

Abstract
Aims
The plasma clearance of theobromine (TB; 3,7-dimethylxanthine) is known to be induced in cigarette smokers. To determine whether TB may serve as a model substrate for cytochrome P450 (CYP) 1A2, or possibly other isoforms, studies were undertaken to identify the individual human liver microsomal CYP isoforms responsible for the conversion of TB to its primary metabolites.
Methods
The kinetics of formation of the primary TB metabolites 3-methylxanthine (3-MX), 7-methylxanthine (7-MX) and 3,7-dimethyluric acid (3,7-DMU) by human liver microsomes were characterized using a specific hplc procedure. Effects of CYP isoform-selective xenobiotic inhibitor/substrate probes on each pathway were determined and confirmatory studies with recombinant enzymes were performed to define the contribution of individual isoforms to 3-MX, 7-MX and 3,7-DMU formation.
Results
The CYP1A2 inhibitor furafylline variably inhibited (0–65%) 7-MX formation, but had no effect on other pathways. Diethyldithiocarbamate and 4-nitrophenol, probes for CYP2E1, inhibited the formation of 3-MX, 7-MX and 3,7-DMU by ≈55–60%, 35–55% and 85%, respectively. Consistent with the microsomal studies, recombinant CYP1A2 and CYP2E1 exhibited similar apparent Km values for 7-MX formation and CYP2E1 was further shown to have the capacity to convert TB to both 3-MX and 3,7-DMU.
Conclusions
Given the contribution of multiple isoforms to 3-MX and 7-MX formation and the negligible formation of 3,7-DMU in vivo, TB is of little value as a CYP isoform-selective substrate in humans.
Keywords: CYP1A2, CYP2E1, cytochrome P450, substrate selectivity, theobromine
Introduction
In recent years considerable attention has focused on cytochromes P450 (CYP) 1A1 and 1A2, the two isoforms which comprise the CYP1 A subfamily, given the capacity of these enzymes to activate a range of procarcinogenic xenobiotics to mutagenic species. CYP1A1 has the capacity to activate polycyclic aromatic hydrocarbons, such as benzo [a] pyrene, while CYP1A2 has been implicated in the metabolic activation of aflatoxin B1, 2-acetylaminofluorene and a number of arylamines and food-derived aminoimidazoazarenes [1, 2]. In addition to the toxicological importance of CYP1A2, there is increasing awareness of the contribution of this isoform to the hepatic biotransformation of drugs. Thus, CYP1A2 contributes to the metabolism of caffeine, clomipramine, clozapine, cyclobenzaprine, imipramine, lisofylline, mianserin, olanzapine, ropinirole, tacrine, theophylline and R-warfarin [3].
CYP1A2 is expressed constitutively in human liver [4, 5], but induction is known to occur following exposure to a variety of xenobiotics [1, 3]. Notable in this regard are the polycyclic aromatic hydrocarbons, and compounds of this class are believed to contribute to the increased clearance of CYP1A2 substrates frequently observed in cigarette smokers. Typical of inducible P450s, wide interindividual variability has been reported in the expression of CYP1A2 in humans [3]. For example, CYP1A2 mRNA content was found to vary 43-fold in 21 livers [5]. Genetic factors have been proposed as a cause of the variability in CYP1A2 activity, but results are conflicting and vary according to the metabolic parameter investigated [3, 6].
Given the toxicological and pharmacological importance of CYP1A2, a number of compounds have been utilized as substrate probes for this enzyme in order to investigate regulation in vivo. The methylxanthines caffeine and theophylline have been employed most widely in this regard [3, 7, 8]. However, problems exist with both compounds. Caffeine metabolism is complex and application of the more conveniently measured putative markers of metabolic clearance (e.g. urinary metabolite ratios) appear to be flawed theoretically [6]. Despite its simpler metabolism compared with caffeine, theophylline is less convenient to administer and has a narrower therapeutic index.
There is evidence to suggest that CYP1A2 contributes to the elimination of theobromine (TB; 3,7-dimethylxanthine), a methylxanthine structurally related to caffeine and theophylline. On average, plasma TB clearance was found to be 33% higher in smokers than in nonsmokers [9]. Pathways of TB metabolism in humans have been well characterized (Figure 1) [9, 10]. TB undergoes CYP catalysed N3-and N7-demethylation to form 7-methylxanthine (7-MX) and 3-methylxanthine (3-MX), respectively. Once formed, 7-MX is oxidized further by xanthine oxidase to give the corresponding monomethylurate (7-methyluric acid). 7-MX and 3-MX formation account for about 60% and 20% of TB clearance, respectively. The conversion of TB to 3,7-dimethyluric acid (3,7-DMU) and 6-amino-5-[N-methylformylamino]-1-methyluracil (3,7-DAU; a diaminouracil) and renal clearance of unchanged compound account for the remainder of TB elimination.
Figure 1.
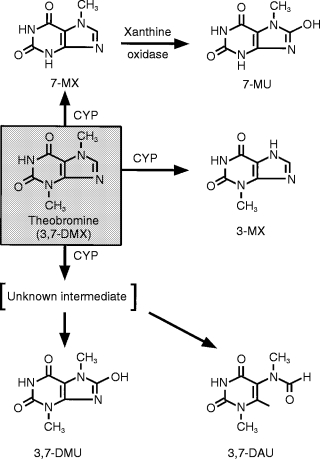
Pathways of theobromine metabolism in humans.
Cigarette smoking induced both the formation of 3-MX and 7-MX [10], the major metabolic pathways of TB, suggesting that hepatic TB N3-and N7-demethylation activities may serve as a surrogate measure of CYP1A2 activity in vivo and in vitro. As a constituent of cocoa, TB is consumed by a large proportion of the population in chocolate products. In contrast to caffeine and theophylline, TB possesses little pharmacological activity and is almost devoid of effects on the CNS and cardiovascular system. TB is well absorbed after oral administration, has a relatively short half-life, and compared to caffeine, routes of metabolism are reasonably noncomplex [9–11]. Collectively, these features make TB a potentially useful substrate for the measurement of CYP activity in population studies. Here we describe kinetic and inhibitor studies undertaken with human liver microsomes and recombinant P450s which aimed to define the contribution of CYP1A2 and other isoforms to the major primary pathways of TB metabolism in humans.
Methods
Chemicals and reagents
Sources of chemicals used were: TB, Hamilton Laboratories (Adelaide, SA); 3-MX, 7-MX, and 3,7-DMU, Fluka AG (Buchs, Switzerland); 1,3-dimethyluric acid (1,3-DMU), Adams Chemical Co (Round Lake, IL, USA); coumarin, diethyldithiocarbamate, glucose-6-phosphate, glucose-6-phosphate dehydrogenase, NADP, 4-nitrophenol, and troleandomycin, Sigma Chemical Co (St Louis, MO, USA); furafylline, Dr R. Gasser (Hoffman La Roche, Basel, Switzerland); mephenytoin, Sandoz Ltd (Basel, Switzerland); quinidine sulphate, Burroughs Wellcome (Sydney, Australia); and sulphaphenazole, CIBA Geigy (Sydney, Australia). All other chemicals and solvents were of analytical reagent grade.
Human liver samples and cDNAs
Human liver samples were obtained from renal transplant donors with the approval of next-of-kin and the Flinders Medical Centre Committee on Clinical Investigation. Upon collection, liver samples were immediately sectioned into 10–20 g portions, frozen in liquid nitrogen and then stored at −70° C until required. Hepatic microsomes were prepared by differential centrifugation, suspended (typically 20–25 mg ml−1) in 0.1 m phosphate buffer (pH 7.4) containing glycerol (20% v/v) and stored at −70° C until use. (When diluted to concentrations below 1% v/v in microsomal incubations, glycerol has been shown to have a negligible effect on the activity of CYP2E1 [12].) Microsomal and COS cell lysate protein concentrations were measured according to Lowry et al. [13], with bovine serum albumin as the standard.
The CYP1A2 and CYP2E1 cDNAs were isolated, subcloned into the expression vector pCMV4, and transfected separately into COS-7 cells as described previously [14, 15]. Cells were harvested 48 h post-transfection, re-suspended in 0.1 m phosphate buffer (pH 7.4) containing glycerol (20% v/v), and stored at −70° C until used. Cells transfected with the respective cDNA in the reverse orientation served as negative controls in the studies investigating TB metabolism by recombinant CYP1A2 and CYP2E1.
Measurement of 3-MX, 7-MX and 3,7-DMU formation by human liver microsomes and cDNA-expressed CYP1 A2 and CYP2E1
Standard 1 ml incubations contained human liver microsomal (1 mg) or COS cell lysate (1.5 mg) protein, NADPH generating system (1 mm NADP, 100 mm glucose-6-phosphate, 2 IU glucose-6-phosphate dehydrogenase and 5 mm MgCl2), and TB (0.1–5 mm) in phosphate buffer (0.1 m, pH 7.4). Reactions were initiated by the addition of NADPH generating system and were carried out at 37° C for 120 (human liver microsomes) or 150 (COS cell lysate) min. Incubations were terminated by cooling on ice. After the addition of 1,3-DMU (20 μl of a 100 μm aqueous solution), the assay internal standard, reaction mixtures were transferred to 15 ml glass culture tubes containing ammonium sulphate (1.2 g) and extracted with dichloromethane-isopropanol (4:1 v/v, 8.5 ml) using a vortex mixer. Following centrifugation (3000 g for 10 min), an 8 ml aliquot of the organic phase was transferred to a conical tipped glass tube and then evaporated to dryness under a stream of N2. Dried extracts were reconstituted in 0.2–1.0 ml of the mobile phase and an aliquot was injected onto the column.
The h.p.l.c. system employed comprised an ICI model LC 1110 solvent delivery system (ICI Instruments, Melbourne, Australia), a Waters μ-Bondapak reversed-phase column (C18, 10 micron particle size, 30 cm ×3.9 mm id; Waters-Millipore, Milford, MA, USA), and an ICI LC 1200 uv-vis absorbance detector operating at 275 nm. The column was eluted with 10 mm acetate buffer (pH 4.5) containing 2.5% methanol at a flow rate of 2 ml min−1. Under these conditions retention times for 7-MX, 3-MX, 3,7-DMU and 1,3-DMU were 10.4, 11.8, 14.6 and 21.0 min, respectively. Despite the screening of a range of commercial column packings and varying mobile phase composition, it was not possible to separate 3,7-DAU from the solvent front without adversely affecting the resolution of other analytes. Standard curves for 3-MX, 7-MX and 3,7-DMU were linear over the concentration range 0.2–10 μm and passed through the origin. Unknown concentrations of each of these compounds were calculated by comparison of metabolite to 1,3-DMU peak height ratios with those of standard curves.
Extraction efficiencies for 3-MX, 7-MX, 3,7-DMU and 1,3-DMU at added concentrations of 0.5 μm and 5.0 μm were >85%, with coefficients of variation <5%. Overall assay within-day precision was assessed from the measurement of 3-MX, 7-MX and 3,7-DMU formation in 10 separate incubations of the same batch of human liver microsomes at two substrate (TB) concentrations, 0.1 mm and 3.0 mm. Coefficients of variation were <6% for the formation of all three metabolites at both the low and high substrate concentrations. Rates of formation of 3-MX, 7-MX and 3,7-DMU were linear with respect to incubation time to at least 150 min and with respect to human liver microsomal protein concentration to 1.5 mg ml−1.
Kinetic and inhibitor studies
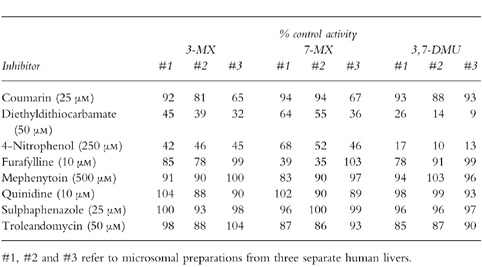
Formation of 3-MX, 7-MX and 3,7-DMU was determined over the TB concentration range 0.1–5.0 mm. The substrate concentration range employed for the kinetic studies with cDNA-expressed CYP1A2 and CYP2E1 was 0.5–5.0 mm. All incubations were performed in duplicate and experimental points represent mean values. CYP isoform selective inhibitors/substrates [16, 17] were screened for effects on human liver microsomal TB metabolism. Compounds employed included coumarin (CYP2A6), diethyldithiocarbamate and 4-nitrophenol (CYP2E1), furafylline (CYP1A2), mephenytoin (CYP2C19), quinidine (CYP2D6), sulphaphenazole (CYP2C9) and troleandomycin (CYP3A4). Inhibitory effects were investigated at substrate concentrations of 3 mm, using microsomes from three livers, and 0.1 mm, using microsomes from two livers. Concentrations of inhibitors are given in Table 1. Except for quinidine and diethyldithiocarbamate, which were added to incubations as aqueous solutions, xenobiotic inhibitors were dissolved in DMSO such that the final concentration in incubations was 0.5% v/v. Control incubations contained an equal volume of DMSO, which was shown to have a negligible effect on the formation of each metabolite. A 10 min preincubation (prior to addition of TB) was utilized for the mechanism- based inhibitors diethyldithiocarbamate, furafylline and troleandomycin.
Table 1.
Effect of CYP isoform selective probes on the conversion of TB (3 mm) to 3-MX, 7-MX and 3,7-DMU in microsomes from three separate human livers.
Analysis of results
The Michaelis-Menten parameters apparent Km and Vmax were calculated using MK Model, an extended least-squares modelling program.
Results
Kinetics of 3-MX, 7-MX and 3,7-DMU formation by human liver microsomes
The kinetics of formation of 3-MX, 7-MX and 3,7-DMU were investigated in microsomes from four separate livers. Michaelis-Menten kinetics were observed for both the N3-and N7-demethylation of TB in all four livers (Figure 2). Mean (±s.d.) apparent Km values for 3-MX and 7-MX formation were 2.7±0.5 mm and 3.9±0.8 mm, respectively. The mean Vmax values for 3-MX and 7-MX formation were 53±19 pmol min−1 mg−1 and 102±42 pmol min−1 mg−1, respectively. In contrast to the N-demethylations Eadie-Hofstee plots for 3,7-DMU formation were curvi-linear (Figure 2).
Figure 2.
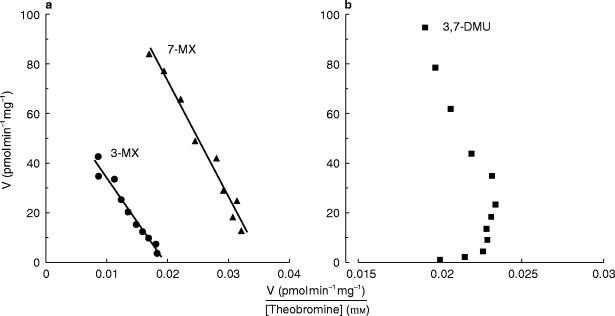
Representative Eadie-Hofstee plots for the conversion of TB to 3-methylxanthine and, 7-methylxanthine (panel a), and 3,7-dimethyluric acid (panel b) by human liver microsomes (liver #1). Points are experimentally derived values while the solid lines in panel a show the model-generated curves of best fit.
Inhibition by CYP isoform selective probes
The effects of CYP isoform selective probes on the conversion of TB (3 mm) to 3-MX, 7-MX and 3,7-DMU in microsomes from three separate human livers are given in Table 1. Consistent inhibition of 3-MX formation in all three livers was observed only with the CYP2E1 probes diethyldithiocarbamate and 4-nitrophenol. Inhibition by diethyldithiocarbamate and 4-nitrophenol ranged from 55–68% and 54–58%, respectively. Substantial inhibition (35%) of 3-MX by coumarin occurred in liver #3. The greater inhibition of 3-MX (and 7-MX, see below) formation by diethyldithiocarbamate compared with 4-nitrophenol in this liver may reflect an effect on CYP2A6 activity, since diethyldithiocarbamate has been reported to inhibit this isoform in addition to CYP2E1 [18]. Inhibition by the other isoform-selective probes was generally <20%.
Both diethyldithiocarbamate and 4-nitrophenol affected 7-MX formation, with inhibition ranging from 36–64% and 32–54%, respectively (Table 1). Furafylline inhibited 7-MX formation by more than 60% in livers #1 and #2, but was without effect in liver #3. Like 3-MX formation, substantial inhibition (33%) of the conversion of TB to 7-MX by coumarin occurred in liver #3 and inhibition by other isoform-selective probes was insignificant (<14%).
Potent inhibition of 3,7-DMU by diethyldithiocarbamate and 4-nitrophenol was observed in all three livers (Table 1). Inhibition by diethyldithiocarbamate and 4-nitrophenol ranged from 74–91% and 83–90%, respectively. In contrast to 3-MX and 7-MX formation, coumarin had no effect on 3,7-DMU formation in liver #3.
The inhibitory effects of the CYP isoform selective probes were also investigated at a low substrate concentration (viz 0.1 mm) using microsomes from two livers (#1 and #3). Inhibition at the lower substrate concentration was undertaken to confirm the inhibitory profile observed for the higher TB concentration (Table 1) and preclude the possible contribution of a high activity enzyme(s) to 3-MX and 7-MX formation not differentiated by the kinetic experiments (Figure 2). The inhibitory profiles observed for livers #1 and #3 at the low substrate (TB) concentration were essentially identical to those reported in Table 1 (data not shown).
TB metabolism by cDNA-expressed CYP1A2 and CYP2E1
Recombinant CYP1A2 and CYP2E1 both catalysed the conversion of TB to 7-MX, with apparent Km values of 4.2 mm and 3.4 mm, respectively (Figure 3). Conversion of TB to 3-MX and 3,7-DMU by CYP2E1, but not CYP1A2, was also observed at high substrate concentration (>2 mm). However, limitations of assay sensitivity precluded formal kinetic analyses of these reactions.
Figure 3.
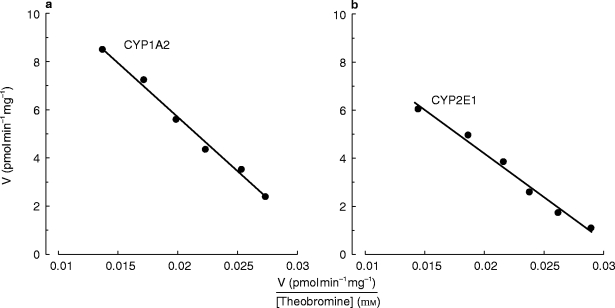
Eadie-Hofstee plots for the conversion of TB to 7-MX by recombinant CYP1A2 (panel a) and CYP2E1 (panel b). Points are experimentally derived values while the solid lines show the model-generated curves of best fit.
Discussion
Results presented here indicate that CYP2E1 is essentially solely responsible for the conversion of TB to 3,7-DMU and additionally contributes to the formation of 3-MX and 7-MX. CYP1A2 also catalyses the conversion of TB to 7-MX, but not in all livers.
Diethyldithiocarbamate, an inhibitor of CYP2E1, and 4-nitrophenol, a highly selective CYP2E1 substrate, both decreased the formation of 3,7-DMU in human liver microsomes by ≈85%. These compounds also decreased the conversion of TB to 3-MX and 7-MX by about 55–60% and 35–55%, respectively. Furafylline, a specific inhibitor of CYP1A2, reduced the formation of 7-MX by more than 60% in two livers but was without effect in a third. Collectively, these data suggest that CYP2E1 and CYP1A2 are the major catalysts for 7-MX formation, although wide interindividual variability may occur in the contribution of the latter, and that CYP2E1 is responsible for ≈50% of the conversion of TB to 3-MX. Confirmatory evidence was provided by the use of recombinant enzymes. Expressed CYP1A2 and CYP2E1 exhibited similar apparent Km values for 7-MX formation, and apparent Km values of the recombinant enzymes were similar to the apparent Km observed for human liver microsomal TB N3-demethylation. CYP2E1 was further shown to have the capacity to convert TB to both 3-MX and 3,7-DMU.
The marked variability in the contribution of CYP1A2 to 7-MX formation warrants comment. The immunoreactive CYP1A2 content and caffeine N3-demethylase activity of liver #3, determined previously [19], are about 50% those of livers #1 and #2, while the 4-nitrophenol hydroxylase activity (a marker for CYP2E1 [15]) of liver #3 is 2–3 fold higher than those for livers #1 and #2. Even given these differences, an apparent complete lack of contribution of CYP1A2 to TB N3-demethylation (i.e. 7-MX formation) in liver #3 is surprising. Inhibition of 7-MX formation by coumarin in liver #3 suggests that CYP2A6 may be responsible in part for TB N3-demethylation. Coumarin also inhibited 3-MX formation in liver #3 and it might be speculated that, comparatively, the CYP2A6 content of this liver was high. The CYP2A6 activities of the livers investigated here are not, however, available. Recombinant CYP2A6 has been shown previously to have the capacity to convert TB to 3-MX, but not 7-MX [20].
The identity of the isoform(s), other than CYP2E1, which is responsible for 3-MX formation is unclear. Coumarin inhibition data suggest that CYP2A6 may contribute to 3-MX formation in some livers and, as noted above, recombinant CYP2A6 has been shown to possess TB N7-demethylase activity. Possible contributions of enzymes such as CYP 1B1, 2B6, 2C8 and 2C18 were not investigated here. The observation of Michaelis-Menten kinetics for human liver microsomal TB N7-demethylation suggests a similar affinity of TB for all enzymes contributing to this reaction.
Of some interest are the atypical TB 8-hydroxylatioin (i.e. 3,7-DMU formation) kinetics observed here (Figure 2). Eadie-Hofstee plots were curvi-linear, suggestive of the substrate activation observed for numerous CYP3A4 substrates, including the 8-hydroxylation of the related methylxanthine caffeine [14, 21]. However, CYP2E1 appears to be almost solely responsible for 3,7-DMU formation, with little or no contribution from CYP3A4. Troleandomycin was essentially without effect on TB 8-hydroxylation (Table 1) and additional experiments undertaken in two livers with erythromycin similarly precluded a contribution of CYP3A4 (data not shown). Substrate activation is a phenomenon not usually associated with CYP2E1 and an alternative explanation for the nonlinear TB 8-hydroxylation kinetics is provided by the mechanism of formation of 3,7-DMU (Figure 1). 3,7-DMU and 3,7-DAU are known to be derived from a common intermediate of unknown structure 22]. In the presence of cellular thiols, the intermediate is reduced to form 3,7-DAU but following oxidation of the available thiol pool, as might be expected to occur with increasing substrate concentration in vitro, 3,7-DMU becomes the major reaction product. Since 3,7-DMU and 3,7-DAU are derived from a common intermediate whose degradation is independent of CYP [22], it may be assumed that CYP2E1 also catalyses the conversion of TB to 3,7-DAU. These data emphasize the importance of a knowledge of reaction mechanisms in interpreting the kinetics of metabolite formation.
As noted in the introduction, TB has numerous features which make it a potentially useful substrate for the measurement of enzyme activity in population studies. Given the higher plasma TB clearance and metabolic clearances by N3-and N7-demethylation (i.e. 7-MX and 3-MX formation) reported in cigarette smokers [10] and marked induction of TB metabolism in 3-methylcholanthrene-treated rats [22], it was assumed that CYP1A2 would be the principal contributor to human hepatic TB metabolism. CYP1A2 is known to be responsible for the N-demethylations of the related methylxanthines caffeine and theophylline [3, 7, 8, 14, 19, 20, 23]. Although a contributory role for CYP1A2 in TB N3-demethylation was demonstrated here, it is clear that TB is unsuitable for use as a substrate probe for this enzyme. CYP2E1 catalyses all TB metabolic pathways, but given the contributions of other enzymes to both N-demethylations and the minimal formation of 3,7-DMU in vivo 9], TB is similarly of little value as a marker for CYP2E1 activity. With respect to the induction of TB elimination in smokers, it has been reported recently that CYP2E1 transcription and translation in kidney are increased in mice exposed to tobacco smoke [24]. However, induction of human CYP2E1 by cigarette smoke is yet to be demonstrated.
Acknowledgments
Support from the National Health and Medical Research Council of Australia and technical assistance from Karen Lillywhite is gratefully acknowledged.
References
- 1.Eaton DL, Gallagher EP, Bammler TK, Kunze KL. Role of cytochrome P4501A2 in chemical carcinogenesis: implications for human variability in expression and enzyme activity. Pharmacogenetics. 1995;5:259–274. doi: 10.1097/00008571-199510000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Guengerich FP. Metabolic activation of carcinogens. Pharmacol Ther. 1992;54:17–61. doi: 10.1016/0163-7258(92)90050-a. [DOI] [PubMed] [Google Scholar]
- 3.Miners JO, McKinnon RA. Cytochrome P4501A. In: Levy RH, Thummel KE, Trager WF, Hansten PD, Eichelbaum M, editors. Metabolic Drug Interactions. Philadelphia: Lippincott-Raven; (in press) [Google Scholar]
- 4.McKinnon RA, Hall PM, Quattrochi LC, Tukey RH, McManus ME. Localization of CYP1A1 and CYP1A2 mRNA in normal liver and hepatocellular carcinoma by in situ hybridization. Hepatology. 1991;14:848–856. doi: 10.1002/hep.1840140517. [DOI] [PubMed] [Google Scholar]
- 5.Schweik H, Taylor JA, Kitareewan S, Linko P, Nagorney D, Goldstein JA. Expression of CYP1A1 and CYP1A2 genes in human liver. Pharmacogenetics. 1993;3:239–249. doi: 10.1097/00008571-199310000-00003. [DOI] [PubMed] [Google Scholar]
- 6.Rostami-Hodjegan A, Nurminsen S, Jackson PR, Tucker GT. Caffeine urinary metabolic ratios as markers of enzyme activity: a theoretical assessment. Pharmacogenetics. 1996;6:121–149. doi: 10.1097/00008571-199604000-00001. [DOI] [PubMed] [Google Scholar]
- 7.Miners JO, Birkett DJ. The use of caffeine as a metabolic probe for human drug metabolising enzymes. Gen Pharmacol. 1996;27:245–249. doi: 10.1016/0306-3623(95)02014-4. [DOI] [PubMed] [Google Scholar]
- 8.Kalow W, Tang B-K. Use of caffeine for enzyme assays: a critical appraisal. Clin Pharmacol Ther. 1993;53:503–514. doi: 10.1038/clpt.1993.63. [DOI] [PubMed] [Google Scholar]
- 9.Miners JO, Attwood J, Birkett DJ. Theobromine metabolism in man. Drug Metab Disp. 1982;10:672–675. [PubMed] [Google Scholar]
- 10.Miners JO, Attwood J, Wing LMH, Birkett DJ. Influence of cimetidine, sulfinpyrazone and cigarette smoking on theobromine metabolism in man. Drug Metab Disp. 1985;13:598–601. [PubMed] [Google Scholar]
- 11.Lelo A, Birkett DJ, Robson RA, Miners JO. Comparative pharmacokinetics of caffeine and its primary demethylated metabolites paraxanthine, theobromine and theophylline. Br J Clin Pharmacol. 1986;22:177–182. doi: 10.1111/j.1365-2125.1986.tb05246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tassaneeyakul W, Veronese ME, Birkett DJ, Miners JO. High performance liquid chromatographic assay for 4-nitrophenol hydroxylation, a putative cytochrome P4502E1 activity, in human liver microsomes. J Chromatogr. 1993;616:73–78. doi: 10.1016/0378-4347(93)80473-h. [DOI] [PubMed] [Google Scholar]
- 13.Lowry OH, Rosebrough NJ, Farr AL, Randall AJ. Protein measurement with Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 14.Tassaneeyakul W, Mohamed Z, Birkett DJ, et al. Caffeine as a probe for human cytochromes P450; validation using cDNA-expression, immunoinhibition and microsomal kinetic and inhibitor techniques. Pharmacogenetics. 1992;2:173–183. doi: 10.1097/00008571-199208000-00004. [DOI] [PubMed] [Google Scholar]
- 15.Tassaneeyakul W, Veronese ME, Birkett DJ, Gonzalez FJ, Miners JO. Validation of 4-nitrophenol as an in vitro substrate probe for human liver CYP2E1 using cDNA-expression and microsomal kinetic techniques. Biochem Pharmacol. 1993;46:1975–1981. doi: 10.1016/0006-2952(93)90639-e. [DOI] [PubMed] [Google Scholar]
- 16.Birkett DJ, Mackenzie PI, Veronese ME, Miners JO. In vitro techniques can predict human drug metabolism. Trends Pharmacol Sci. 1993;14:291–294. doi: 10.1016/0165-6147(93)90043-j. [DOI] [PubMed] [Google Scholar]
- 17.Miners JO, Veronese ME, Birkett DJ. In vitro approaches for the prediction of human drug metabolism. Ann Reports Med Chem. 1994;29:307–316. [Google Scholar]
- 18.Chang TKH, Gonzalez FJ, Waxman DJ. Evaluation of triacetyloleandomycin, α-naphthoflavone and diethyldithiocarbamate as selective chemical probes for the inhibition of human cytochromes P450. Arch Biochem Biophys. 1994;311:437–442. doi: 10.1006/abbi.1994.1259. [DOI] [PubMed] [Google Scholar]
- 19.Tassaneeyakul W, Birkett DJ, McManus ME, et al. Caffeine metabolism by human hepatic cytochromes P450: contributions of 1A2, 2E1 and 3A isoforms. Biochem Pharmacol. 1994;47:1767–1776. doi: 10.1016/0006-2952(94)90304-2. [DOI] [PubMed] [Google Scholar]
- 20.Gu L, Gonzalez FJ, Kalow W, Tang B-K. Biotransformation of caffeine, paraxanthine, theobromine and theophylline by cDNA-expressed human CYP1A2 and CYP2E1. Pharmacogenetics. 1992;2:73–77. doi: 10.1097/00008571-199204000-00004. [DOI] [PubMed] [Google Scholar]
- 21.Andersson T, Miners JO, Veronese ME, Birkett DJ. Diazepam metabolism by human liver microsomes is mediated by both S-mephenytoin hydroxylase and CYP3A isoforms. Br J Clin Pharmacol. 1994;38:131–137. doi: 10.1111/j.1365-2125.1994.tb04336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lelo A, Birkett DJ, Miners JO. Mechanism of formation of 6-amino-5-(N-methylformylamino) -1-methyluracil and 3,7–dimethyluric acid from theobromine in the rat in vitro; involvement of cytochrome P450 and a cellular thiol. Xenobiotica. 1990;20:823–833. doi: 10.3109/00498259009046896. [DOI] [PubMed] [Google Scholar]
- 23.Robson RA, Miners JO, Matthews AP, et al. Characterisation of theophylline metabolism by human liver microsomes: Inhibition and immunochemical studies. Biochem Pharmacol. 1998;37:1651–1659. doi: 10.1016/0006-2952(88)90423-6. [DOI] [PubMed] [Google Scholar]
- 24.Seree EM, Villard PH, Re JL, et al. High inducibility of mouse renal CYP2E1 gene by tobacco smoke and its possible effect on DNA single strand breaks. Biochem Biophys Res Commun. 1996;219:429–434. doi: 10.1006/bbrc.1996.0250. [DOI] [PubMed] [Google Scholar]