

Abstract
Aims
In patients with lower urinary tract symptoms suggestive of benign prostatic obstruction the α1-adrenoceptor antagonist terazosin lowers blood pressure whereas only very small if any alterations were reported with the α1-adrenoceptor antagonist tamsulosin. Therefore, we have compared the vascular α1-adrenoceptor antagonism of tamsulosin and terazosin directly.
Methods
Ten healthy subjects were investigated in a randomized, single-blind, three-way cross-over design and received a single dose of 0.4 mg tamsulosin, 5 mg terazosin or placebo on 3 study days at least 1 week apart. Before and 1, 3, 5, 7, 10 and 23.5 h after drug intake, alterations of diastolic blood pressure and other haemodynamic parameters in response to a graded infusion of the α1-adrenoceptor agonist phenylephrine were determined non-invasively.
Results
At most time points tamsulosin inhibited phenylephrine-induced diastolic blood pressure elevations significantly less than terazosin (5 h time point: median difference in inhibition 35%, 95% CI: 18.7–50.3%). On the other hand, phenylephrine-induced changes of cardiac output, heart rate and stroke volume were similar during both active treatments.
Conclusions
In doses equi-effective for treatment of lower urinary tract symptoms tamsulosin causes less inhibition of vasoconstriction than terazosin.
Keywords: tamsulosin, terazosin, α1-adrenoceptor, vasoconstriction
Introduction
The sympathetic nervous system plays an important role in the maintenance of vascular tone and thus peripheral vascular resistance. The vasoconstricting effects of the sympathetic nervous system are mainly mediated by α-adrenoceptors. While both α1- and α2-adrenoceptors can mediate vasoconstriction, only α1-adrenoceptor antagonists are used for blood pressure lowering in clinical practice [1]. We have recently demonstrated that the vasoconstriction following systemic infusion of noradrenaline in man is mediated mostly by α1-adrenoceptors with an only small, if any, contribution of α2-adrenoceptors [2].
In recent years α1-adrenoceptor antagonists originally developed as anti-hypertensives (e.g. doxazosin and terazosin) are increasingly used for symptomatic treatment of lower urinary tract symptoms (LUTS) suggestive of benign prostatic obstruction (BPO) [3, 4]. As expected they lower blood pressure and sometimes produce symptoms of orthostatic hypotension in patients with LUTS [5–7]. Tamsulosin, a new α1-adrenoceptor antagonist developed primarily for LUTS treatment, lacked clinically relevant hypotensive effects in phase III placebo-controlled studies [8–10]. Even in patients with concomitant antihypertensive treatment (diuretics, β-adrenoceptor antagonists, Ca2+ entry blockers, angiotensin converting enzyme inhibitors) only marginal blood pressure alterations were seen [11]. It has been postulated that the lack of hypotensive effects may explain why tamsulosin, in contrast to doxazosin and terazosin, did not exhibit significantly more side effects attributable to the cardiovascular system than placebo in clinical trials although all three drugs were similarly effective in symptom relief of patients with LUTS suggestive of BPO [4]. However, no comparative haemodynamic studies of tamsulosin vs standard α1-adrenoceptor antagonists have been reported in man.
In trying to understand the differential effects of tamsulosin vs doxazosin and terazosin, pharmacokinetic and pharmacodynamic reasons can be envisioned. Doxazosin and terazosin typically reach peak plasma concentrations within 1–2 h following oral ingestion [12–14], while tamsulosin in its commercially available modified release formulation reaches peak plasma concentrations after 5–6 h [15, 16]. Tamsulosin also differs from the others on a pharmacodynamic basis. Thus, at least three subtypes of α1-adrenoceptors exist which have been designated α1A, α1B and α1D [17]. Most studies have reported that tamsulosin has somewhat, i.e. 12–15 fold, greater affinity for α1A- than for α1B-adrenoceptors with intermediate values at α1D-adrenoceptors, while doxazosin and terazosin have similar affinity for all α1-adrenoceptor subtypes [18]. In vitro data with animal blood vessels have suggested that all three subtypes of α1-adrenoceptors may participate in mediating vasoconstriction depending on the vascular bed and species under investigation [19]. Whether pharmacokinetic or pharmacodynamic factors underly the differential blood pressure effects of tamsulosin and conventional α1-adrenoceptor antagonists in man, is unknown.
Due to the marked pharmacokinetic differences between the α1-adrenoceptor antagonists, a reasonable analysis of their differential haemodynamic profile has to include multiple time points including the peak and trough levels of the respective drugs. Therefore, we have performed graded infusions of the α1-adrenoceptor agonist phenylephrine in healthy volunteers before and at six time points after oral ingestion of single doses of 0.4 mg tamsulosin or 5 mg terazosin in a single-blind, three-way cross-over, placebo-controlled study.
Methods
Subjects and study protocol
The following study protocol was approved by the ethics committee of the University of Essen Medical School. Study conduct and data verification procedures complied with the principles of Good Clinical Research Practise as specified by the European Guidelines and Boehringer Ingelheim standard operating procedures. Twelve male subjects (median age: 27 years, range: 22–36 years) judged to be healthy on the basis of medical history, physical examination, electrocardiogram and routine laboratory screening, participated after having given written informed consent. Exclusion criteria included a resting heart rate <45 beats min−1, and the following haemodynamic changes during a standard orthostatic test: an increase of heart rate >20 beats min−1, a decrease of systolic blood pressure >20 mm Hg or a standing diastolic blood pressure <60 mm Hg. None of the subjects was on any regular medication.
The study was a partially single-blind, randomized cross-over in 12 subjects. Double blinding was not performed for safety reasons, i.e. to allow adjustment of infused agonist doses against treatment (see below). The subjects completed 3 study days during which they each received one tablet of terazosin 5 mg (purchased as Flotrin® from a German pharmacy), one capsule of tamsulosin 0.4 mg (provided by Yamanouchi Europe B.V., Leiderdorp, Netherlands), or one capsule of placebo matching tamsulosin. Study days were at least 7 days apart. According to the original protocol the first two subjects received 10 mg terazosin and 0.8 mg tamsulosin. The inhibition of phenylephrine-induced vasoconstriction by these antagonist doses was too pronounced, particularly with terazosin, to allow further analysis. Therefore, the dosing was lowered to 0.4 mg tamsulosin and 5 mg terazosin for the remaining 10 subjects. Only these 10 subjects are included in the analysis.
On each study day subjects reported to the laboratory at 07.00 h after an overnight fast. Two indwelling catheters were placed into forearm veins which were used for phenylephrine infusions (obtained as Neo Synephrine® from Sanofi Winthrop, New York, NY, USA) and blood withdrawals. The subjects remained in the supine position until after the last phenylephrine infusion on the next morning. Following a resting period to allow stabilization of baseline values, the first graded intravenous phenylephrine infusion was performed. Two hours after start of this infusion the subjects received their medication. Additional phenylephrine infusions were performed 1, 3, 5, 7, 10 and 23.5 h after drug intake; immediately prior to each infusion blood samples were taken for the determination of plasma drug concentrations and radioreceptor assays [16]. A light snack (single serving of cereal with 200 ml of skimmed milk) was given after the 7 h infusion, and a pizza or pasta dish of the subject’s choice after the 10 h infusion. While drinking of water was allowed ad libitum, no other food intake was permitted during study days to avoid interference with the haemodynamic measurements.
Haemodynamic measurements
The duration of each phenylephrine infusion was approximately 60 min. During that time up to 5 consecutive dosages were chosen out of 7 possible incremental steps (0.25, 0.5, 1, 1.5, 2, 3 and 4 μg kg−1 min−1) each lasting 10 min. The incremental steps started at 0.25 μg kg−1 min−1 if the subject had taken placebo, but were allowed to start at 0.5 or 1 μg kg−1 min−1 if the subjects had taken active medication.
Increase in diastolic blood pressure (DBP) was chosen as the primary parameter of vasoconstriction. During the phenylephrine dose-response profiles, cardiovascular function (heart rate, blood pressure, and impedance cardiographic estimates of stroke volume) was measured. Heart rate and blood pressure were measured 5 times at 1 min intervals within the 5 min preceding the start of the infusion and every min during the last 5 min of each phenylephrine dosage step. The other cardiovascular variables were measured just before starting the infusion and at the end of each dosage step immediately following the last blood pressure measurement. We aimed for an elevation of DBP of ≈20 mm Hg. When that was obtained or when the 4 μg kg−1 min−1 dose limit was reached, no further dosage steps were investigated. For safety reasons, the infusions were stopped if DBP increased from baseline by more than 30 mm Hg or heart rate decreased to values <36 beats min−1.
Blood pressure (mmHg) was measured with a standard mercury sphygmomanometer with DBP taken as the disappearance of Korotkoff’s sound. Total peripheral resistance (dyn s cm−5) was calculated as mean arterial pressure divided by cardiac output multiplied by 80, where mean blood pressure was defined as DBP plus one third of the pulse pressure. Cardiac output was measured non-invasively by impedance cardiography using the standard approach with circular tape electrodes and graphical signal analysis [20]. Estimates of stroke volume (ml) were calculated from the maximum change in transthoracic impedance and the duration of left ventricular ejection time (measured from the carotid pulse tracing) by use of Kubicek’s equation [20]. Cardiac output (l min−1) was then calculated as the product of heart rate and stroke volume divided by 1000. Heart rate was calculated from the RR-Interval of the ECG.
Data analysis
We have quantified the degree of inhibition by the antagonists in three ways: Firstly we determined ‘responder rates’ for each assessment time. A responder was defined as a subject in which phenylephrine infusion increased DBP by more than twice the standard deviation of basal (pre-phenylephrine) values, i.e. 7.6 mm Hg. Secondly and thirdly we determined two inhibition indices designated ‘Iall’ and ‘Iact’. They were defined as the percentage inhibition of DBP elevation during antagonist treatment relative to the change in DBP seen in the same subject at the same time point on placebo. The calculation used the highest phenylephrine dose achieved in a given subject common to all three experimental days (Iall, almost always 1 μg kg−1 min−1) or the highest phenylephrine dose achieved common to active treatment days as compared to the highest phenylephrine dose on placebo (Iact).
To allow for points when phenylephrine decreased rather than increased DBP (presumably due to β-adrenoceptor agonism) all inhibition indices were transformed with the sigmoidal function
with a=p/(100-p), c=2×log(a)/Δplacebo and p=90. This transformation guarantees that all inhibition indices lie between 0% and 100%. The parameter p was chosen such that Δtest=0 (complete inhibition) corresponds to a calculated value of 90%, and Δtest=Δplacebo (no inhibition) corresponds to a value of 10%. In the range between 20% and 80% inhibition transformed and untransformed indices are nearly identical.
Since DBP is a composite measure of peripheral vasoconstriction, vascular compliance and cardiac output, we have compared the haemodynamic effects of tamsulosin and terazosin in more detail using the 5 h time point as an example. At this time point phenylephrine-induced alterations of DBP, mean arterial pressure, total peripheral resistance, cardiac output, heart rate and stroke volume were compared during both active treatments using the phenylephrine dose which was also used in the Iact calculations. The 5 h time point was chosen because it represents the tmax for tamsulosin but is already 4 h after the tmax for terazosin [16]; thus, this time point should be particularly powerful in elucidating differences between the two treatments.
All quantitative data are shown as median with upper and lower quartiles in parentheses. Comparisons between tamsulosin and terazosin treatments were done by Wilcoxon’s signed rank test separately for each time point. If this indicated a statistically significant difference between treatments, the precision of comparison is presented as median difference with 95% confidence intervals. For the additional analyses at the 5 h time point, the same test was applied to compare phenylephrine effects in the presence of tamsulosin vs those in the presence of terazosin. A P<0.05 was considered as significant. No adjustments for multiple comparisons were made. Thus, all resulting P values are to be interpreted in a descriptive manner.
Results
Infusion of phenylephrine in the absence of antagonist dose-dependently increased DBP. The median increase caused by 1 μg kg−1 min−1 phenylephrine 2 h before administration of test medication was 16.8 mm Hg; data from a representative subject 5 h after administration of test medication are shown in Figure 1. There were no major differences in phenylephrine responses at the −2 h time point between the three study days, and the elevations of DBP reached by the highest phenylephrine dose allowed in the protocol did not undergo major diurnal changes on the placebo day (data not shown).
Figure 1.
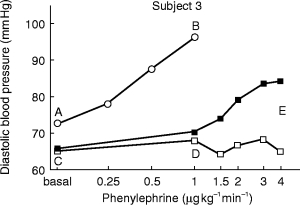
Diastolic blood pressure elevations induced by infusion of phenylephrine. The figure shows original data from a representative subject obtained 5 h after intake of placebo (open circles), 0.4 mg tamsulosin (filled squares) or 5 mg terazosin (open squares). The inhibition index Iall (Figure 3) was calculated as [1—(D-C)/(B-A)]×100, while the inhibition index Iact (Figure 4) was calculated as [1—(E-C)/(B-A)]×100.
On the placebo day the number of responders with regard to DBP was 10 out of 10 subjects at all time points (Figure 2). On terazosin there were no responders for DBP from 1 to 3 h after administration, while on tamsulosin at least 8 subjects were responders at all time points (Figure 2). The inhibition index Iall for DBP peaked at 90% 1 h after terazosin intake and remained above 66% even after 23.5 h (Figure 3). In contrast the Iall for DBP following tamsulosin intake peaked at 82% after 5 h and declined to median values of less than 20% after 23.5 h (Figure 3). Thus, Iall was significantly different between treatments at the 1, 3, 5 and 23.5 h time points; the median difference at the 5 h time point was 6.9% (95% CI: 4.3–15.9%). The inhibition index Iact, which discriminates treatments more effectively (Figure 1), peaked at 93% after 1 h in terazosin-treated subjects and was 59% even after 23.5 h (Figure 4). In tamsulosin-treated subjects Iact peaked at 39% after 5 h and declined to 13% after 23.5 h (Figure 4). The difference between tamsulosin and terazosin with regard to Iact was statistically significant at all time points; the median difference at the 5 h time point was 35.0% (95% CI: 18.7–50.3%).
Figure 2.
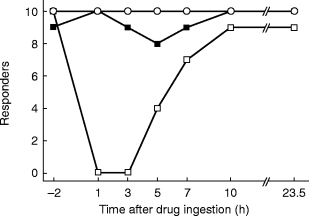
Number of responders with regard to diastolic blood pressure upon infusion of phenylephrine. Responders were defined as subjects with an increase of >7.6 mm Hg in diastolic pressure upon the highest agonist dose allowed in the protocol. Shown are the number of responders out of 10 subjects studied at the indicated times following intake of placebo (open circles), 0.4 mg tamsulosin (filled squares) or 5 mg terazosin (open squares).
Figure 3.
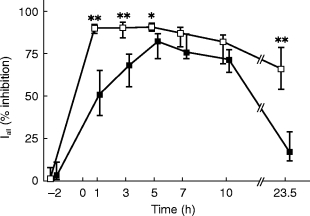
Inhibition of phenylephrine infusion-induced elevations of diastolic blood pressure by 0.4 mg tamsulosin (filled squares) or 5 mg terazosin (open squares). Inhibition was calculated at each time point relative to the highest phenylephrine dose which was reached on all study days including the placebo day (‘Iall’, 1 μg kg−1 min−1 in almost every case, for calculation see Figure 1) and is shown as medians with upper and lower quartiles. * and **: P<0.05 and <0.01, respectively, vs tamsulosin in a Wilcoxon signed rank test.
Figure 4.
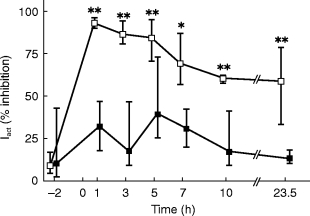
Inhibition of phenylephrine infusion-induced elevations of diastolic blood pressure by 0.4 mg tamsulosin (filled squares) or 5 mg terazosin (open squares). Inhibition was calculated at each time point relative to the highest phenylephrine dose which was reached on both study days during active treatment and compared with the highest phenylephrine dose on the placebo day (‘Iact’, for calculation see Figure 1) and is shown as medians with upper and lower quartiles. * and **: P<0.05 and <0.01, respectively, vs tamsulosin in a Wilcoxon signed rank test.
A more detailed haemodynamic analysis at the 5 h time point demonstrated that phenylephrine caused significantly smaller elevations of DBP, mean arterial pressure and total peripheral resistance in terazosin- than in tamsulosin treated subsjects (Figure 5); the median differences were 13.0 (95% CI: 9.2–16.0) mm Hg, 12.5 (95% CI: 7.1–14.8) mm Hg, and 295 (95% CI: 103–359) dyn s cm−5, respectively. However, phenylephrine increased cardiac output and stroke volume and reduced heart rate to a similar extent in tamsulosin- and terazosin-treated subjects (Figure 6).
Figure 5.
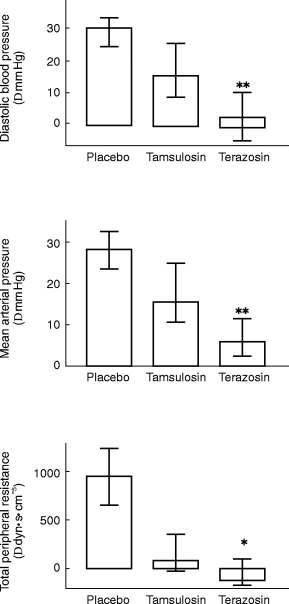
Effects of 0.4 mg tamsulosin and 5 mg terazosin on phenylephrine-induced alterations of diastolic blood pressure (upper panel), mean arterial pressure (middle panel) and total peripheral resistance (lower panel) as determined 5 h following medication. Data are based on the highest phenylephrine dose achieved during both active treatments (‘Iact’, for calculation see Figure 1) and are shown as medians with upper and lower quartiles. * and **: P<0.05 and <0.01 vs tamsulosin in a Wilcoxon signed rank test. Phenylephrine effects during placebo treatment are shown for comparison; they were not compared statistically to the data obtained during active treatment since they were determined at a different phenylephrine dose.
Figure 6.
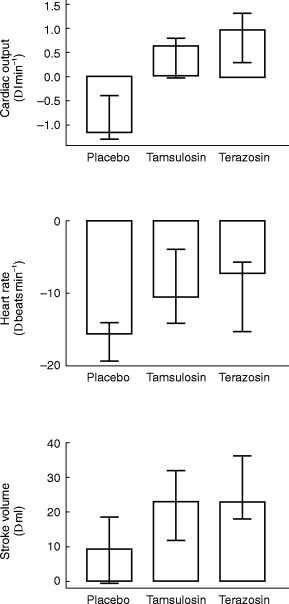
Effects of 0.4 mg tamsulosin and 5 mg terazosin on phenylephrine-induced alterations of cardiac output (upper panel), heart rate (middle panel) and stroke volume (lower panel) as determined 5 h following medication. Data are based on the highest phenylephrine dose achieved during both active treatments (‘Iact’, for calculation see Figure 1) and are shown as medians with upper and lower quartiles. Effects of tamsulosin and terazosin were not significantly different in a Wilcoxon signed rank test. Phenylephrine effects during placebo treatment are shown for comparison; they were not compared statistically to the data obtained during active treatment since they were determined at a different phenylephrine dose.
Discussion
Methodological considerations
The aim of the present study was to compare the haemodynamic effects of two α1-adrenoceptor antagonists, tamsulosin and terazosin, which have previously been shown to differentially affect blood pressure in the treatment of patients with LUTS suggestive of BPO [6–9]. A head-to-head comparison of both antagonists faces two obstacles. Firstly, both drugs have distinct pharmacokinetic properties. To address the role of this factor, we have investigated six different time points in the first 24 h following drug intake. Secondly, vascular α1-adrenoceptor antagonism is dose-dependent. Therefore, the comparison of two drugs requires that both are studied at doses which are clinically equi-effective. Our choice of antagonist doses was based on the following considerations: In dose-finding studies in patients with LUTS suggestive of BPO 0.4 mg once daily was found to be the maximum effective dose of tamsulosin [8, 10]. In similar studies with terazosin maximum effective doses of terazosin were 10 mg once daily, while 5 mg once daily had only submaximal effects [21]. Placebo-controlled studies in patients with LUTS suggestive of BPO have reported a similar overall efficacy of tamsulosin (fixed dose of 0.4 mg [8, 9]) and terazosin (fixed dose of 10 mg [22] or dose-escalation studies with 1–10 mg terazosin, in which the median final dose was 10 mg [6, 7]). Finally, a direct comparative study in patients with LUTS suggestive of BPO has reported similar efficacy for 0.2 mg day−1 tamsulosin and 5 mg day−1 terazosin [23]. Therefore, we have chosen to compare 0.4 mg tamsulosin with 5 mg terazosin in the present study. If anything this dosage choice would be expected to cause a bias to detect less vascular inhibition by terazosin relative to tamsulosin. The bias was introduced on purpose to enhance the power of our study in light of the previous reports on lack of blood pressure lowering with tamsulosin [9, 23, 24].
The classical method for analysis of receptor antagonism in vivo is the administration of increasing doses of agonist with measurement of shifts in the dose-response curve in the presence of antagonist. For studies of α1-adrenoceptor antagonists this usually involves the agonist phenylephrine with measurements of DBP elevations as an indicator of arterial vasoconstriction [25–28]. When standard therapeutic doses of tamsulosin (0.4 mg) and terazosin (5 mg) were given to fasting, healthy subjects, the extent of inhibition of DBP responses at several time points was so pronounced, especially for terazosin, that shifts of the phenylephrine dose-response curve could not formally be calculated. Higher phenylephrine doses might theoretically have overcome the inhibition, but were not tested due to cardiac stimulation by those doses of phenylephrine, presumably via β-adrenoceptor-stimulating properties of the agonist. Therefore, we have assessed the extent of antagonism by tamsulosin and terazosin by three other means. Firstly, we have determined responder rates, which are robust but only semiquantitative parameters. Second, we have calculated an inhibition index designated Iall; while this accurately reflects the degree of inhibition by both antagonists, it may underestimate a possible difference between them (Figure 1). Third, we have calculated an inhibition index designated Iact; while this may underestimate the degree of inhibition it is more sensitive in the detection of differences between the antagonists. While this approach may be less powerful than classical dose-response curve shifts, we feel that the combination of all three forms of quantification of antagonism yields a reliable means to compare the haemodynamic properties of tamsulosin and terazosin.
Haemodynamic analysis
In the present study tamsulosin caused significantly less inhibition of vasoconstriction than terazosin at most time points, regardless of how inhibition was assessed. The extent of differentiation between the two antagonists was largest for responder rates and smallest for Iall; while these differences reflect the specific advantages and disadvantages of the three inhibition indices, it cannot be decided from the present data which reflects the clinical situation with endogenous agonist more closely. Despite the quantitative differences between the inhibition indices, all three consistently differentiated between tamsulosin and terazosin. This is remarkable since we have compared a dose of terazosin which is submaximally effective in patients with LUTS against a dose of tamsulosin which is maximally effective in such patients, i.e. our study was biased to detect less inhibition by terazosin (see above). Moreover, this differential inhibition was evident even at the 5 h time point, i.e. when tamsulosin blood concentrations peaked while terazosin blood concentrations had already declined for 4 h [13–16]. A more detailed haemodynamic analysis at the 5 h time point demonstrated that three parameters of vasoconstriction, i.e. phenylephrine-induced elevations of DBP, mean arterial pressure and total peripheral resistance, were significantly smaller in terazosin- than in tamsulosin-treated subjects. On the other hand, phenylephrine-induced changes of parameters of cardiac function, i.e. cardiac output, heart rate and stroke volume, were similar during both treatments. Thus, the differential effects of tamsulosin and terazosin on DBP are due to quantitatively different antagonism at the vascular level. These observations are consistent with the results from placebo-controlled clinical studies with tamsulosin in patients with LUTS suggestive of BPO, in which no clinically relevant blood pressure reductions were observed [8, 9]. Moreover, in a direct comparison 0.2 mg day−1 tamsulosin did not alter blood pressure while 5 mg day−1 terazosin significantly lowered it, despite the fact that both drugs caused similar improvements of LUTS [23].
A differentiation was also found for inhibition of finger tip arterial and dorsal hand vein vasoconstriction in a comparison of tamsulosin with another non-subtype-selective α1-adrenoceptor antagonist, doxazosin, although only a single time point was investigated in that study [29]. Another study by the same group of investigators has also reported a differential inhibition of finger tip vasoconstriction by the non-selective prazosin vs the sligthly α1A-selective urapidil over a full time course [30]. Thus, our data demonstrate unequivocally that in LUTS-equivalent doses tamsulosin causes less inhibition of vasoconstriction than terazosin. The comparison with published data on doxazosin, prazosin and urapidil indicated that this might relate to its relative selectivity for the α1A-adrenoceptor subtype. To test this hypothesis directly, we have determined ex vivo occupancy of human α1A-, α1B-and α1D-adrenoceptors using a radioreceptor assay at all time points [16]. However, we were unable to define the α1-adrenoceptor subtype predominantly mediating phenylephrine-induced DBP elevations with this approach (data not shown). Therefore, identification of the α1-adrenoceptor subtype mediating vasoconstriction in man will require future studies with alternative approaches, e.g. the use of antagonists with greater subtype selectivity.
In vitro studies on a human conductance vessel, the isolated iliac artery, have implicated α1B-adrenoceptors in vasoconstriction [31], but results with human resistance arteries have not been communicated to our knowledge. Most studies on rat mesenteric or renal resistance vessels have found a predominant involvement of α1A-adrenoceptors [32–35]. Studies in other blood vessels of experimental animals demonstrate that all three α1-adrenoceptor subtypes can mediate vasoconstriction depending on the vascular bed and species under investigation [19]. Thus, human vasoconstriction may also involve a mixture of α1-adrenoceptor subtypes.
In conclusion, we have shown that the α1-adrenoceptor antagonists, tamsulosin and terazosin, when given in similarly effective doses with regard to the treatment of LUTS suggestive of BPO, differ markedly in their haemodynamic effects. Thus, tamsulosin caused some inhibition of vasoconstriction under our experimental conditions which involved young healthy volunteers, fasting drug intake, and administration of exogenous agonist. However, tamsulosin produced considerably less inhibition of α1-adrenoceptor-mediated vasoconstriction than terazosin at most time points irrespective of the distinct pharmacokinetic profile of both drugs. Whether this differential haemodynamic profile is due to the modest selectivity of tamsulosin for α1A-adrenoceptors, remains to be studied.
Acknowledgments
This work was supported in part by grants from Boehringer Ingelheim and the Deutsche Forschungsgemeinschaft (Ph 49/1–3).
References
- 1.van Zwieten PA. α-Adrenoceptor blocking agents in the treatment of hypertension. In: Laragh JH, Brenner BM, editors. Hypertension: Pathophysiology, Diagnosis and Management. 2. New York: Raven Press; 1995. pp. 2917–2935. [Google Scholar]
- 2.Schäfers RF, Poller U, Pönicke K, et al. Influence of adrenoceptor and muscarinic receptor blockade on the cardiovascular effects of exogenous noradrenaline and of endogenous noradrenaline released by infused tyramine. Naunyn-Schmiedeberg’s Arch Pharmacol. 1997;357:239–249. doi: 10.1007/pl00004938. [DOI] [PubMed] [Google Scholar]
- 3.Eri LM, Tveter KJ. α-Blockade in the treatment of symptomatic benign prostatic hyperplasia. J Urol. 1995;154:923–934. [PubMed] [Google Scholar]
- 4.Chapple CR. Selective α1-adrenoceptor antagonists in benign prostatic hyperplasia: rationale and clinical experience. Eur Urol. 1996;29:129–144. [PubMed] [Google Scholar]
- 5.Kirby RS. Doxazosin in benign prostatic hyperplasia: effects on blood pressure and urinary flow in normotensive and hypertensive men. Urology. 1995;46:182–186. doi: 10.1016/s0090-4295(99)80191-5. [DOI] [PubMed] [Google Scholar]
- 6.Brawer MK, Adams G, Epstein H. Terazosin in the treatment of benign prostatic hyperplasia. Arch Fam Med. 1993;2:929–935. doi: 10.1001/archfami.2.9.929. [DOI] [PubMed] [Google Scholar]
- 7.Roehrborn CG, Oesterling JE, Auerbach S, et al. The Hytrin community assessment trial study: a one-year study of terazosin versus placebo in the treatment of patients with symptomatic benign prostatic hyerplasia (BPH) Urology. 1996;47:159–168. doi: 10.1016/s0090-4295(99)80409-9. [DOI] [PubMed] [Google Scholar]
- 8.Lepor H. Phase III multicenter placebo-controlled study of tamsulosin in benign prostatic hyperplasia. Urology. 1998;51:892–900. doi: 10.1016/s0090-4295(98)00126-5. [DOI] [PubMed] [Google Scholar]
- 9.Chapple CR, Wyndaele JJ, Nordling J, Boeminghaus F, Ypma AFGVM, Abrams P. Tamsulosin, the first prostate-selective α1A-adrenoceptor antagonist. A meta-analysis of two randomized, placebo-controlled multicentre studies in patients with benign prostatic obstruction (symptomatic BPH) Eur Urol. 1996;29:155–167. [PubMed] [Google Scholar]
- 10.Abrams P, Speakman M, Stott M, Arkell D, Pocock R. A dose-ranging study of the efficacy and safety of tamsulosin, the first prostate-selective α1A-adrenoceptor antagonist, in patients with benign prostatic obstruction (symptomatic benign prostatic hyperplasia) Br J Urol. 1997;80:587–596. doi: 10.1046/j.1464-410x.1997.00380.x. [DOI] [PubMed] [Google Scholar]
- 11.Michel MC, Mehlburger L, Bressel H-U, Schumacher H, Schäfers RF, Goepel M. Tamsulosin treatment of 19 365 patients with lower urinary tract symptoms: does comorbidity alter tolerability? J Urol. 1998;160:784–791. doi: 10.1016/S0022-5347(01)62787-3. [DOI] [PubMed] [Google Scholar]
- 12.Fulton B, Wagstaff AJ, Sorkin EM. Doxazosin. An update of its clinical pharmacology and therapeutic applications in hypertension and benign prostatic hyperplasia. Drugs. 1995;49:295–320. doi: 10.2165/00003495-199549020-00011. [DOI] [PubMed] [Google Scholar]
- 13.Patterson SE. Terazosin kinetics after oral and intravenous doses. Clin Pharmacol Ther. 1985;38:423–427. doi: 10.1038/clpt.1985.198. [DOI] [PubMed] [Google Scholar]
- 14.Sonders RC. Pharmacokinetics of terazosin. Am J Med. 1986;80(Suppl. 5b):20–24. doi: 10.1016/0002-9343(86)90847-8. [DOI] [PubMed] [Google Scholar]
- 15.Wilde MI, McTavish D. Tamsulosin—a review of its pharmacological properties and therapeutic potential in the management of benign prostatic hyperplasia. Drugs. 1996;52:883–896. doi: 10.2165/00003495-199652060-00012. [DOI] [PubMed] [Google Scholar]
- 16.Taguchi K, Schäfers RF, Michel MC. Radioreceptor assay analysis of tamsulosin and terazosin pharmacokinetics. Br J Clin Pharmacol. 1998;45:49–55. doi: 10.1046/j.1365-2125.1998.00636.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Michel MC, Kenny BA, Schwinn DA. Classification of α1-adrenoceptor subtypes. Naunyn-Schmiedeberg’s Arch Pharmacol. 1995;352:1–10. doi: 10.1007/BF00169183. [DOI] [PubMed] [Google Scholar]
- 18.Michel MC, Grübbel B, Taguchi K, Verfürth F, Otto T, Kröpfl D. Drugs for treatment of benign prostatic hyperplasia: affinity comparison at cloned α1-adrenoceptor subtypes and in human prostate. J Auton Pharmacol. 1996;16:21–28. doi: 10.1111/j.1474-8673.1996.tb00352.x. [DOI] [PubMed] [Google Scholar]
- 19.Vargas HM, Gorman AJ. Vascular alpha-1 adrenergic receptor subtypes in the regulation of arterial pressure. Life Sci. 1995;57:2291–2308. doi: 10.1016/0024-3205(95)02224-7. [DOI] [PubMed] [Google Scholar]
- 20.Kubicek WG, Karnegis JN, Patterson RP, Witsoe DA, Mattson RH. Development and evaluation of an impedance cardiac output system. Aerospace Medicine. 1966;37:1208–1212. [PubMed] [Google Scholar]
- 21.Lepor H, Auerbach S, Puras-Baez A, et al. A randomized, placebo-controlled multicenter study of the efficacy and safety of terazosin in the treatment of benign prostatic hyperplasia. J Urol. 1992;148:1467–1474. doi: 10.1016/s0022-5347(17)36941-0. [DOI] [PubMed] [Google Scholar]
- 22.Lepor H, Williford WO, Barry MJ, et al. The efficacy of terazosin, finasteride, or both in benign prostatic hyperplasia. New Engl J Med. 1996;335:533–539. doi: 10.1056/NEJM199608223350801. [DOI] [PubMed] [Google Scholar]
- 23.Lee E, Lee C. Clinical comparison of selective and non-selective α1A-adrenoreceptor antagonists in benign prostatic hyperplasia: studies on tamsulosin in a fixed dose and terazosin in increasing doses. Br J Urol. 1997;80:606–611. doi: 10.1046/j.1464-410x.1997.00411.x. [DOI] [PubMed] [Google Scholar]
- 24.Buzelin JM, Fonteyne E, Kontturi MJ, Witjes WPJ, Khan A. Comparison of tamsulosin with alfuzosin in the treatment of patients with lower urinary tract symptoms sugestive of bladder outlet obstruction (symptomatic benign prostatic hyperplasia) Br J Urol. 1997;80:597–605. doi: 10.1046/j.1464-410x.1997.00205.x. [DOI] [PubMed] [Google Scholar]
- 25.Sumner DJ, Elliott HL, Reid JL. Analysis of the pressor dose response. Clin Pharmacol Ther. 1982;32:450–458. doi: 10.1038/clpt.1982.188. [DOI] [PubMed] [Google Scholar]
- 26.Schäfers RF, Elliott HL, Howie CA, Reid JL. An evaluation of the α-adrenoceptor antagonism produced by SK&F 86466 in healthy normotensive males. Br J Clin Pharmacol. 1990;30:884–888. doi: 10.1111/j.1365-2125.1990.tb05455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schäfers RF, Elliott HL, Howie CA, Reid JL. Studies with abanoquil (UK-52,046) a novel quinoline α1-adrenoceptor antagonist: I. Effects on blood pressure, heart rate and pressor responsiveness in normotensive subjects. Br J Clin Pharmacol. 1991;32:599–604. doi: 10.1111/j.1365-2125.1991.tb03958.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tham TCK, McKaigue JP, Guy S, Shanks RG, Riddell JG. The dose dependency of the α-adrenoceptor antagonist and β-adrenoceptor partial agonist activity of dilevalol and labetalol in man. Br J Clin Pharmacol. 1993;36:251–256. doi: 10.1111/j.1365-2125.1993.tb04225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harada K, Ohmori M, Fujimura A. Comparison of the antagonistic activity of tamsulosin and doxazosin at vascular α1-adrenoceptors in humans. Naunyn-Schmiedeberg’s Arch Pharmacol. 1996;354:557–561. doi: 10.1007/BF00170828. [DOI] [PubMed] [Google Scholar]
- 30.Harada K, Ohashi K, Fujimura A, Kumagai Y, Ebihara A. Effect of α1-adrenoceptor antagonists, prazosin and urapidil, on a finger skin vasoconstrictor response to cold stimulation. Eur J Clin Pharmacol. 1996;49:371–375. doi: 10.1007/s002280050034. [DOI] [PubMed] [Google Scholar]
- 31.Hatano A, Takahashi H, Tamaki M, et al. Pharmacological evidence of distinct α1-adrenoceptor subtypes mediating the contraction of human prostatic urethra and peripheral artery. Br J Pharmacol. 1994;113:723–728. doi: 10.1111/j.1476-5381.1994.tb17053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Williams TJ, Clarke DE. Characterization of α1-adrenoceptors mediating vasoconstriction to noradrenaline and nerve stimulation in the isolated perfused mesentery of rat. Br J Pharmacol. 1995;114:531–536. doi: 10.1111/j.1476-5381.1995.tb13259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blue DR, Bonhaus DW, Ford APDW, et al. Functional evidence equating the pharmacologically-defined α1A-and the cloned α1C-adrenoceptor: studies in the isolated perfused rat kidney. Br J Pharmacol. 1995;115:283–294. doi: 10.1111/j.1476-5381.1995.tb15875.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen H, Fetscher C, Schäfers RF, Wambach G, Philipp T, Michel MC. Effects of noradrenaline and neuropeptide Y on rat mesenteric microvessel contraction. Naunyn-Schmiedeberg’s Arch Pharmacol. 1996;353:314–323. doi: 10.1007/BF00168634. [DOI] [PubMed] [Google Scholar]
- 35.Chen H, Bischoff A, Schäfers RF, Wambach G, Philipp T, Michel MC. Vasoconstriction of rat renal interlobar arteries by noradrenaline and neuropeptide Y. J Auton Pharmacol. 1997;17:137–146. doi: 10.1046/j.1365-2680.1997.00452.x. [DOI] [PubMed] [Google Scholar]