

Abstract
Aims
The aim of this study was to determine the pharmacokinetic parameters of flutamide, a nonsteroidal antiandrogenic compound, and its pharmacologically active metabolite, hydroxyflutamide, in renal insufficiency. Haemodialysis (HD) clearance of flutamide and hydroxyflutamide was also determined.
Methods
Pharmacokinetic parameters were assessed for flutamide and hydroxyflutamide in 26 male subjects with normal renal function (creatinine clearance by 24 h urine collection, CLcr, greater than 80 ml min−1 1.73 m−2; n = 6) or reduced renal function; CLcr = 50–80 (n = 7), 30–49 (n = 3), 5–29 (n = 4), and <5 ml min−1 1.73 m−2-HD (n = 6), following a single, oral 250 mg flutamide dose. Subjects undergoing HD received a second 250 mg dose of flutamide 4 h prior to HD; blood and dialysate were collected during HD to determine dialysability of flutamide and hydroxyflutamide.
Results
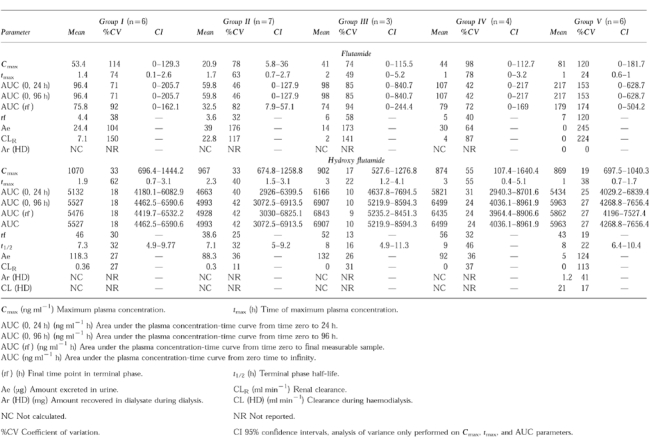
Cmax, tmax, AUC, t1/2, and renal clearance of flutamide and hydroxyflutamide did not differ between groups. Less than 1% of the dose appeared in dialysate as hydroxyflutamide. No serious adverse events were observed.
Conclusions
Renal function did not affect flutamide nor hydroxyflutamide disposition. HD did not alter hydroxyflutamide pharmacokinetics. Dosing adjustments for renal impairment or HD are not indicated for flutamide.
Keywords: pharmacokinetics, flutamide, renal failure
Introduction
Flutamide is a nonsteroidal, toluidine derivative which possesses antiandrogenic effects and is efficacious for palliative treatment of metastatic prostatic cancer [1, 2]. Flutamide is readily absorbed from the gastrointestinal tract and rapidly metabolized into an active metabolite, hydroxyflutamide [3–5]. Both flutamide and hydroxyflutamide are highly protein bound (>94%) and are eliminated from the body, in part, by renal excretion. Given the potential for drug accumulation in patients with reduced renal function, this study was undertaken to evaluate the pharmacokinetics of flutamide and hydroxyflutamide in subjects with reduced renal function. Additionally, the dialysability of flutamide and hydroxyflutamide was determined in subjects undergoing chronic hemodialysis to establish whether dosage supplementation would be indicated in this patient population.
Methods
Subjects and study design
This was a single-dose, parallel group study involving 26 male subjects with varying degrees of renal function based upon 24 h creatinine clearances (CLcr) normalized for body surface area. Subjects in Group I had normal renal function (CLcr >80 ml min−1 1.73 m−2) while those in Groups II, III, and IV had mild (CLcr = 50–80 ml min−1 1.73 m−2), moderate (CLcr = 30–49 ml min−1 1.73 m−2), and severe (CLcr = 5–29 ml min−1 1.73 m−2) renal impairment, respectively. Subjects in Group V had a CLcr less than 5 ml min−1 1.73 m−2 and received chronic, outpatient haemodialysis treatments thrice weekly.
The study protocol was reviewed and approved by the Institutional Review Board at our facility. All subjects gave written informed consent. Each subject underwent a complete physical examination and provided a medical history within a 2 week period prior to entering the study. Additionally, a complete blood count, chemistry profile, electrocardiogram, routine urinalysis, HIV antibody and hepatitis B surface antigen titres, and a urine drug screen were obtained. Individual subjects were grouped according to their creatinine clearance values quantitated by 24 h urine collections. Subjects were excluded from study participation if they had clinically significant cardiovascular, respiratory, haematologic, gastrointestinal, or hepatic disease. Likewise, renal insufficiency in Group II-V had to be stable (unchanged in previous 3 months). Any infectious disorders within the 4 weeks prior to study enrolment excluded potential participants. Illicit drug use or a history of narcotic addiction excluded subjects as did allergies to antiandrogens, oestrogens, and flutamide. Subjects with positive HIV or hepatitis B surface antigen titres were excluded.
Each subject in Groups I-IV received an oral, 250 mg dose (two 125 mg capsules) of flutamide (Schering-Plough; Kenilworth, NJ). Haemodialysis subjects (Group V) received the same dose on two different study days; once immediately following a haemodialysis treatment (interdialytic period) and once 4 h prior to a haemodialysis treatment (intradialytic period). The washout period between the inter- and intradialytic periods was 14 days. Subjects were confined to the clinical research unit 12 h prior to dosing and remained for 48 h following dosing. All subjects fasted, with the exception of water, for 10 h prior to dosing and 4 h post-dose. Prescription medications were also held for the same duration, pre- and post-dose.
For Groups I—IV, blood samples for flutamide and hydroxyflutamide determinations were collected in heparinized vacutainer syringes immediately prior to drug administration (0 h) and then at 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 14, 16, 24, 30, 36, 48, 60, 72, and 96 h post-dose. Samples were centrifuged, cells discarded, and plasma frozen at −20° C. Urine samples were collected just prior to drug administration (0 h) and then in block samples at 0–4, 4–8, 8–12, 12–16, 16–24, 24–36, 36–48, 48–72, and 72–96 h following dosing.
To evaluate intradialytic pharmacokinetic parameters in Group V, venous samples were drawn immediately prior to dose administration (0 h) and then at 0.5, 1, 2, and 3 h post-dose. Arterial and venous samples were then collected at the start of dialysis (4 h post-dose) and at 5, 6, 7, and 8 h or the end of the dialysis treatment. Venous blood samples were also collected at 8.16, 8.33, 8.5, 8.75, 9, 10, 12, 14, 16, 24, 30, 36, and 48 h post-dose. Dialysate was collected, pre- and post-filter, prior to the start of hemodialysis (0 h) and then in block samples at 0–1, 1–2, 2–3, and 3–4 h after dialysis was initiated.
Analytical procedures and pharmacokinetic analysis
Concentrations of flutamide and hydroxyflutamide were measured in plasma, urine, and dialysate samples with a gas chromatograph equipped with a DB17 mega-bore column with electron capture (63Ni) detection. The respective linear ranges for flutamide and hydroxyflutamide in plasma were 2.0–300 ng ml−1 and 2.0–800 ng ml−1; in urine, were 3.0–500 ng ml−1 and 3.0–1200 ng ml−1; and in dialysate were 3.0–500 ng ml−1 and 3.0–1200 ng ml−1. Precision and accuracy data for flutamide and hydroxyflutamide assays in plasma, urine and dialysate demonstrated intraday coefficients of variation (%CV) less than 15% over a concentration range of 10 to 1000 ng ml−1 (%CV range [flutamide] 2–12% plasma, 1–6% urine, 2–8% dialysate; %CV range (hydroxyflutamide) 3–14% plasma, 2–11% urine, 1–4% dialysate). The level of quantitiation (LOQ) values for flutamide and hydroxyflutamide were the same within each matrix: 2.0 ng ml−1 in plasma and 3.0 ng ml−1 in urine and dialysate. Flutamide and hydroxyflutamide concentrations in samples were determined by using the ratio peak area of each analyte to that of the internal standard. Concentrations were calculated using the best fit straight line relationship between concentration of the analyte and the observed peak area ratio.
The maximum concentration in plasma (Cmax) was defined as the highest observed plasma concentration. The terminal rate constant (λz) was calculated as the negative of the slope of the log-linear terminal portion of the plasma concentration-time curve using linear regression. The terminal phase half-life (t1/2) was calculated as 0.693/λz. For hydroxyflutamide, area under the plasma concentration-time curve to the time of final measurable sample [AUC(tf)] was calculated using the linear trapezoidal method and was extrapolated to infinity (I) according to the equation: AUC(I) = AUC(tf)+Ctf/k, where Ctf is the estimated concentration at tf (time of final sample). AUC(0, 24 h) and AUC(0,96 h) were also calculated using the linear trapezoidal method. For all groups except Group V, renal clearance was calculated as CLR = Ae(0, 96 h)/AUC(0,96 h) where Ae was the amount of flutamide or hydroxyflutamide excreted in the urine from 0–96 h. In haemodialysis patients, renal clearance of hydroxyflutamide was calculated as follows: CLR = Ae(0, 48)/AUC(0,48 h), where Ae was the amount of hydroxyflutamide excreted in urine for the given interval.
Clearance of hydroxyflutamide following haemodialysis [CL(HD)] was calculated as follows: CL(HD) = Ae(HD)/AUC(HD), where Ae(HD) was the amount of hydroxyflutamide recovered in the dialysate during haemodialysis, and AUC(HD) was the area under the arterial plasma concentration-time curve during haemodialysis.
Results
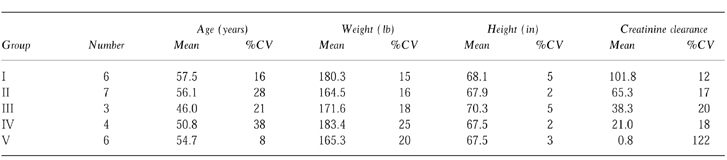
Twenty-six subjects between the ages of 18 and 75 years (mean = 54) were enrolled and completed the study. Demographic profiles of all participants are presented in Table 1. No serious adverse events were reported.
Table 1.
Mean demographic data for subjects grouped by stage of renal disease.
Nineteen of 26 subjects reported one or more minor adverse events during the course of the study. These adverse events were single occurrences, mild in nature, and did not require medical intervention. Dizziness (15%), fatigue (12%), and headache (12%) were the most frequently experienced adverse events. One subject experienced an increase in lactic dehydrogenase (LDH) from a pre-dose concentration of 144 IU l−1 to 237 IU l−1 at follow-up (normal range: 85–220IU l−1) which was considered to be possibly related to flutamide administration.
Concentration vs time relationships for flutamide and hydroxyflutamide are illustrated in Figures 1 and 2. The mean pharmacokinetic parameters of flutamide and hydroxyflutamide are shown in Table 2. In the majority of subjects, plasma concentrations of flutamide could not be determined beyond 4 h post-dose and high variability in the mean data was present in all groups.
Figure 1.
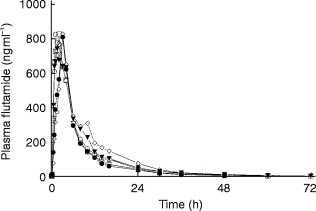
Mean plasma concentration-time profiles of flutamide in subjects with varying stages of renal failure following administration of a single 250 mg dose of flutamide. □ group 1, • group II, ◊ group III, ▾ group IV, ○ group V.
Figure 2.
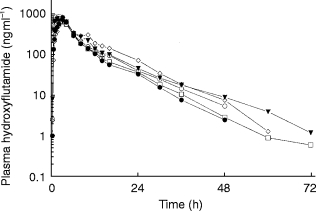
Mean plasma concentration-time profiles of hydroxy-flutamide in subjects with varying stages of renal failure following administration of a single 250 mg dose of flutamide. □ group 1, • group II, ◊ group III, ▾ group IV, ○ group V.
Table 2.
Summary of mean pharmacokinetic parameters following single administration of 250 mg flutamide.
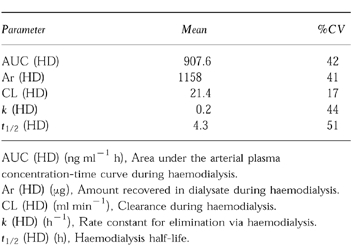
Two weeks after the initial 250 mg dose of flutamide, subjects in Group V received a second 250 mg dose. Following the 4 h blood collection after the second dose, hemodialysis was initiated for 2–4 h. Only two subjects out of six demonstrated appreciable concentrations of flutamide in their plasma on the dialysis day. No flutamide and less than 1% of the dose was recovered as hydroxyflutamide in dialysate. Mean pharmacokinetic parameters of flutamide and hydroxyflutamide are summarized in Table 3.
Table 3.
Summary of mean pharmacokinetic parameters following single administration of 250 mg flutamide on dialysis day.
Discussion
Following a single, oral 250 mg flutamide dose in subjects with normal as well as impaired renal function, no clinically meaningful differences in the incidence, frequency, or severity of adverse events were noted among the subjects based on degree of renal dysfunction.
Given the high variability in the mean data and the inability to detect flutamide in the majority of subjects beyond 4 h post-dose, rigorous pharmacokinetic analysis of fluatmide concentrate could not be performed. The urinary excretion of flutamide was also highly variable. The small amount of drug excreted in the urine (<1% of the dose) illustrates that renal clearance of this compound does not represent a significant elimination route.
Less variability was observed with the hydroxyflutamide data. The degree of renal impairment did not alter Cmax, tmax, or AUC values for hydroxyflutamide. The terminal phase half-life in Groups I-III ranged from 7.3 to 7.6 h while being slightly greater in Group IV (9.3 h) and Group V (8.4 h). Likewise, the degree of renal impairment did not have an apparent effect on the amount of hydroxyflutamide excreted in the urine (91–118 μg) nor on renal clearance of the drug. The pharmacokinetic parameters for hydroxyflutamide for Group V subjects were not significantly different clinically from other groups despite an elimination half-life during dialysis of 4.3 h vs 8.4 h on non-dialysis days. The lack of statistical significance between these values is due, in part, to considerable variability (22–51%CV).
In conclusion, flutamide was safe and well-tolerated in subjects with normal renal function as well as in those with impaired renal function and those requiring chronic haemodialysis therapy when administered as a single oral 250 mg dose. While the effect of renal impairment on the plasma pharmacokinetics of flutamide could not be determined due to high intersubject variability, renal impairment had no significant effect on hydroxyflutamide plasma pharmacokinetics. Based on plasma hydroxyflutamide concentrations, dose reduction is not required in patients with chronic renal insufficiency. Neither flutamide nor hydroxyflutamide were significantly removed by haemodialysis, making dosage supplementation after dialytic therapy unnecessary.
Acknowledgments
The authors would like to thank Anita Goth for her assistance in manuscript preparation.
References
- 1.Neri RO, Koziol P, Kung T. SCH 15423 (α, α, α-trifluoro-2-methyl-4′-nitro-M-lactotoluidide), a potent anti androgen. Endocrinology. 1975;96(Suppl):298–298. [Google Scholar]
- 2.Peets E, Henson F, Neri R. Effect of non-steroidal anti androgen SCH 15423 (α, α, α-trifluoro-2-methyl-4′-nitro-M-lactotoluidide) on androgen disposition in rats. Endocrinology. 1975;96(Suppl):306–306. [Google Scholar]
- 3.Schulz M, Schmoldt A, Donn F, Becker H. The pharmacokinetics of flutamide and its major metabolites after a single oral dose and during chronic treatment. Eur J Clin Pharmacol. 1988;34:633–636. doi: 10.1007/BF00615229. [DOI] [PubMed] [Google Scholar]
- 4.Neri R. Pharmacology and pharmacokinetics of flutamide. Urology. 1989;34:19–21. doi: 10.1016/0090-4295(89)90230-6. [DOI] [PubMed] [Google Scholar]
- 5.Radwanski E, Perentesis G, Symchowicz S, Zampaglione N. Single and multiple dose pharmacokinetic evaluation of flutamide in normal geriatric volunteers. J Clin Pharmacol. 1989;29:554–558. doi: 10.1002/j.1552-4604.1989.tb03381.x. [DOI] [PubMed] [Google Scholar]