

Abstract
Aims
Uncertainty as to relative under-reporting plagues the comparisons of spontaneous reporting rates as a tool for decision-making in pharmacovigilance. However, it is generally accepted that under-reporting should be reasonably similar for similar drugs sharing the same indication, country and period of marketing. To test this, we compared the adverse drug reaction reporting rates to the French regional pharmacovigilance centres for six pairs of identical drug marketed at the same time by different companies under different brand names (co-marketing).
Methods
All reaction reports were related to sales, to compute reporting rate; within each pair, the reporting rate ratio and its confidence interval were calculated.
Results
The rate ratios were all between 0.76 and 1.33. Two of them were significantly different from 1 (1.28; 95% C.I. [1.01; 1.60] and 1.33; 95% C.I. [1.06; 1.74]).
Conclusions
These small differences in reporting rates would not warrant regulatory action and support the usual assumption of similar reporting for similar drugs.
Keywords: pharmacovigilance, spontaneous reporting, decision-making, pharmacoepidemiology, drugs
Introduction
Most estimations and decisions made in pharmacovigilance are based on spontaneously reported data [1, 2]. Due to the size of the surveyed population and to the prompt reporting of serious and unexpected effects, this first-line surveillance method is particularly efficient in identifying new adverse drug reactions (ADRs) [3]. However, it suffers from many limitations when one intends to quantify the risk associated with a given drug, mostly because of an inescapable under-reporting, the magnitude of which remains unknown [4]. When comparing the risk associated with drugs, under-reporting whatever its magnitude does not affect the validity of conclusions if it is assumed that it is more or less identical for the compared drugs. It is generally admitted that this could be the case for two drugs belonging to the same therapeutic class, with the same indications, marketed in the same country, during the same period of time [5]. Under these conditions, observing a significant difference in reporting rates would mean that the risks associated with these drugs could differ.
To test this hypothesis, we compared the reporting rates for twelve co-marketed drugs (six pairs) in France. In order to share development costs or for other reasons, two companies may decide to market simultaneously the same compound, under different brand-names. This situation represents a kind of gold standard for similitude: same active ingredient(s), same recommended dose, same presentation, same approved indications and same launching date in the same country.
Methods
For this study, we selected all co-marketed drug pairs meeting the following criteria:
(i) new chemical entities or new associations of known active principles, (ii) launched in France between 1988 and 1993, with (iii) a market life of at least 3 years, for which (iv) a minimal number of thirty ADR cases were reported to the French pharmacovigilance centres over the period considered. We only considered ADR cases, whatever the type, assessed as ‘reasonably associated’ by the reporter. Moreover, in order to eliminate the influence of possible differences in the efficiency in the surveillance system of companies, we only considered ADRs directly reported to the pharmacovigilance centres (around 50% of the whole number of cases collected per year in France).
For each drug of each pair, we estimated a reporting rate considering:
(i) as numerator, the number of reports of ADR (as defined above) found in the French pharmacovigilance database
(ii) as denominator, the corresponding number of patient-months calculated by dividing the number of units (e.g.tablets, capsules …) sold during the same period of time by the average daily dose and by 30.4 (the average number of days in a month).
These data were obtained from the French Medicines Agency.
For each pair, we calculated a reporting rate ratio (RRR), by dividing the reporting rate estimated for the drug marketed by the company which originally developed the compound by the reporting rate estimated for the drug marketed by the associated company. The two-sided ninety-five percent confidence interval (95% C.I.) for this ratio was calculated according to the normal approximation of the binomial distribution as suggested by Tubert et al. [6].
Results
Six pairs of co-marketed drugs met the criteria considered for this study:
Pair 1: an angiotensin-converting enzyme inhibitor launched in 1988
Pair 2: another angiotensin-converting enzyme inhibitor associated with a thiazide diuretic (1988)
Pair 3: an HMG-CoA reductase inhibitor (1989)
Pair 4: a third angiotensin-converting enzyme inhibitor (1990)
Pair 5: another HMG-CoA reductase inhibitor (1991)
Pair 6: a proton-pump inhibitor (1991)
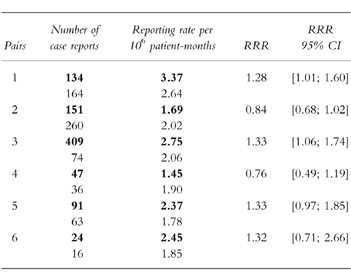
The results are given in Table 1.
Table 1.
Numbers of reported cases and reporting rates for 12 drugs marketed in France between 1988 and 1993. RRR is the ratio of the reporting rates for the two drugs of the same pair. The bold figures correspond to the drug marketed by the company which originally developed the compound.
Discussion
Due to the selection criteria used in this study (i.e. new chemical entities launched between 1988 and 1993), three pairs corresponded to ACE inhibitors and two to lipid-lowering drugs. However, this over-representation does not per se constitute a limitation for the purpose of the present analysis.
It can be seen in Table 1 that for all studied pairs, the RRR value differed slightly from 1. However, the difference was statistically significant only in two cases (pairs 1 and 3), for which the corresponding 95% C.I. for RRR did not include 1. In both cases, significance is borderline (lower bounds being 1.01 and 1.06, respectively).
It is very improbable that a regulatory decision would be undertaken on the basis of such low difference, even if statistically significant.
The most relevant point is that the observed differences remained low: the RRR value never exceeded 1.33 (the reverse of the RRR observed in pair 4, i.e. 0.76, is 1.32). Although the upper 95% confidence interval did exceed this in four cases, this is reassuring for the validity of comparisons made in the framework of spontaneous reporting because confirming that the magnitude of under-reporting is the same for two drugs of the same therapeutic class marketed concurrently. In the conditions of this study, it is reasonable to state that the actual risks of ADRs associated with each drug of the same pair are more or less the same. Thus, under the hypothesis of an under-reporting of equal magnitude, the RRR are expected to be close to 1 which was effectively the case in our study: the ratio of reporting rates never exceeded the low value of 1.33.
These results tend to validate the basic assumption currently accepted in spontaneous reporting: the ratio of reporting rates approximates the ratio of actual risks (which would have been calculated on the basis of the actual number of cases) given that the two compared drugs belong to the same therapeutic class and are used in similar conditions. This reinforces the credibility of calculations and comparisons made in this context, even if decision-making based on the results of an ad hoc pharmacoepidemiologic study remains always preferable.
Acknowledgments
The authors wish to acknowledge the assistance of Mr Patrick de Roef (French Medicines Agency).
References
- 1.Rawlins MD. Spontaneous reporting of adverse drug reactions. I: the data. Br J Clin Pharmacol. 1988;26:1–5. doi: 10.1111/j.1365-2125.1988.tb03356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith CC, Bennet PM, Pearce HM, et al. Adverse drug reactions in a hospital general medical unit meriting notification to the Committee on Safety of Medicines. Br J Clin Pharmacol. 1996;42:423–429. doi: 10.1046/j.1365-2125.1996.04376.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Praus M, Schindel F, Fescharek R, et al. Alert system for post-marketing surveillance of adverse drug reactions. Stat Med. 1993;12:2383–2393. doi: 10.1002/sim.4780122414. [DOI] [PubMed] [Google Scholar]
- 4.Lumley CE, Walker SR, Hall GC, et al. The under-reporting of adverse drug reactions seen in general practice. Pharm Med. 1986;1:205–212. [Google Scholar]
- 5.Bégaud B, Tubert P, Haramburu F, et al. Comparing toxicity of drugs: use and misuse of spontaneous reporting. Post-Marketing Surveillance. 1991;5:69–76. [Google Scholar]
- 6.Tubert-Bitter P, Bégaud B, Moride Y, et al. Comparing the toxicity of two drugs in the framework of spontaneous reporting: a confidence interval approach. J Clin Epidemiol. 1996;49:121–123. doi: 10.1016/0895-4356(95)00537-4. [DOI] [PubMed] [Google Scholar]