a direct substrate for xanthine oxidase; it requires an intermediate metabolic step (guanase catalysed 2-deamination).
A number of compounds which could be coadministered with thiopurine drugs can potentially influence TPMT activity. After a therapeutic dose of aspirin the plasma concentrations of salicylic acid are within the range for TPMT inhibition [57]. Sulphasalazine and its metabolite 5-aminosalicylic acid inhibit TPMT. In a large long-term clinical study of inflammatory bowel disease MP was withdrawn in 10% of patients because of the occurrence of adverse reactions. Over half the patients in that study were treated simultaneously with MP and sulphasalazine [58]. A possible drug–drug interaction was reported in a patient with refractory Crohn’s disease who developed bone marrow suppression whilst receiving daily MP and olsalazine (two molecules of 5-aminosalicylic acid) [59]. Concurrent frusemide therapy could influence the S-methylation of thiopurines as frusemide inhibits TPMT with an IC50 of 170 μm [60], a concentration within the therapeutic range for frusemide. In addition, TPMT could interfere with disulfiram treatment in alcoholism [61]. TPMT S-methylates the diethyldithiocarbamate metabolite involved in disulfiram activation.
The MP prodrug azathioprine induces warfarin resistance, and impairs the anticoagulant effect of other vitamin-K antagonists [62, 63]. The discontinuation of azathioprine in patients concomitantly treated with oral anticoagulants may increase the risk of severe bleeding. The mechanism behind this interaction has not been established, but it could be due to a MP mediated increase in prothrombin, although MP effects on warfarin absorption and metabolism cannot be ruled out [63].
Methotrexate (MTX)
What to measure
MTX exerts its cytotoxic effects by competitively inhibiting dihydrofolate reductase (DHFR), the enzyme responsible for converting folates to tetrahydrofolate, the reduced folate carriers which function in the transfer of carbon units. These carbon units are required for de novo purine synthesis and the methylation of uracil to thymine in DNA synthesis. To avoid excessive destruction of host cells, leucovorin (a folate analogue) is used to rescue cells from MTX inhibition. Leucovorin is a synthetic substrate for DHFR and allows the resumption of thymine formation and the reinitiation of DNA synthesis.
There are two major routes of MTX metabolism, 7-hydroxylation and polyglutamation, (Figure 3) [64, 65]. Aldehyde oxidase catalysed 7-hydroxylation produces 7-hydroxy MTX, a less effective inhibitor of DHFR but the metabolite responsible for the nephrotoxicity which occurs during high dose MTX therapy [66]. Both MTX and 7-hydroxy MTX undergo intracellular polyglutamate formation, a metabolic route which allows selective retention of the antifolate within the cell [65]. Polyglutamation is an important determinant of toxicity [67], and these metabolites are as potent as MTX as DHFR inhibitors [64]. The accumulation of high levels of MTX polyglutamates in malignant hyperdiploid cells vs diploid cells may be related to the more favourable prognosis that hyperdiploidy confers in B-lineage ALL [67].
Figure 3.
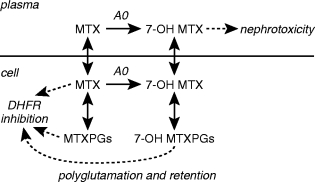
Methotrexate (MTX) metabolism. Oxidation to 7-hydroxy MTX (7-OH MTX) is catalysed by aldehyde oxidase (AO). MTX, MTX polyglutamates (MTXPGs), and 7-OH MTX polyglutamates (7-OH MTXPGs) inhibit dihydrofolate reductase (DHFR).
Evidence also suggests that it is the differential accumulation of MTX polyglutamates in malignant cells vs the rapidly dividing normal cell which makes MTX an effective anticancer agent, and partially accounts for the selectivity of leucovorin rescue for normal cells [68]. For example, normal rapidly dividing epithelial tissue does not accumulate intracellular MTX polyglutamates. The epithelial cell possesses a hydrolyase which swiftly degrades these polyglutamates and the MTX produced is extruded from the cell by carrier mediated transport [68].
Methods of analysis
MTX can be measured in biological fluids by a variety of techniques [68]. A number of commercial kits are available for MTX quantification and they can all measure MTX in plasma, serum, urine and CSF. These kits are based on radioimmune, radioenzymic, enzyme immuno-[69] and fluorescence polarization immuno-[70] assay. Both the fluorescence polarization (TDx) and enzyme immunoassays (EMIT) have the advantage of using similar technology to other assays in routine clinical use. They are fully automated, use small sample volumes (50 μl serum), and have lower limits of 0.05 μm and 0.1 μm plasma, respectively. Time, a consequence of the more manual technology, and larger sample volumes (300 μl plasma) are drawbacks for both the radioenzymic and radioimmune assays, but they both have a lower limit of detection (0.01 μm plasma). The lowest sensitivity (1 nm) is offered by a labour intensive radio-ligand binding assay [71]. To quantify MTX metabolites or to achieve the specificity coupled with sensitivity required for TDM in low dose oral therapy, high performance liquid chromatography (h.p.l.c.) is the preferred assay technique. Many methods have been published [72–76] with lower limits of ≈0.02–0.03 μm, but none offers the one step, fully automated advantages of EMIT. H.p.l.c. has been coupled with a radioenzymic assay [77] to quantify intracellular MTX polyglutamate metabolites in target tissues, this combination of techniques couples the selectivity of h.p.l.c. with the speed of quantitating the separated polyglutamates by radio assay.
Therapeutic drug monitoring
MTX is a nonspecific cytotoxic and prolongation of blood levels appropriate to killing tumour cells may lead to severe unwanted side-effects such as myelosuppression, gastrointestinal mucositosis and hepatic cirrhosis. The aim of high dose MTX therapy is to keep plasma MTX levels high (10−4–10−5 m) for prolonged periods (12–36 h) [65]. TDM forms the basis for decisions for the timing of initiation and continuation of leucovorin rescue in high-dose MTX schedules, and a wide variety of dosage schedules exist [65]. The optimum period for measuring plasma MTX concentrations and determining the risk of toxicity is between 24 and 48 h after the initiation of MTX [68]. Over the 24–48 h interval MTX plasma levels are generally comparable for patients receiving different MTX dosage protocols at ≈10−6 m and 1−6 m at 24 and 48 h, respectively [68]. MTX levels above 5×10−7 m at 48 h require additional rescue, e.g. 1×10−6 m MTX requires 15 mg m−2 leucovorin every 6 h×8 doses [65]. MTX concentrations are measured at 48 h intervals thereafter with the leucovorin dose adjusted until MTX falls below 5×10−8 m [65]. Guidelines for adjusting leucovorin rescue are discussed in Evans et al. 1992 [68]. In the treatment of childhood ALL adjusting the MTX dose to account for the child’s ability to clear the drug improves outcome without increasing toxicity [9]. Five cycles of high dose MTX (initially 1500 mg m−2 over 24 h), with leucovorin rescue at 48 h after the start of the infusion, are given over the first year of therapy. In the first cycle MTX clearance is estimated on the basis of MTX plasma concentrations at 1 and 6 h from the start of the infusion, with dose adjustment at 8 h to achieve the target range of exposure (580–950 μm MTX). This is repeated for each MTX cycle [9].
The clinical pharmacological studies of MTX have been expanded to include the formation of MTX active metabolites in target tissues. Blast cell concentrations of long-chain MTX polyglutamates (MTXPGs) were studied during single agent ‘window’ therapy immediately prior to conventional remission induction. Higher MTXPGs within leukaemic blasts (>500 pmol 10−9 blasts) equated with a greater antileukaemic response and these high metabolite concentrations were formed more readily from HDMTX [77]. There is no practical method by which intracellular MTX concentrations can be routinely measured in target cells in clinical specimens [68].
Drug interactions
The purine catabolic enzyme xanthine oxidase can also be inhibited by folate compounds, including methotrexate. Both MP and MTX are used together in the prolonged continuing chemotherapy phase of treatment for childhood ALL. Concurrent MTX results in a significant 26% increase in MP plasma levels. In light of the wide intrapatient variability in MP plasma levels at constant dosages this statistically significant interaction was thought not to be of clinical importance at low oral MTX dosages [78]. In addition, MTX inhibits folate dependent enzymes involved in de novo purine synthesis. The reduction in supply of endogenous substrates promotes the use of the competing thiopurine MP in many biochemical reactions, and increases the formation of intracellular thionucleotide metabolites [79]. Thus, low dose MTX may promote the formation of TGNs from MP.
Fluorouracil (FU)
What to measure
FU is metabolized intracellularly to a nucleotide metabolite (fluorodeoxyuridine monophosphate, FdUMP) which inhibits thymidylate synthesis. FU undergoes rapid hepatic metabolism principally via dihydropyridine dehydrogenase (DPD). DPD is the first, and rate limiting enzyme in pyrimidine catabolism and is responsible for the degradation of >80% of administered FU [80]. DPD activities and plasma FU concentrations have been investigated as TDM tools [81, 82]. FU metabolism is summarized in Figure 4.
Figure 4.
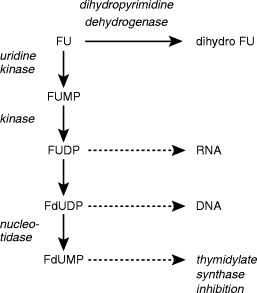
The intracellular metabolism of fluorouracil (FU). FU can either be catabolized by dihydropyrimidine dehydrogenase (DPD) to form dihydro FU or phosphoribosylated to form active nucleotide metabolites. The nucleotide metabolites depicted above are fluorouridine monophosphate and diphosphate, and fluoro-deoxyuridine diphosphate and monophosphate (FUMP, FUDP, FdUDP, FdUMP, respectively). Intracellular metabolism of FU and its nucleotides is analogous to uracil metabolism ]14[. FU is converted to FUMP via 5′-fluorouridine in a two step reaction catalysed by nucleoside phosphorylase and uridine kinase. It is possible that FUMP could be formed directly from the FU base via pyrimidine phosphoribosyltransferase. Phosphorylation of FUMP proceeds by nucleoside monophosphate kinase (1). Nucleoside 5′-diphosphate reductase (ribonucleotide reductase) catalyses the production of 2′-deoxribonucleotide (FdUDP) from the ribonucleotide (FUMP). Phosphate is removed from nucleotides by 5′-nucleotidases (2).
Methods of analysis
Plasma concentrations of FU, quantified by h.p.l.c., have been used to predict FU toxicity [82]. FU plasma concentrations after continuous i.v. infusion are in the μg 1−1 range in patient plasma. This is easily measured by reverse-phase h.p.l.c. with u.v. detection [83], the initial published assay having a lower limit of 23 μg l−1 plasma. H.p.l.c. technology offers lower limits of detection of 1 ng using precolumn fluorescence derivatization [84]. This methodology has been used for the measurement of FU in scalp hair as an index of patient compliance with oral adjuvant therapy (200 mg FU day−1). The lower limits of detection are 0.006 ng FU per patient hair strand or 0.01 p.p.m. [85]. In vivo fluorine-19 nuclear magnetic resonance spectroscopy (19F NMRS) has been used to estimate FU localization and metabolism inside human tumours after i.v. bolus doses of 600 mg m2 [86]. FU ‘trapping’ inside tumours can be related to clinical response [86], and 19F NMRS offers a noninvasive investigational route albeit one confined to a number of specialist centres.
Tissue FU concentrations are regulated by DPD catalysed breakdown. DPD activity is measured in peripheral blood mononuclear cells [80], and this has been developed into a sensitive, accurate and precise assay for large scale screening in cancer patients. The mononuclear cells are isolated by standard techniques, if not used immediately for the assay of DPD activity they can be stored at −70° C in foetal calf serum [80]. Enzyme activity is determined by the use of tritiated FU in a radioassay, and the FU catabolites formed are quantified by reverse-phase h.p.l.c. [87]. Plasma uracil concentrations, which are markedly elevated in DPD deficiency, have been used as a clinical marker for the deficiency syndrome [88].
Therapeutic drug monitoring
For the same FU dosage plasma drug concentrations show a large interpatient variability [82]. Response rate and patient survival are correlated with FU plasma concentrations but not FU dose [89]. FU area under the plasma concentration curve (AUC) for a whole 5 day cycle or for half a cycle can be used to predict drug cytotoxicity, and nomograms have been developed to adjust FU dosage based on its plasma concentration [82]. Comparing a standard dosage group (1000 mg m−2 24 h−1 continuous i.v. infusion) against a treatment monitored group in which the FU dosage for the second part of the cycle (3–5 days) was adjusted in the light of the AUC (0, 3 days), the incidence of moderate to severe toxicity (e.g. FU induced stomatitis) was significantly reduced in the monitored group. Clinical response (complete remission) was significantly higher in the treatment monitored group [82].
DPD activity can regulate the amount of FU available for active metabolite formation and is correlated with FU pharmacokinetics [81, 82]. PBMC-DPD deficiency is associated with minimal FU catabolism and severe FU cytotoxicity [80]. In subjects with normal PBMC-DPD activity the t1/2 of FU is 6–13 min and ≈10% of the dose is excreted in the urine. In subjects with DPD deficiency the t1/2 increases to 159 min and up to 90% of the dose is excreted unchanged [88]. In deficient patients low PBMC-DPD activity correlates with liver DPD activity [80]. In patients with head and neck cancer those showing a complete response to FU therapy had lower tumour DPD activities as compared with partial or nonresponding patients, thus tumour DPD activity is a determining factor for FU responsiveness in cancer patients [90]. Clinical studies have shown a significant relationship between peripheral blood mononuclear cell (PBMC) DPD activity and FU clearance, but this weak correlation is not sufficiently accurate to predict FU toxicity in head and neck cancer patients [90]. A pharmacologically based FU dose adaptation is therefore recommended rather than dose adjustment based on at diagnosis PBMC-DPD activity [90].
Interpretive problems
There is a circadian rhythm in PBMC-DPD activity. Initial studies indicated that there is a 24 h periodicity with peak activities occurring, on average, at 1.00 h [81]. Further studies have indicated that the time of day that peak PBMC-DPD activity occurs varies between subjects and is essentially randomly distributed over 24 h [91], with a five-fold difference between peak and trough enzyme activities (21 vs 4 pmol min−1 per 106 cells, respectively). Such variations in DPD activity could influence FU tolerance over time [91].
Drug interactions
Inhibition of DPD may enhance the therapeutic effect of FU [92], and one potent DPD inhibitor, 5-ethynyluracil, is currently in clinical trials in combination with low dose FU [93, 94]. In Japanese patients fatal potentiation of FU toxicity occurred after a few days of dosing with the antiviral agent sorivudine. Bromovinyluracil, a major sorivudine metabolite, inhibits DPD [92]. This drug–drug interaction emphasizes the influence of DPD on the therapeutic effect of FU and it has recently been extensively reviewed [95].
Cytosine arabinoside (Ara-C)
What to measure
Ara-C cytotoxicity is dependent upon intracellular phosphorylation, via cytidine kinase, to its active metabolite cytosine arabinoside triphosphate (Ara-CTP), the nucleotide responsible for the inhibition of DNA polymerase (Figure 5). Intracellular Ara-CTP retention time correlates with the likelihood of achieving complete remission after high dose Ara-C therapy [96]. Ara-C is inactivated to uridine arabinoside (Ara-U) by cytidine deaminase. The plasma ratio of Ara-U/Ara-C has been used to analyse the phenotype for Ara-C deamination [97].
Figure 5.
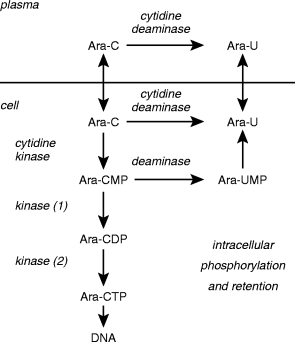
Cytosine arabinoside (Ara-C) metabolism. Deamination to uracil arabinoside (Ara-U) and the formation of the intracellular nucleotides cytosine arabinoside mono-, di-and tri-phosphates and arabinofuranosyl-uridine monophosphate (Ara-CMP, Ara-CDP, Ara-CTP and Ara-UMP, respectively), analogues of the endogenous deoxynucleotides. The deamination of Ara-C is catalysed by (deoxy)cytidine deaminase and Ara-CMP by dCMP deaminase. The formation of Ara-CMP is catalysed by (deoxy)cytidine kinase. Subsequent phosphorylation of the monophosphate proceeds by nucleoside monophosphate kinase (1) and nucleoside diphosphate kinase (2).
Methods of analysis
Assays have been published for the measurement of Ara-C and Ara-CTP concentrations in both leukaemic blasts and normal mononuclear blood cells [98]. H.p.l.c. techniques are used to measure Ara-C and Ara-U plasma concentrations, with a lower limit of detection of 10 ng ml−1 Ara-C and leukaemic blast/mononuclear cell Ara-CTP levels with a lower limit of 5 pmol on the column [98, 99]. Ion-pair h.p.l.c., coupled with solid phase scintillation detection, has been used as a rapid assay technique for the determination of ]3H[-Ara-C and its major metabolites including Ara-U, Ara-UTP and Ara-CTP. The absolute, on column, limit of detection ranges from 40 to 200 pg [100]. Quantification of Ara-C incorporation into leukaemic cell DNA [101] has been described, the assay involves breakdown of DNA by enzymatic digestion and radioimmunoassay of Ara-C. The DNA (25–100 μg) is dissolved in tris buffer to give a sensitivity of 2 pmol ml−1 Ara-C (≈0.03 pmol Ara-C/μg DNA). Ara-CTP is the only intracellular active metabolite that can be measured in vivo after Ara-C administration, and intracellular Ara-CTP pharmacokinetics have been used to optimize Ara-C therapy. The clinical use of Ara-C/DNA levels remains to be evaluated [101].
Therapeutic drug monitoring
Ara-C is given parenterally not orally because on absorption it would be rapidly deaminated to its inactive metabolite Ara-U by the intestinal mucosa [102]. The plasma ratio of Ara-U/Ara-C has been used to analyse the phenotype for Ara-C deamination, slow (ratio <14) and fast (ratio >14) deaminators exist [97]. In 56 subjects the ratio of slow:fast deaminators of Ara-C was 70:30%. In a study of leukaemia patients those showing complete or partial remission were slow deaminators [97].
Plasma Ara-C pharmacokinetics are unable to predict the cellular behaviour of Ara-CTP [98, 103], but there is a strong correlation between the ability to accumulate and retain Ara-CTP by leukaemic blast cells and response to therapy [98]. Marked interpatient variation in Ara-CTP concentrations [98] have lead to the concept of individualized treatment strategies by directing the administered dose of Ara-C to achieve Ara-CTP concentrations in leukaemic blasts of >100–150 μmol l−1 (≈185–275 ng per 107 blasts) over a prolonged period of time [104].
Non-leukaemic mononuclear cells eliminate Ara-C more rapidly than do leukaemic blast cells, and so accumulate less Ara-CTP over a period of time. In leukaemic children receiving Ara-C normal mononuclear cells showed little interpatient variation in Ara-CTP accumulation. This was not the case for leukaemic blast cells [98]. In addition, the blast cells continued to accumulate Ara-CTP after the Ara-C infusion, whereas in normal cells Ara-CTP levels declined rapidly at the end of Ara-C therapy. These pharmacokinetic differences between normal and leukaemic cells provided the basis for optimized scheduling of Ara-C therapy, maintaining cytotoxic Ara-CTP levels in leukaemic cells whilst allowing an intermittent drop of Ara-CTP in normal cells. The modified schedule consists of eight intermittent 45 min infusions separated by a 135 min treatment free interval rather than a prolonged twice daily infusion [98].
Investigations of optimal Ara-C schedules in leukaemic patients have shown that the ability of circulating blasts to accumulate Ara-CTP in vivo during a continuous Ara-C infusion was similar to the Ara-CTP accumulation rate measured in an in vitro assay, thus the latter can be used to reflect the former [105]. Ara-CTP accumulation differs amongst patient groups as defined by the immunophenotype at the time of disease diagnosis. Non-T ALL blasts accumulate significantly more Ara-CTP than T-ALL or AML blasts. This could have implications for dosage strategies [105].
Interpretive problems
In the paediatric population the rate of conversion of Ara-C to Ara-U increases with patient age and this will influence the interpretation of deamination phenotype. Infants have a lower Ara-C clearance, hence a lower Ara-U to Ara-C ratio, than older children [106]. Resistance to Ara-C is an important cause of treatment failure in childhood acute leukaemias, and resistance mechanisms have been found in leukaemic blasts covering all aspects of Ara-C metabolism [107]. In vitro sensitivity tests correlate well with clinical responses and a number of treatment strategies have been developed, but have not been evaluated in a prospective clinical study [107].
Conclusion
Effective TDM of cytotoxic agents could prevent the consequences of treatment failure in drug sensitive cancers. Treatment failure reduces the chances of long-term disease-free survival for the cancer patient. Successful first time chemotherapy is of particular importance in childhood ALL, a disease for which a significant cure rate is seen. Treatment failure and subsequent intensive second remission therapy carries the risk of the secondary malignancies associated with alkylating agents and epipodophylotoxins, coupled with the potential of late cardiac toxicity endowed by the anthracyclines [108, 109]. In addition to the TDM of the cytotoxic drug or active metabolite per se, routine measurement and TDM of polymorphic enzyme activities must be seriously considered. At least one enzyme (TPMT) must be added to the list of agents which show a concentration effect relationship.
Acknowledgments
LL is supported by the Leukaemia Research Fund of Great Britain.
References
- 1.Pratt WB, Ruddon RW, Ensminger WD, Mayboum J, editors. The Anticancer Drugs. 2. Oxford: Oxford University Press; 1994. [Google Scholar]
- 2.Slattery JT, Sanders JE, Buckner CD, et al. Graft-rejection and toxicity following bone-marrow transplantation in relation to busulfan pharmacokinetics. Bone Marrow Trans. 1995;16:31–42. [PubMed] [Google Scholar]
- 3.Demirer T, Buckner CD, Appelbaum FR, et al. Busulfan, cyclophosphamide and fractionated total-body irradiation for autologous or syngenic marrow transplantation for acute and chronic myelogenous leukaemia—phase I dose escalation of busulfan based on targeted plasma levels. Bone Marrow Trans. 1996;17:491–495. [PubMed] [Google Scholar]
- 4.Newell DR, Pearson ADJ, Balmanno K, et al. Carboplatin pharmacokinetics in children: the development of a pediatric dosing formula. J Clin Oncol. 1993;11:2314–2323. doi: 10.1200/JCO.1993.11.12.2314. [DOI] [PubMed] [Google Scholar]
- 5.Chatelut E, Boddy AV, Peng B, et al. Population pharmacokinetics of carboplatin in children. Clin Pharmacol Ther. 1996;59:436–443. doi: 10.1016/S0009-9236(96)90113-7. [DOI] [PubMed] [Google Scholar]
- 6.Minami H, Shimokata K, Saka H, et al. Phase I clinical and pharmacokinetic study of a 14 day infusion of etoposide in patients with lung cancer. J Clin Oncol. 1993;11:1602–1608. doi: 10.1200/JCO.1993.11.8.1602. [DOI] [PubMed] [Google Scholar]
- 7.Relling MV, McLeod HL, Bowman LC, Santana VM. Etoposide pharmacokinetics and pharmacodynamics after acute exposure to cisplatin. Clin Pharmacol Ther. 1994;56:503–511. doi: 10.1038/clpt.1994.171. [DOI] [PubMed] [Google Scholar]
- 8.Lilleyman JS, Lennard L. Mercaptopurine metabolism and risk of relapse in childhood lymphoblastic leukaemia. Lancet. 1994;343:1188–1190. doi: 10.1016/s0140-6736(94)92400-7. [DOI] [PubMed] [Google Scholar]
- 9.Evans WE, Relling MV, Rodman JH, Crom WR, Boyett JM, Pui C-H. Conventional compared with individualized chemotherapy for childhood acute lymphoblastic leukaemia. N Engl J Med. 1998;338:499–505. doi: 10.1056/NEJM199802193380803. [DOI] [PubMed] [Google Scholar]
- 10.Chessells JM. Acute leukaemia in childhood. In: Peckman M, Pinedo H, Veronesi U, editors. Oxford Textbook of Clinical Oncology Vol2. Oxford UK: Oxford University Press; 1995. pp. 1628–1647. [Google Scholar]
- 11.Chessells JM. Relapsed lymphoblastic leukaemia in children: a continuing challenge. Br J Haematol. 1998;102:423–438. doi: 10.1046/j.1365-2141.1998.00776.x. [DOI] [PubMed] [Google Scholar]
- 12.Eden OB, Lilleyman JS, Richards S, Shaw M, Peto J. Results of Medical Research Council childhood leukaemia trial UKALL VIII. Br J Haematol. 1991;78:187–196. doi: 10.1111/j.1365-2141.1991.tb04415.x. [DOI] [PubMed] [Google Scholar]
- 13.Lennard L, Lilleyman JS. Are children with lymphoblastic leukaemia given enough 6-mercaptopurine? Lancet. 1987;ii:785–787. doi: 10.1016/s0140-6736(87)92511-6. [DOI] [PubMed] [Google Scholar]
- 14.Cory JG. Purine and pyrimidine nucleotide metabolism. In: Devlin TM, editor. Textbook of Biochemistry with Clinical Correlations. 4. New York: Wiley-Liss Inc; 1998. pp. 489–521. [Google Scholar]
- 15.Lennard L. The clinical pharmacology of 6-mercaptopurine. Eur J Clin Pharmacol. 1992;43:329–339. doi: 10.1007/BF02220605. [DOI] [PubMed] [Google Scholar]
- 16.Parks DA, Granger DN. Xanthine oxidase: biochemistry, distribution and physiology. Acta Physiol Scand Suppl. 1986;548:89–99. [PubMed] [Google Scholar]
- 17.Guerciolini R, Szumlanski C, Weinshilboum RM. Human liver xanthine oxidase: nature and extent of individual variation. Clin Pharmacol Ther. 1991;50:663–672. doi: 10.1038/clpt.1991.205. [DOI] [PubMed] [Google Scholar]
- 18.Relling MV, Lin J-S, Ayers G, Evans WE. Racial and gender differences in N-acetyltransferase, xanthine oxidase, and CYP1A2 activities. Clin Pharmacol Ther. 1992;52:643–658. doi: 10.1038/clpt.1992.203. [DOI] [PubMed] [Google Scholar]
- 19.Zimm S, Collins JM, O’Niell D, Chabner BA, Poplack DG. Inhibition of first-pass metabolism in cancer chemotherapy. Interaction of 6-mercaptopurine and allopurinol. Clin Pharmacol Ther. 1983;34:810–817. doi: 10.1038/clpt.1983.254. [DOI] [PubMed] [Google Scholar]
- 20.Rundles RW, Elion GB. Mercaptopurine ‘bioavailability’. N Engl J Med. 1984;310:929. doi: 10.1056/NEJM198404053101421. [DOI] [PubMed] [Google Scholar]
- 21.Tidd DM, Paterson ARP. A biochemical mechanism for the delayed cytotoxic reactions of 6-mercaptopurine. Cancer Res. 1974;34:738–746. [PubMed] [Google Scholar]
- 22.Warren DJ, Andersen A, Slordal L. Quantitation of 6-thioguanine residues in peripheral blood leukocyte DNA obtained from patients receiving 6-mercaptopurine based maintenance therapy. Cancer Res. 1995;55:1670–1674. [PubMed] [Google Scholar]
- 23.Bokkerink JPM, Stet EH, de Abreu RRA, et al. 6-mercaptopurine: Cytotoxicity and biochemical pharmacology in human malignant T-lymphoblasts. Biochem Pharmacol. 1993;45:1455–1463. doi: 10.1016/0006-2952(93)90045-x. [DOI] [PubMed] [Google Scholar]
- 24.Weinshilboum RM, Sladek SL. Mercaptopurine pharmacogenetics: Monogenic inheritance of erythrocyte thiopurine methyltransferase activity. Am J Hum Genet. 1980;32:651–662. [PMC free article] [PubMed] [Google Scholar]
- 25.Weinshilboum RM. Methylation pharmacogenetics: thiopurine methyltransferase as a model system. Xenobiotica. 1992;22:1055–1071. doi: 10.3109/00498259209051860. [DOI] [PubMed] [Google Scholar]
- 26.McLeod HL, Relling MV, Liu Q, Pui C-H, Evans WE. Polymorphic thiopurine methyltransferase in erythrocytes is indicative of activity in leukemic blasts from children with acute lymphoblastic leukemia. Blood. 1995;7:1897–1902. [PubMed] [Google Scholar]
- 27.Vuchetich JP, Weinshilboum RM, Price RA. Segregation analysis of human red blood cell thiopurine methyltransferase activity. Genetic Epidemiol. 1995;12:1–11. doi: 10.1002/gepi.1370120102. [DOI] [PubMed] [Google Scholar]
- 28.Lennard L, Lilleyman JS, Van Loon JA, Weinshilboum RM. Genetic variation in response to 6-mercaptopurine for childhood acute lymphoblastic leukaemia. Lancet. 1990;336:225–229. doi: 10.1016/0140-6736(90)91745-v. [DOI] [PubMed] [Google Scholar]
- 29.Lennard L, Van Loon JA, Weinshilboum RM. Pharmacogenetics of acute azathioprine toxicity: Relationship to thiopurine methyltransferase genetic polymorphism. Clin Pharmacol Ther. 1989;46:149–154. doi: 10.1038/clpt.1989.119. [DOI] [PubMed] [Google Scholar]
- 30.Evans WE, Horner M, Chu YQ, Kalwinsky D, Roberts WM. Altered mercaptopurine metabolism, toxic effects, and dosage requirement in a thiopurine methyltransferase deficient child with acute lymphoblastic leukaemia. J Pediatr. 1991;119:985–989. doi: 10.1016/s0022-3476(05)83063-x. [DOI] [PubMed] [Google Scholar]
- 31.Lennard L, Gibson BES, Nicole T, Lilleyman JS. Congenital thiopurine methyltransferase deficiency and 6-mercaptopurine toxicity during treatment for acute lymphoblastic leukaemia. Arch Dis Childh. 1993;69:577–579. doi: 10.1136/adc.69.5.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schutz E, Gummert J, Mohr F, Oellerich M. Azathioprine-induced myelosuppression in thiopurine methyltransferase deficient heart transplant recipient. Lancet. 1993;341:436. doi: 10.1016/0140-6736(93)93028-y. [DOI] [PubMed] [Google Scholar]
- 33.Otterness DM, Szumlanski C, Lennard L, et al. Human thiopurine methyltransferase pharmacogenetics: Gene sequence polymorphisms. Clin Pharmacol Ther. 1997;62:60–73. doi: 10.1016/S0009-9236(97)90152-1. [DOI] [PubMed] [Google Scholar]
- 34.Andersen JB, Szumlanski C, Weinshilboum RM, Schmiegelow K. Pharmacogenetics, dose adjustments and 6-mercaptopurine/methotrexate drug interactions in two patients with thiopurine methyltransferase deficiency. Acta Paediatr. 1998;87:108–111. doi: 10.1080/08035259850158001. [DOI] [PubMed] [Google Scholar]
- 35.Lennard L, Singleton HJ. High performance liquid chromatographic assay of the methyl and nucleotide metabolites of 6-mercaptopurine: quantitation of red blood cell 6-thioguanine nucleotide, 6-thioinosinic acid and 6-methylmercaptopurine metabolites in a single sample. J Chromatog. 1992;583:83–90. doi: 10.1016/0378-4347(92)80347-s. [DOI] [PubMed] [Google Scholar]
- 36.Lennard L, Singleton HJ. High performance liquid chromatographic assay of human red blood cell thiopurine methyltransferase activity. J Chromatogr B. 1994;661:25–33. doi: 10.1016/0378-4347(94)00327-0. [DOI] [PubMed] [Google Scholar]
- 37.Weinshilboum RM, Raymond FA, Pazmino PA. Human erythrocyte thiopurine methyltransferase: Radiochemical microassay and biochemical properties. Clin Chim Acta. 1978;85:323–333. doi: 10.1016/0009-8981(78)90311-x. [DOI] [PubMed] [Google Scholar]
- 38.Lafolie P, Hayder S, Bjork O, Peterson C. Intraindividual variation in 6-mercaptopurine pharmacokinetics during oral maintenance therapy of children with acute lymphoblastic leukaemia. Eur J Clin Pharmacol. 1991;40:599–601. doi: 10.1007/BF00279977. [DOI] [PubMed] [Google Scholar]
- 39.Zimm S, Ettinger LJ, Holcenberg JS, et al. Phase I and clinical pharmacological study of mercaptopurine administered as a prolonged intravenous infusion. Cancer Res. 1985;45:1869–1873. [PubMed] [Google Scholar]
- 40.Endresen L, Lie S, Stom-Methisen I, Rugstad HE, Stokke O. Pharmacokinetics of oral 6-mercaptopurine: Relationship between plasma levels and urine excretion of parent drug. Ther Drug Monit. 1990;12:227–234. [PubMed] [Google Scholar]
- 41.Bergan S, Bentdal O, Sodal G, Brun A, Rugstad HE, Stokke O. Patterns of azathioprine metabolites in neutrophils, lymphocytes, reticulocytes, and erythrocytes: relevance to toxicity and monitoring in recipients of renal allografts. Therap Drug Monit. 1997;19:502–509. doi: 10.1097/00007691-199710000-00003. [DOI] [PubMed] [Google Scholar]
- 42.Lavi LE, Holcenberg JS. A rapid high performance liquid chromatographic assay for 6-mercaptopurine metabolites in red blood cells. Analytical Biochem. 1985;144:514–521. doi: 10.1016/0003-2697(85)90148-4. [DOI] [PubMed] [Google Scholar]
- 43.Hayder S, Lafolie P, Bjork O, Peterson C. 6-Mercaptopurine plasma levels in children with acute lymphoblastic leukaemia: Relation to relapse risk and myelotoxicity. Ther Drug Monit. 1989;11:617–622. doi: 10.1097/00007691-198911000-00001. [DOI] [PubMed] [Google Scholar]
- 44.Koren G, Ferrazini G, Sulh H, et al. Systemic exposure to mercaptopurine as a prognostic factor in acute lymphocytic leukaemia in children. N Engl J Med. 1990;323:17–21. doi: 10.1056/NEJM199007053230104. [DOI] [PubMed] [Google Scholar]
- 45.Erdmann GR, Chan GLC, Canafax DM. HPLC determination of 6-thiouric and 6-mercaptopurine in organ transplant patient serum. J Liquid Chromatog. 1988;11:971–981. [Google Scholar]
- 46.Szumlanski CL, Honchel R, Scott MC, Weinshilboum RM. Human liver thiopurine methyltransferase pharmacogenetics: biochemical properties, liver-erythrocyte correlation and presence of isozymes. Pharmacogenetics. 1992;2:148–159. [PubMed] [Google Scholar]
- 47.Medard Y, Nafa S, Jacqz-Aigrain E. Thiopurine methyltransferase activity: new high-performance liquid chromatographic assay conditions. J Chromatogr B. 1997;700:275–277. doi: 10.1016/s0378-4347(97)00287-9. [DOI] [PubMed] [Google Scholar]
- 48.Mayo Foundation Laboratory Medicine Bulletin. Thiopurine methyltransferase measurement. Dec 6. 1991;14:1. [Google Scholar]
- 49.Szumlanski C, Otterness D, Her C, et al. Thiopurine methyltransferase pharmacogenetics: human gene cloning and characterization of a common genetic polymorphism. DNA and Cell Biology. 1996;15:17–30. doi: 10.1089/dna.1996.15.17. [DOI] [PubMed] [Google Scholar]
- 50.Krynetski EY, Tai H-L, Yates CR, et al. Genetic polymorphism of thiopurine S-methyltransferase: clinical importance and molecular mechanisms. Pharmacogenetics. 1996;6:279–290. doi: 10.1097/00008571-199608000-00001. [DOI] [PubMed] [Google Scholar]
- 51.Lennard L, Welch J, Lilleyman JS. Intracellular metabolites of 6-mercaptopurine in children with lymphoblastic leukaemia: a possible indicator of non-compliance. Br J Cancer. 1995;72:1004–1006. doi: 10.1038/bjc.1995.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schmeigelow K, Bruunshuus I. 6-Thioguanine nucleotide accumulation in red blood cells during maintenance chemotherapy for childhood acute lymphoblastic leukaemia, and its relation to leukopenia. Cancer Chemother Pharmacol. 1990;26:288–292. doi: 10.1007/BF02897232. [DOI] [PubMed] [Google Scholar]
- 53.Schmeigelow K, Schroder H, Gustafsson G, et al. Risk of relapse in childhood acute lymphoblastic leukaemia is related to RBC methotrexate and mercaptopurine metabolites during maintenance chemotherapy. J Clin Oncol. 1995;13:345–351. doi: 10.1200/JCO.1995.13.2.345. [DOI] [PubMed] [Google Scholar]
- 54.Lennard L, Welch J, Lilleyman JS. Thiopurine drugs in the treatment of childhood leukaemia: the influence of inherited thiopurine methyltransferase activity on drug metabolism and cytotoxicity. Br J Clin Pharmacol. 1997;44:455–461. doi: 10.1046/j.1365-2125.1997.t01-1-00607.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lennard L, Lewis IJ, Michelangnoli M, Lilleyman JS. Thiopurine methyltransferase deficiency in childhood lymphoblastic leukaemia: 6-mercaptopurine dosage strategies. Med Ped Oncol. 1997;29:252–255. doi: 10.1002/(sici)1096-911x(199710)29:4<252::aid-mpo3>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 56.Cummins D, Sekar M, Halil O, Banner N. Myelosuppression associated with azathioprine–allopurinol interaction after heart and lung transplantation. Transplantation. 1996;61:1661–1662. doi: 10.1097/00007890-199606150-00023. [DOI] [PubMed] [Google Scholar]
- 57.Woodson LC, Ames MM, Selassie CD, Hansch C, Weinshilboum RM. Thiopurine methyltransferase: aromatic thiol substrates and inhibition by benzoic acid derivatives. Mol Pharmacol. 1983;24:471–478. [PubMed] [Google Scholar]
- 58.Present DH, Korelitz BI, Wisch N, Glass JL, Sacher DB, Pasternack BS. Treatment of Crohn’s disease with 6-mercaptopurine: a long-term randomised double blind study. N Engl J Med. 1980;302:981–987. doi: 10.1056/NEJM198005013021801. [DOI] [PubMed] [Google Scholar]
- 59.Lewis LD, Benin A, Szumlanski CL, Otterness DM, Lennard L, Weinshilboum RM, Nierenberg DW. Olsalazine and 6-mercaptopurine-related bone marrow suppression: a possible drug–drug interaction. Clin Pharmacol Ther. 1997;62:464–475. doi: 10.1016/S0009-9236(97)90125-9. [DOI] [PubMed] [Google Scholar]
- 60.Lysaa RA, Giverhaug T, Wold HL, Aarbakke J. Inhibition of human thiopurine methyltransferase by furosemide, bendroflumethiazide and trichlormethiazide. Eur J Clin Pharmacol. 1996;49:393–396. doi: 10.1007/s002280050038. [DOI] [PubMed] [Google Scholar]
- 61.Glauser TA, Nelson MD, Zembower DE, Lipsky JJ, Weinshilboum RM. Diethylthiocarbamate S-methylation: Evidence for catalysis by human liver thiol methyltransferase and thiopurine methyltransferase. J Pharmacol Exp Ther. 1993;266:23–32. [PubMed] [Google Scholar]
- 62.Rivier G, Khamashta MA, Hughes GRV. Warfarin and azathioprine: a drug interaction does exist. Am J Med. 1993;94:342. doi: 10.1016/0002-9343(93)90292-w. [DOI] [PubMed] [Google Scholar]
- 63.Jeppesen U, Rasmussen JM, Brosen K. Clinically important interaction between azathioprine (Imurel) and phenprocoumon (Marcoumar) Eur J Clin Pharmacol. 1997;52:503–504. doi: 10.1007/s002280050326. [DOI] [PubMed] [Google Scholar]
- 64.Peters GJ. Antimetabolites. In: Peckham M, Pinedo HM, Veronesi U, editors. Oxford Textbook of Oncology. Vol. 1. Oxford: University Press Oxford; 1995. pp. 524–552. [Google Scholar]
- 65.Jolivet J, Cowan KH, Curt GA, Clendenin NJ, Chabner BA. The pharmacolgy and clinical use of methotrexate. N Engl J Med. 1983;309:1094–1104. doi: 10.1056/NEJM198311033091805. [DOI] [PubMed] [Google Scholar]
- 66.Seidel H, Moe PJ, Nygaard R, Nygaard K, Brede W, Borsi J. Evaluation of serious adverse events in patients treated with protocols including methotrexate infusions. Ped Hem Oncol. 1994;11:165–172. doi: 10.3109/08880019409141652. [DOI] [PubMed] [Google Scholar]
- 67.Whitehead VM, Vuchich MJ, Lauer SJ, et al. Accumulation of high levels of methotrexate polyglutamates in lymphoblasts from children with hyperdiploid (>50 chromosomes) B-lineage acute lymphoblastic leukaemia: a Pediatric Oncology Group study. Blood. 1992;80:1316–1323. [PubMed] [Google Scholar]
- 68.Crom WR, Evans WE. Methotrexate. In: Evans WE, Schentag JJ, Jusko WJ, editors. Applied Pharmacokinetics, Principles of Therapeutic Drug Monitoring. 3. Vancouver: Applied Therapeutics Inc.; 1992. pp. 29-1–29-42. [Google Scholar]
- 69.Buice R, Evans WE, Karos J, et al. Evaluation of enzyme immunoassay, radioassay and radioimmunoassay of serum methotrexate as compared with liquid chromatography. Clin Chem. 1980;26:1902–1904. [PubMed] [Google Scholar]
- 70.Dandiker WB, De Saussure VA. Fluorescence polarization immunoassay. Theory and experimental method. Immunochemistry. 1973;10:219. doi: 10.1016/0019-2791(73)90198-5. [DOI] [PubMed] [Google Scholar]
- 71.Kamen BA, Takach PL, Vatev R, Caston JD. A rapid, radio-ligand binding assay for methotrexate. Anal Biochem. 1976;70:54–63. doi: 10.1016/s0003-2697(76)80047-4. [DOI] [PubMed] [Google Scholar]
- 72.Aboleneen H, Simpson J, Backes D. Determination of methotrexate in serum by high performance liquid chromatograpy. J Chromatogr B. 1996;681:317–322. doi: 10.1016/0378-4347(95)00580-3. [DOI] [PubMed] [Google Scholar]
- 73.Beck O, Seideman P, Wennberg M, Peterson C. Trace analysis of methotrexate and 7-hydroxymethotrexate in human plasma and urine by a novel high performance liquid chromatographic method. Ther Drug Monit. 1991;13:528–532. doi: 10.1097/00007691-199111000-00011. [DOI] [PubMed] [Google Scholar]
- 74.Cociglio M, Hillaire-Buys D, Alric C. Determination of methotrexate and 7-hydroxymethotrexate by liquid chromatography for routine monitoring of plasma levels. J Chromatogr. 1995;674:101–110. doi: 10.1016/0378-4347(95)00301-x. [DOI] [PubMed] [Google Scholar]
- 75.Najjar TAO, Matar KM, Alfawaz IM. Comparison of a new high performance liquid chromatography method with fluorescence polarization immunoassay for analysis of methotrexate. Ther Drug Monit. 1992;14:142–146. doi: 10.1097/00007691-199204000-00012. [DOI] [PubMed] [Google Scholar]
- 76.Slordal L, Prytz PS, Pettersen I, Aarbakke J. Methotrexate measurements in plasma: comparison of enyme multiplied immunoassay technique, TDx fluorescence polarization immunoassay and high performance liquid chromatography. Ther Drug Monit. 1986;8:368–372. [PubMed] [Google Scholar]
- 77.Masson E, Relling MV, Synold TW, et al. Accumulation of methotrexate polyglutamates in lymphoblasts is a determinant of antileukemic effects in vivo. J Clin Invest. 1996;97:73–80. doi: 10.1172/JCI118409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Balis FM, Holcenberg JS, Zimm S, et al. The effect of methotrexate on the bioavailability of oral 6-mercaptopurine. Clin Pharmacol Ther. 1987;41:384–387. doi: 10.1038/clpt.1987.45. [DOI] [PubMed] [Google Scholar]
- 79.Bokkerink JPM, DeAbreu RA, Bakker MAH, Hulscher TW, van Baal JM, DeVaan GAM. Dose-related effects of methotrexate on purine and pyrimidine nucleotides and on cell-kinetic parameters in Molt-4 malignant human T-lymphoblasts. Biochem Pharmacol. 1986;35:3557–3564. doi: 10.1016/0006-2952(86)90626-x. [DOI] [PubMed] [Google Scholar]
- 80.Lu Z, Zhang R, Diasio RB. Dihydropyridine dehydrogenase activity in human peripheral blood mononuclear cells and liver: population characteristics, newly identified deficient pateints, and clinical implications in 5-flurouracil chemotherapy. Cancer Res. 1993;53:5433–5438. [PubMed] [Google Scholar]
- 81.Harris BE, Song R, Soong S-j, Diassio RB. Relationshio between dihydropyridine dehydrogenase activity and plasma 5-fluorouracil levels with evidence for circadian variation of enzyme activity and plasma drug levels in cancer patients receiving 5-fluorouracil by protracted continuous infusion. Cancer Res. 1990;50:197–201. [PubMed] [Google Scholar]
- 82.Santini J, Milano G, Thyss A, et al. 5-FU therapeutic monitoring with dose adjustments leads to an improved therapeutic index in head and neck cancer. Br J Cancer. 1989;59:287–289. doi: 10.1038/bjc.1989.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Christophidis N, Mihaly G, Vajda F, Louis W. Comparison of liquid- and gas-chromatographic assays of 5-fluorouracil in plasma. Clin Chem. 1979;25:83–86. [PubMed] [Google Scholar]
- 84.Kindberg CG, Riley CM, Stobauch JF. Analysis of 5-fluorouracil in plasma by precolumn derivatization with 4-bromomethyl-7-methoxycoumarin, followed by multi-dimentional high performance liquid chromatography. J Chromatogr. 1989;473:431–444. doi: 10.1016/s0021-9673(00)91327-x. [DOI] [PubMed] [Google Scholar]
- 85.Uematsu T, Nakashima M, Fujii M, et al. Measurement of 5-fluorouracil in scalp hair: a possible index of patient compliance with oral adjuvant chemotherapy. Eur J Clin Pharmacol. 1996;50:109–113. doi: 10.1007/s002280050077. [DOI] [PubMed] [Google Scholar]
- 86.Presant CA, Wolf W, Waluch V, et al. Association of intratumoral pharmacokinetics of fluorouracil with clinical response. Lancet. 1994;343:1184–1187. doi: 10.1016/s0140-6736(94)92399-x. [DOI] [PubMed] [Google Scholar]
- 87.Sommadossi J-P, Gewirtz D, Diasio RB, Aubert C, Cano JP, Goldman ID. Rapid catabolism of 5-fluorouracil in freashly isolated hepatocytes as analyzed by high performance liquid chromatography. J Biol Chem. 1982;257:8171–8176. [PubMed] [Google Scholar]
- 88.Diasio RB, Beavers TL, Carpenter JT. Familial deficiency in dihydropyrimidine dehydrogenase: biochemical basis for familial pyridinemia and severe 5-fluorouracil-induced toxicity. J Clin Invest. 1988;81:47–51. doi: 10.1172/JCI113308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Milano G, Etienne MC, Thyss RA, et al. Relationship between fluorouracil systemic exposure and tumour response and patient survival. J Clin Oncol. 1994;12:1291–1295. doi: 10.1200/JCO.1994.12.6.1291. [DOI] [PubMed] [Google Scholar]
- 90.Milano G, Etienne MC. Individualizing therapy with 5-fluorouracil related to dihydropyridine dehydrogenase: theory and limits. Ther Drug Monit. 1996;18:335–340. doi: 10.1097/00007691-199608000-00004. [DOI] [PubMed] [Google Scholar]
- 91.Grem JL, Yee LK, Venzon DJ, Takimoto CH, Allegra CJ. Inter- and intraindividual variation in dihydropyridine dehydrogenase activity in peripheral blood mononuclear cells. Cancer Chemother Pharmacol. 1997;40:117–125. doi: 10.1007/s002800050635. [DOI] [PubMed] [Google Scholar]
- 92.Peck R, Wiggs R, Callaghan J, et al. Inhibition of dihydropyrimidine dehydrogenase by 5-propynyluracil, a metabolite of the anti-varicella zoster virus agent netivudine. Clin Pharmacol Ther. 1996;59:22–31. doi: 10.1016/S0009-9236(96)90020-X. [DOI] [PubMed] [Google Scholar]
- 93.Baccanari DP, Davis ST, Knick VC, Spector T. 5-Ethynyluracil (776C85): a potent modulator of the pharmacokinetics and antitumour efficacy of 5-fluorouracil. Proc Natl Acad Sci. 1993;90:11064–11068. doi: 10.1073/pnas.90.23.11064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Khor SP, Lucas S, Schilsky R, et al. A phase I pharmacokinetic study of 5-ethynyluracil plus 5-fluorouracil in cancer pateints with solid tumors. ]abstract[ Proc Am Assoc Cancer Res. 1995;36:1439. [Google Scholar]
- 95.Diasio RB. Sorvudine and 5-fluorouracil; a clinically sugnificant drug–drug interaction due to inhibition of dihydropyrimidine dehydrogenase. Br J Clin Pharmacol. 1998;46:1–4. doi: 10.1046/j.1365-2125.1998.00050.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Estey E, Plunkett W, Dixon D, Keating M, McCredie K, Freirich EJ. Variables predicting response to high-dose cytosine arabinoside therapy in patients with refractory acute leukaemia. Leukaemia. 1997;8:580–583. [PubMed] [Google Scholar]
- 97.Kreis W, Lesser M, Budman DR, et al. Phenotypic analysis of 1–D arabinofuranosylcytosine deamination in patients treated with high doses and correlation with response. Cancer Chemother Pharmacol. 1992;30:126–130. doi: 10.1007/BF00686404. [DOI] [PubMed] [Google Scholar]
- 98.Hiddemann W, Schleyer E, Unterhalt M, Kern W, Buchner T. Optimizing therapy for acute myeloid leukaemia based on diferences in intracellular metabolism of cytosine arabinoside and between leukaemic blasts and normal mononuclear blood cells. Ther Drug Monit. 1996;18:341–349. doi: 10.1097/00007691-199608000-00005. [DOI] [PubMed] [Google Scholar]
- 99.Schleyer E, Ehninger G, Zuhlsdorf M, Proksch B, Hiddemann W. Detection and separation of intracellular Ara-CTP by ion-pair high performance liquid chromatography: a sensitive, isocratic, highly reproducible and rapid method. J Chromatogr. 1989;497:109–120. doi: 10.1016/0378-4347(89)80010-6. [DOI] [PubMed] [Google Scholar]
- 100.Braess J, Pforntner J, Kaufmann CC, et al. Detection and Determination of the major metabolites of H3 cytosine arabinoside by high performance liquid chromatography. J Chromatogr B. 1996;676:131–140. doi: 10.1016/0378-4347(95)00372-x. [DOI] [PubMed] [Google Scholar]
- 101.Colly LP, Richel DJ, Arentsen-Honders W, Starrenburg IWJ, Edelbroek PM, Willemze R. A simplified assay for measurement of cytosine arabinoside incorporation into DNA in Ara-C-sensitive and resistant leukemic cells. Cancer Chemother Pharmacol. 1990;27:151–156. doi: 10.1007/BF00689101. [DOI] [PubMed] [Google Scholar]
- 102.Capizzi RL, Agrawal K. Drugs useful in the chemotherapy of acute leukaemias. Handbook Exp Pharm. 1992;101:523–564. [Google Scholar]
- 103.Plunkett W, Gandi V. Pharmacokinetics of arabinosylcytosine. J Infusional Chemotherapy. 1992;2:169–176. [Google Scholar]
- 104.Heinemann V, Estey E, Keating MJ, Plunkett W. Patient-specific dose rate for continuous infusion high-dose cytarabine in relapsed myelogenous leukaemia. J Clin Oncol. 1989;7:622–628. doi: 10.1200/JCO.1989.7.5.622. [DOI] [PubMed] [Google Scholar]
- 105.Boos J, Hohenlochter B, Schulzewesthoff P, et al. Intracellular retention of cytosine-arabinoside triphosphate in blast cells from children with acute myelogenous and lymphoblastic leukaemia. Med Ped Oncol. 1996;26:397–404. doi: 10.1002/(SICI)1096-911X(199606)26:6<397::AID-MPO5>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 106.Periclou AP, Avramis VI. Nonmem population pharmacogenetic studies of cytosine arabinoside after high dose and after loading bolus followed by continuous infusion of the drug in pediatric patients with leukaemias. Cancer Chemother Pharmacol. 1996;39:42–50. doi: 10.1007/s002800050536. [DOI] [PubMed] [Google Scholar]
- 107.Zuhlsdorf M, Vormoor J, Boos J. Cytosine arabinoside resistance in childhood leukaemia. Int J Ped Hem/Oncol. 1997;4:565–581. [Google Scholar]
- 108.Boffetta P, Kaldor JM. Secondary malignancies following cancer chemotherapy. Acta Oncol. 1994;33:591–598. doi: 10.3109/02841869409121767. [DOI] [PubMed] [Google Scholar]
- 109.Pui C-H, Behm FG, Raimondi SC, et al. Secondary acute myeloid leukaemia in children treated for acute leukaemia. N Engl J Med. 1989;321:136–142. doi: 10.1056/NEJM198907203210302. [DOI] [PubMed] [Google Scholar]