

Abstract
Aims
To investigate the kinetics of CYP-mediated N-demethylation of methadone in human liver microsomes, and examine the role of stereoselectivity and CYP isoforms involved.
Methods
The kinetics of 2-ethylidene-1,5-dimethyl-3,3-diphenylpyrrolidine (EDDP) formation via N-demethylation of rac-, (R)- and (S)-methadone in human liver microsomes prepared from six liver samples were determined by h.p.l.c., and inhibition of metabolic function was studied using isoform-specific chemical inhibitors and monoclonal antibodies. Microsomes containing expressed CYP3A4, CYP2D6 and CYP2C19 were also used to examine the formation of EDDP.
Results
The Vmax, Km, and CLint values for the formation of EDDP from rac-, (R)- and (S)-methadone were in the ranges of 20–77 nmol mg−1 protein h−1, 125–252 μm, and 91–494 ml h−1g−1 protein. Km and CLint values for (R)- and (S)-methadone were not statistically significantly different (P>0.05), while Vmax values for (S)-methadone were 15% (P=0.045) lower than for (R)-methadone. Expressed CYP3A4 and CYP2C19 showed similar reaction rates for both (R)- and (S)-methadone, while CYP2D6 did not catalyse this reaction. Selective chemical inhibitors of CYP3A (troleandomycin, ketoconazole) and monoclonal human CYP3A4 antibodies significantly inhibited (P<0.05) the formation of EDDP in a concentration dependent manner by up to 80%. Sulphaphenazole (CYP2C9) also significantly inhibited (P<0.05) EDDP formation (range 14–25%). There were no statistically significant differences in the inhibition observed between the three substrates. Selective inhibitors of CYP1A2 (furafylline), CYP2A6 (coumarin), CYP2C19 ((S)-mephenytoin), CYP2D6 (quinidine) and CYP2E1 (diethyldithiocarbamic acid sodium salt and monoclonal human CYP2E1 antibodies) had no significant (P>0.05) effect.
Conclusions
The N-demethylation of methadone in human liver microsomes is not markedly stereoselective, and is mediated mainly by CYP3A4 with the possible involvement of CYP2C9 and CYP2C19. Thus, the large interindividual variation reported for methadone pharmacokinetics may be due to variability in the expression of these CYP isoforms, and the reported stereoselectivity in the systemic clearance of methadone in vivo is not due to stereoselectivity in N-demethylation.
Keywords: CYP3A4, EDDP formation, human liver microsomes, methadone N-demethylation, stereoselectivity
Introduction
Rac-methadone is commonly used in the management of chronic pain and as maintenance therapy for the pharmacological treatment of opioid dependence. Rac-methadone (6-dimethylamino-4,4-diphenyl-3-heptanone) is a chiral molecule (Figure 1) of (R)- and (S)-enantiomeric forms in which (R)-methadone has a 10-fold higher affinity at μ and δ opioid receptors [1] and possesses up to 50 times the analgesic activity of (S)-methadone in human and animal models of pain [2]. (R)-methadone has also been shown to prevent the occurrence of opioid withdrawal symptoms, while (S)-methadone is ineffective [3]. Despite this high eudismic ratio, methadone is used as the racemate in most countries. The two enantiomers differ significantly in their disposition in humans, with (R)-methadone having a greater volume of distribution [4], longer half-life [4], higher total systemic clearance [4] and lower plasma protein binding [5, 6]. It is not known whether the stereoselectivity in systemic clearance is due to metabolism (intrinsic clearance) and/or protein binding.
Figure 1.
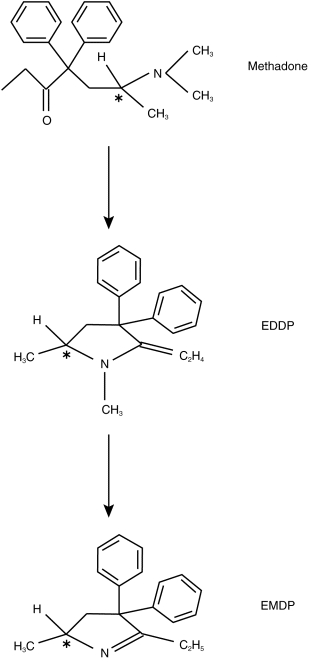
Chemical structure and N-demethylation pathway of rac-methadone (*indicates chiral carbon atom).
Methadone metabolism in humans is complex, with nine metabolites identified [7–9] although the data have been qualitative rather than quantitative. Mono N-demethylation, a primary metabolic pathway, results in the formation of a highly unstable compound which then undergoes spontaneous cyclization and dehydration to form 2-ethylidene-1,5-dimethyl-3,3-diphenylpyrrolidine (EDDP, Figure 1), which may then be further N-demethylated to 2-ethyl-5-methyl-3,3-diphenylpyrroline (EMDP, Figure 1) [8].p-hydroxylation of one aromatic ring has also been reported, resulting in the formation of four diastereomeric compounds collectively referred to as p-hydroxy methadone [8, 10]. After oral dosing, unchanged methadone and EDDP are found in similar amounts in the urine of humans and account for up to 50% of the dose [8, 10], with only trace amounts of EMDP; other metabolites appear to contribute relatively little to the overall metabolism of methadone [8, 10], although this has not been well studied, since complete mass balance has not been achieved.
The in vitro oxidative metabolism of rac-methadone in human liver microsomes has only recently been described [11–14]. Iribarne and coworkers [11] reported that rac-methadone was N-demethylated by CYP3A4, and that selective inhibitors of CYP2D6, CYP1A2, CYP2C9 had no effect. Based upon experiments with heterologously expressed CYP enzymes they did not rule out the contribution of other CYP450 isoforms. The role of stereoselectivity was not examined.
The aims of the present study were to investigate the CYP-mediated N-demethylation of methadone in human liver microsomes, and to examine the role of stereoselectivity and CYP isoforms involved in each reaction.
Methods
Chemicals
Rac-methadone as the hydrochloride salt, (R)- and (S)-methadone as the free bases (4R,6S)- and (4S,6S)-p hydroxylated methadone (p-hydroxy methadone) as the hydrochloride salts were obtained from the National Institute of Drug Abuse (Rockville, MD, USA). 2-Ethylidene-1,5-dimethyl-3,3-diphenylpyrrolidine (EDDP) as the hydroiodide salt and 2-ethyl-5-methyl-3, 3-diphenylpyrroline (EMDP) as the hydrochloride salt were purchased from Alltech-Applied Science Labs (State College, PA, USA). Other materials were obtained from the following sources: furafylline and (S)-mephenytoin (Ultrafine Chemicals, Manchester, England), bovine serum albumin (fraction V), butylated hydroxytoluene, chlorzoxazone, coumarin, diethyldithiocarbamic acid sodium salt (DDC), DL-isocitric acid tri-sodium salt, folin-ciocalteau reagent, isocitrate dehydrogenase (NADP, type IV), quinidine sulphate, sulphaphenazole, triethylamine and troleandomycin were obtained from Sigma Chemical Company (St Louis, Mo, USA). Omeprazole was supplied by Astra Pharmaceuticals (Sydney, Australia), fluoxetine was a gift from Dr W. Hooper (Department of Medicine, Royal Brisbane Hospital, Brisbane, Australia) and diazepam and ketoconazole were gifts from Professor J. Miners (Department of Clinical Pharmacology, Flinders Medical Centre, Bedford Park, Adelaide, Australia). Monoclonal antibodies to CYP3A4 and CYP2E1, and microsomes from human lymphoblastoid cells containing expressed CYP3A4, CYP2D6 and CYP2C19 were purchased from Gentest Corporation (Woburn, MA, USA). All other reagents and chemicals were obtained from commercial sources and were of analytical grade quality.
Human liver microsomes
Ethical approval was obtained from the Committee on the Ethics of Human Experimentation of the University of Adelaide and the Human Ethics Committee of the Royal Adelaide Hospital. All patients gave written informed consent for their liver tissue to be used. Liver tissue (HLS# 5, 16, 21, 22, 23, 31) was obtained from six patients undergoing partial hepatectomy for hepatic tumours. Donors ranged in age from 25 to 73 years, three were female and all had normal clinical chemistry and haematology prior to surgery except donor 31 with hypoalbuminaemia (33 g l−1) and anaemia (haemoglobin 9.7 g l−1). All tissue samples used were normal on gross morphology. Microsomes were prepared by differential centrifugation of liver homogenates [15], and total microsomal protein and cytochrome P450 content of the microsomes were quantified using the methods of Lowry and coworkers [16] and Omura & Sato [17], respectively. Liver samples and microsomal preparations were stored at −80° C until used. DNA was extracted from the liver tissue for the determination of CYP2D6 and CYP2C19 genotypes. CYP2D6 alleles screened were CYP2D6*1A, CYP2D6*4A, CYP2D6*4B, CYP2D6*4C, CYP2D6*4D and CYP2D6*5 [18, 19]. CYP2C19 alleles screened were CYP2C19*1, CYP2C19*2 and CYP2C19*3 [20]. Results of the genotyping corresponded to the extensive metaboliser phenotype for both CYP isoforms in all liver samples. Genotyping of HLS# 16 was not possible due to insufficient tissue.
Microsomal incubations
Incubations were performed in duplicate at 37° C in a shaking water bath for 45 min. The incubates of 200 μl final volume contained 50 mm sodium phosphate buffer (pH 7.4), NADPH generating system (1 mm NADP, 1 unit ml−1 isocitrate dehydrogenase, 5 mm magnesium chloride), substrate (minimum of 10 concentrations, final concentration range of 1–1500 μm for rac-methadone and 1–1250 μm for (R)- and (S)-methadone) and 100 μg microsomal protein. The formation of EDDP was stopped by the addition of 100 μl ice cold acetonitrile, samples were then vortexed briefly, centrifuged (10 000 g, 10 min) and a 100 μl aliquot injected on to the h.p.l.c. system. The rate of EDDP formation from 100 μm rac-methadone, (R)-methadone and (S)-methadone was observed to be linear up to 120 min and 400 μg microsomal protein.
EDDP quantification
EDDP was quantified using a reversed phase h.p.l.c. system which comprised a LC-6 A pump (Shimadzu, Kyoto, Japan) at a flow rate of 1.0 ml min−1, a Wisp 710B autoinjector (Waters, Milford, MA, USA), a 8×10 Radial Compression Module (Waters) containing a 100 mm×5 mm Nova-Pak C18 4 μm cartridge (Waters), an UVIDEC-100-V spectrophotometer (Jasco, Tokyo, Japan) set at 210 nm and a C-R6 A Chromatopac integrator (Shimadzu). Optimal separation of the compounds of interest was achieved with a mobile phase of 30% acetonitrile and 0.2% triethylamine in 50 mm NaH2PO4 with final pH adjusted to 4.3 with ortho-phosphoric acid. Retention times for (4R,6S)-p-hydroxy methadone, (4S,6S)-p-hydroxy methadone, EDDP, methadone, and EMDP were 5, 6, 9, 17, and 25 min, respectively. Under these conditions all compounds of interest were baseline resolved, with the exception of the p-hydroxy methadone diastereomers. In preliminary experiments, EMDP and the p-hydroxy methadone diastereomers were not observed following microsomal incubation and no further modifications to the h.p.l.c. system were attempted. Quantification of EDDP was performed with calibration curves consisting of 12 standards over the concentration range 0.25–50 μm. Inter-assay variability was monitored with quality control (QC) samples prepared in duplicate at three concentrations; low (LQC, 0.62 μm), medium (MQC, 1.74 μm) and high (HQC, 12.45 μm). Inter-assay precision (n=6) was 7.4%, 3.6% and 0.7%, while interassay inaccuracy (n=6) was −3.0%, +4.8% and −3.1% for the LQC, MQC and HQC, respectively. Similarly intra-assay precision (n=6) was 6.4%, 6.2% and 3.9%, while intra-assay inaccuracy (n=6) was −8.6%, −0.2% and −3.2% for the LQC, MQC and HQC, respectively. The assay was both precise and accurate at the limit of quantification (0.25 μm) with coefficient of variation and inaccuracy (n=3) below 10%.
Inhibition studies with chemical inhibitors
Microsomes from three human liver samples (HLS# 5, 16 and 21) were used in duplicate to examine the inhibition of EDDP formation from rac-, (R)- and (S)-methadone. Substrates were incubated at a concentration equal to their respective Km values for that particular liver sample. Chemical inhibitors considered to be specific for various isoforms [21] were coumarin (CYP2 A6, 100 μm), DDC (CYP2E1, 100 μm and 10 μm), furafylline (CYP1A2, 100 μm), quinidine (CYP2D6, 1 μm), sulphaphenazole (CYP2C9, 100 μm), troleandomycin (CYP3A, 1, 10 and 100 μm), ketoconazole (CYP3A, 0.1, 1 and 100 μm), and (S)-mephenytoin (CYP2C19, 100 μm). In addition diazepam (CYP3A/2C19, 100 μm [22]) and fluoxetine (CYP2D6/3A, 100 μm [23]) were also tested as they are often coadministered to individuals in methadone maintenance therapy. Chlorzoxazone (100 μm) and omeprazole (100 μm) were also tested. Incubation conditions did not alter from the kinetic study, except that the mechanism-based inhibitors DDC, furafylline and troleandomycin required 20 min preincubation prior to the addition of substrate. Inhibitors were dissolved in various solvents due to differences in solubility. Of those not made up in water, dimethylsulphoxide was used to dissolve furafylline and sulphaphenazole with final dimethylsulfoxide concentrations of 1.7% and 0.5%, respectively; methanol (<0.5% final concentration) was used to dissolve chlorzoxazone, diazepam, fluoxetine, ketoconazole and troleandomycin. Omeprazole was prepared in a mixture of water with pH adjusted to 10.5 with sodium hydroxide, and methanol (final methanol concentration 0.5%). Incubations containing equivalent amounts of diluents were always used as controls. When compared with control reactions containing no diluent, control reactions containing methanol or 0.5% dimethylsulphoxide showed greater than 90% activity, as did the pH 10.5 water containing 0.5% methanol, while those containing 1.7% dimethylsulphoxide showed greater than 80% activity. Control incubations containing inhibitors alone did not produce any chromatographic peaks that interfered with the quantification of EDDP.
Inhibition studies with CYP specific antibodies
Microsomes from a single human liver sample (HLS# 5) were used in duplicate to examine the inhibition of EDDP formation from (R)- and (S)-methadone. Substrates were incubated at a concentration equal to their respective Km values for that particular liver sample. Antibodies specific for CYP3A4 and CYP2E1 were used at two titres (2 μl/100 μg and 8 μl/100 μg microsomal protein) in duplicate. Antibodies were used according to the manufacturers directions. Incubation conditions did not alter from the kinetic study.
Metabolism studies with expressed CYP isoforms
Microsomes from human lymphoblastoid cells containing expressed CYP3A4, CYP2D6 and CYP2C19 were used in duplicate to examine the formation of EDDP from (R)- and (S)-methadone. Microsomes containing expressed CYP isoforms were used according to the manufacturers directions. Incubation conditions did not alter from the kinetic study, except that the CYP enzymes were kept ice cold until addition to the reaction mixture. Amounts of CYP enzyme used were kept constant (6 pmol P450/incubation), which resulted in amounts of protein similar to that used for the human liver microsomes (100 μg). Reaction velocities were compared to that obtained from a single human liver sample (HLS#5), which was incubated at the same time as the expressed enzyme. Substrate concentrations were fixed at 200 μm.
Secondary metabolism of EDDP
The formation of EMDP from EDDP via a second N-demethylation reaction was also examined. EDDP was incubated at a concentration of 1 mm in a single microsomal preparation under conditions identical to those used for the methadone incubations. Analysis of the sample for EMDP was performed as for the methadone incubations.
Data analysis
The rates of EDDP formation from the kinetic studies (V) were expressed as nmol mg−1 protein h−1, and Eadie-Hofstee (V/Substrate concentration vsV) plots were constructed, and a single enzyme Michaelis-Menten kinetics equation was fitted to the unweighted data using nonlinear least-squares regression analysis (Regression, Blackwell Scientific Publications, Oxford, UK) giving estimates of Vmax and Km, where Vmax is the maximum reaction velocity and Km is the substrate concentration at which the reaction rate is half Vmax. Intrinsic clearance (CLint) was calculated as Vmax/Km. Rates of EDDP formation obtained from the studies with expressed CYP isoforms were expressed as pmol EDDP formed pmol−1 P450 h−1. All data are presented as mean±s.d.. Statistically significant differences in Vmax, Km, CLint and percentage inhibition by the chemical inhibitors between rac-, (R)-, and (S)-methadone were assessed using paired t-tests. Statistically significant differences in percentage inhibition of the chemical inhibitors compared to control reactions was assessed with one-tailed t-tests. Differences were considered significant when P<0.05.
Results
Metabolic profile
No chromatographic peaks corresponding to either of the p-hydroxy methadone diastereomers or EMDP were observed in any incubation containing rac-, (R)- or (S)-methadone. The only metabolite observed was EDDP, whose formation was NADP-dependent, and was localized to the microsomal fraction as no EDDP formation was observed in incubations of methadone in the cytosolic liver fraction (preliminary investigations, results not shown).
Kinetics of EDDP formation
Eadie-Hofstee plots were found to be linear in all cases (Figure 2) and a single enzyme Michaelis-Menten kinetic equation was fitted to the data (Table 1). For Vmax there was a statistically significant difference (P=0.045) between (R)-methadone and (S)-methadone (Table 1). For Km there was a statistically significant difference (P=0.041) between (R)-methadone and rac-methadone (Table 1) and for CLint there was a statistically significant difference (P=0.025) between (R)-methadone and rac-methadone (Table 1).
Figure 2.
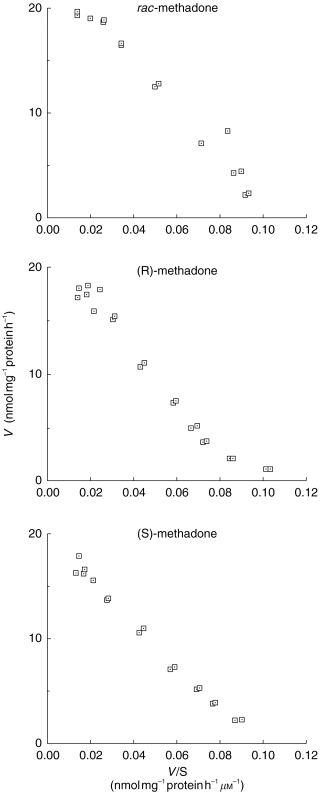
Eadie-Hofstee representations of EDDP formation from rac-, (R)-, and (S)-methadone by microsomes prepared from HLS# 31.
Table 1.
Michaelis-Menten parameters (Vmax and Km) and CLint for EDDP formation from rac-, (R)-, and (S)-methadone (MD) by human liver microsomes.

Inhibition with chemical inhibitors
EDDP formation from rac-, (R)-, and (S)-methadone was significantly (P<0.05) inhibited by ketoconazole at 100 μm (75±18%, 83±11%, 78±4% inhibition), 1 μm (43±12%, 43±10%, 46±14% inhibition) and 0.1 μm (8±2%, 8±3%, 7±5% inhibition); troleandomycin at 100 μm (45±7%, 44±11%, 43±10% inhibition), 10 μm (22±11%, 19±10%, 21±12% inhibition); sulphaphenazole at 100 μm (18±6%, 26±4%, 14±7% inhibition); DDC at 100 μm (44±12%, 44±11%, 45±11% inhibition); omeprazole at 100 μm (29±13%, 29±7.3%, 23±11% inhibition) and fluoxetine at 100 μm (56±10%, 53±6%, 50±13% inhibition) compared with control reactions (Figure 3). In addition EDDP formation from (S)-methadone was significantly (P=0.006) inhibited by chlorzoxazone (15±2% inhibition) (Figure 3). The inhibition of EDDP formation was not statistically significantly different between the three substrates in all cases (P>0.05, Figure 3). The other chemical inhibition experiments did not result in significant (P>0.05) inhibition of the formation of EDDP from rac-, (R)-, and (S)-methadone compared with control reactions (Figure 3).
Figure 3.
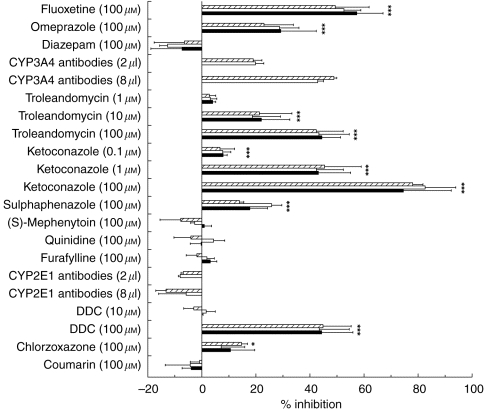
Effect of chemical inhibitors on EDDP formation from (▪) rac-, (□) (R)-, and ( ) (S)-methadone by human liver microsomes (n=3). Error bars indicate s.d. *Indicates statistically significant inhibition compared with control (P<0.05). No statistically significant differences (P0.05) were observed between rac-, (R)-, and (S)-methadone.
) (S)-methadone by human liver microsomes (n=3). Error bars indicate s.d. *Indicates statistically significant inhibition compared with control (P<0.05). No statistically significant differences (P0.05) were observed between rac-, (R)-, and (S)-methadone.
Immunological inhibition
EDDP formation from (R)- and (S)-methadone was inhibited by both the 8 μl (43±2% and 49±1%) and 2 μl (20±3% and 19±3%) titres of the CYP3A4 specific antibodies, respectively. CYP2E1 specific antibodies did not inhibit this reaction.
Metabolism by expressed CYP isoforms
When corrected for CYP450 content, expressed CYP3A4 and CYP2C19 catalysed the N-demethylation of (R)- and (S)-methadone at similar rates, while the formation of EDDP with expressed CYP2D6 was not observed (Figure 4).
Figure 4.
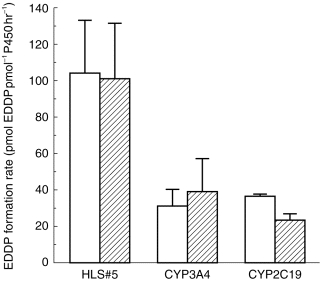
Comparison of EDDP formation from (□) (R)-methadone and ( ) (S)-methadone by microsomes from a human liver (HLS#5) and human lymphoblastoid cells containing the expressed individual CYP450 isoforms CYP3A4 and CYP2C19 (n=2). Error bars indicate s.d.
) (S)-methadone by microsomes from a human liver (HLS#5) and human lymphoblastoid cells containing the expressed individual CYP450 isoforms CYP3A4 and CYP2C19 (n=2). Error bars indicate s.d.
Secondary metabolism of EDDP
Incubation of EDDP at a single high concentration (1 mm) resulted in <1% formation of EMDP.
Discussion
Substantial interindividual variability in rac-methadone disposition has hindered its more widespread use in pain management [24] and can pose difficulties in dosage regimen designs in opioid dependence treatment. The 4–5 fold range in CLint observed for the formation of EDDP from rac-, (R)- and (S)-methadone seen in the present study was mainly a consequence of the variability in Vmax values since the Km values showed little intersubject variability.
To date this is the first report examining the stereoselectivity in the N-demethylation of methadone in human liver microsomes. The Vmax for the formation of EDDP from (S)-methadone was significantly lower than for (R)-methadone, however, the magnitude of the difference was small (mean difference: 6.2 nmol mg−1 protein h−1) and less than 15%. There were no significant differences between (R)- and (S)-methadone observed for Km, even though the chiral carbon is in close proximity to the site of oxidation. Similarly, there was no significant difference between (R)- and (S)-methadone for CLint, indicating that it is unlikely that there would be stereoselectivity in this metabolic pathway in vivo. These data are supported by the expressed enzyme and inhibition data, which also did not indicate a clear difference between the two substrates. Rac-methadone Km and CLint were significantly different (P<0.05) to those of (R)-methadone, but not (S)-methadone, although the magnitude of the differences were small (mean differences ≈15%). Interpretation of these data is difficult, as rac-methadone is a 1:1 mixture of the two enantiomers. As the two individual enantiomers do not differ in their affinity (Km) then one would expect the Km of the racemate to be comparable. The observed greater CLint for rac-methadone compared with (R)-methadone appears to be due to the lower Km, as Vmax values were similar.
The Vmax values are comparable with those reported by Iribarne and coworkers [11] in their study of rac-methadone in a panel of 20 human liver microsomal preparations. However, they estimated the Km for rac-methadone in three livers to be 545±258 μm, a result indicating a low and highly variable affinity for the CYP450 isoform(s) mediating the formation of EDDP. The results from the present study indicate very little variability in six human liver preparations with Km values for rac-, (R)- and (S)-methadone almost 3-fold lower than that reported by Iribarne and coworkers [11]. Reasons for this difference are not apparent.
In the present study p-hydroxy methadone and EMDP were not formed, suggesting that they are likely to be minor metabolites in humans, supporting the data of Sullivan and Due [8].
CYP3A4 has been reported to be involved in the N-demethylation of rac-methadone, although the contribution of other isoforms could not be ruled out [11, 12, 14]. Our results confirm the role of CYP3A4 in this metabolic pathway as troleandomycin, ketoconazole and a selective 3A4 antibody caused concentration dependent inhibition. Approximately 20% inhibition with sulphaphenazole (100 μm) was observed indicating that CYP2C9 may also mediate this reaction, in support of Moody and coworkers [14] who reported a mean of 57% inhibition of this pathway by the same concentration of sulphaphenazole. However, Iribarne and coworkers [11] found no inhibition with sulphaphenazole at 5 μm. The selective chemical inhibitors of CYP1A2, CYP2A6, CYP2C19, CYP2D6 and CYP2E1 had no effect indicating that these isoforms are unlikely to contribute substantially to EDDP formation.
Heterologously expressed human CYP450 proteins have also been examined for their ability to N-demethylate rac-methadone, and only CYP3A4 was considered to be important [11, 14]. The current results agree with those above since formation of EDDP from (R)- and (S)-methadone by microsomes from human lymphoblastoid cells containing expressed CYP3A4 at velocities comparable to those in human liver microsomes were observed. Microsomes containing expressed CYP2C19 were also found to catalyse the N-demethylation of (R)- and (S)-methadone at similar rates to expressed CYP3A4. It is unlikely that CYP2C19 plays a major role in this reaction as, total CYP2C is expressed in the human liver in amounts less than 50% of CYP3A [25, 26], no correlation was found between (S)-mephenytoin hydroxylation and rac-methadone N-demethylation reaction rates [11] and 100 μm (S)-mephenytoin, a CYP2C19 inhibitor, caused no significant inhibition of EDDP formation.
There are several reports of either decreased rac-methadone plasma concentrations and/or concurrent complaints of opioid withdrawal symptoms by patients soon after commencement of therapy with known CYP3A inducers [27–31]. The present findings confirm the involvement of CYP3A4 in the metabolism of methadone and the potential for clinically important drug–drug interactions.
Methadone has been shown to alter the metabolism of several CYP2D6 substrates in vivo and in vitro [32–35]. These results would appear to suggest that methadone is metabolized by CYP2D6. However, the results of Iribarne and coworkers [11], Moody and coworkers [14] and the present study in which quinidine, a potent CYP2D6 inhibitor, elicited no inhibition of methadone N-demethylation and expressed CYP2D6 resulted in very low EDDP formation seem to rule out the role of CYP2D6. CYP isoforms can have high affinity for a substrate but not metabolize the substrate (e.g. quinidine [36]). These data indicate that, although methadone is a potent inhibitor of CYP2D6, the metabolism of methadone is not mediated by this CYP isoform in humans.
There was no inhibition of EDDP formation observed when DDC was used at 10 μm, or with a CYP2E1 selective antibody. DDC inhibited the formation of EDDP from rac-, (R)- and (S)-methadone by greater than 56% at 100 μm, however, this concentration is not selective for CYP2E1, and probably reflects inhibition of CYP3A4 [37]. Iribarne and coworkers [11] reported that catalytic activities of CYP2E1-mediated reactions did not correlate with rac-methadone N-demethylation, and more recently rac-methadone did not inhibit CYP2E1-mediated reactions [12]. Chlorzoxazone (100 μm) had a minimal effect on (S)-methadone N-demethylation, and no effect on rac- or (R)-methadone. These data indicate that CYP2E1 is not involved in methadone N-demethylation, and that care should be taken when selecting the concentrations of chemical inhibitors; the use of antibodies and expressed CYP isoforms are important in determining CYP isoform involvement.
Although diazepam has been reported to competitively inhibit the metabolism of rac-methadone with a Ki of 50 μm in human liver microsomes [11], the present study found no such inhibitory effect at a concentration of 100 μm. This difference in results is hard to reconcile, however, Iribarne and coworkers [11] appeared to base their results on a single liver preparation, and the influence of variation between tissue samples is one possible explanation. Our in vitro findings are consistent with two reports of no effect of diazepam on the disposition of rac-methadone in vivo [38, 39].
Fluoxetine inhibited the formation of EDDP from rac-, (R)- and (S)-methadone by ≈ 50% which is consistent with its known CYP3A4 inhibitory action [23, 40, 41]. Omeprazole was found to inhibit EDDP formation from rac-, (R)- and (S)-methadone by ≈30%. This is consistent with its known affinity for CYP3A4 and CYP2C19 [42–44].
In conclusion, the N-demethylation of methadone in human liver microsomes is not stereoselective, and is mediated predominantly by CYP3A, and possibly CYP2C19 and CYP2C9 to a minor extent. Thus, the large interindividual variation reported for the pharmacokinetics of methadone may be due to variability in the expression of these CYP isoforms, and the reported stereoselectivity in the clearance of methadone in humans is not due to partial intrinsic clearance by the N-demethylation reaction.
Acknowledgments
The results were presented in part at the 29th annual meeting of the Australasian Society of Clinical and Experimental Pharmacologists and Toxicologists (ASCEPT) in December 1995. DJR Foster was a recipient of a Dawes Postgraduate Scholarship from the Royal Adelaide Hospital, Adelaide, South Australia. Funding for this research was provided by the Royal Adelaide Hospital and the University of Adelaide. The authors wish to thank the National Institute of Drug Abuse for supplying the drug compounds, and to acknowledge the contributions of Mr J. A. R. Williams, Head of the Hepatobiliary and Pancreatic Surgery Unit, Royal Adelaide Hospital, who provided the liver tissue, Ms Janet Coller of the Department of Clinical and Experimental Pharmacology, University of Adelaide, and Ms Heather James of the Division of Human Immunology, Institute of Medical and Veterinary Science, Adelaide, who genotyped the liver tissue.
References
- 1.Kristensen K, Christensen CB, Christrup LL. The mu1, mu2, delta, kappa opioid receptor binding profiles of methadone stereoisomers and morphine. Life Sci. 1995;56:45–50. doi: 10.1016/0024-3205(94)00426-s. [DOI] [PubMed] [Google Scholar]
- 2.Scott CC, Robbins EB, Chen KK. Pharmacologic comparison of the optical isomers of methadon. J Pharmacol Exp Ther. 1948;93:282–286. [PubMed] [Google Scholar]
- 3.Isbell H, Eisenman AJ. The addiction liability of some of the drugs of the methadon series. Fed Proc. 1948;7:162. [PubMed] [Google Scholar]
- 4.Kristensen K, Blemmer T, Angelo HR, et al. Stereoselective pharmacokinetics of methadone in chronic pain patients. Ther Drug Monit. 1996;18:221–227. doi: 10.1097/00007691-199606000-00001. [DOI] [PubMed] [Google Scholar]
- 5.Eap CB, Cuendet C, Baumann P. Binding of d-methadone, l-methadone, and dl-methadone to proteins in plasma of healthy volunteers: role of the variants of alpha 1-acid glycoprotein. Clin Pharmacol Ther. 1990;47:338–346. doi: 10.1038/clpt.1990.37. [DOI] [PubMed] [Google Scholar]
- 6.Romach MK, Piafsky KM, Abel JG, Khouw V, Sellers EM. Methadone binding to orosomucoid (alpha 1-acid glycoprotein): determinant of free fraction in plasma. Clin Pharmacol Ther. 1981;29:211–217. doi: 10.1038/clpt.1981.34. [DOI] [PubMed] [Google Scholar]
- 7.Sullivan HR, Smits SE, Due SL, Booher RE, McMahon RE. Metabolism of d-methadone: Isolation and identification of analgesically active metabolites. Life Sci. 1972;11:1093–1104. [Google Scholar]
- 8.Sullivan HR, Due SL. Urinary metabolites of dl-methadone in maintenance subjects. J Med Chem. 1973;16:909–913. doi: 10.1021/jm00266a009. [DOI] [PubMed] [Google Scholar]
- 9.Pond SM, Kreek MJ, Tong TG, Raghunath J, Benowitz NL. Altered methadone pharmacokinetics in methadone-maintained pregnant women. J Pharmacol Exp Ther. 1985;233:1–6. [PubMed] [Google Scholar]
- 10.Änggård E, Gunne LM, Homstrand J, McMahon RE, Sandberg CG, Sullivan HR. Disposition of methadone in methadone maintenance. Clin Pharmacol Ther. 1975;17:258–266. doi: 10.1002/cpt1975173258. [DOI] [PubMed] [Google Scholar]
- 11.Iribarne C, Berthou F, Baird S, et al. Involvement of cytochrome P450 3A4 enzyme in the N-demethylation of methadone in human liver microsomes. Chem Res Toxicol. 1996;9:365–373. doi: 10.1021/tx950116m. [DOI] [PubMed] [Google Scholar]
- 12.Iribarne C, Dreano Y, Bardou LG, Menez JF, Berthou F. Interaction of methadone with substrates of human hepatic cytochrome P450 3A4. Toxicol. 1997;117:13–23. doi: 10.1016/s0300-483x(96)03549-4. [DOI] [PubMed] [Google Scholar]
- 13.Iribarne C, Berthou F, Carlhant D, et al. Inhibition of methadone and buprenorphine N-dealkylations by three HIV- 1 protease inhibitors. Drug Metab Dispos. 1998;26:257–260. [PubMed] [Google Scholar]
- 14.Moody DE, Alburges ME, Parker RJ, Collins JM, Strong JM. The involvement of cytochrome P450 3A4 in the N-demethylation of l-alpha-acetylmethadol (laam), norlaam, and methadone. Drug Metab Dispos. 1997;25:1347–1353. [PubMed] [Google Scholar]
- 15.Zanger UM, Vilbois F, Hardwick JP, Meyer UA. Absence of hepatic cytochrome P450bufI causes genetically deficient debrisoquine oxidation in man. Biochemistry. 1988;27:5447–5454. doi: 10.1021/bi00415a010. [DOI] [PubMed] [Google Scholar]
- 16.Lowry OH, Roseburg NJ, Farr AL, Randall RJ. Measurement with the folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 17.Omura T, Sato R. The carbon monoxide-binding pigment of liver microsomes, II Solubilisation, purification and properties. J Biol Chem. 1964;234:2374–2385. [PubMed] [Google Scholar]
- 18.Steen VM, Andreassen OA, Daly AK, et al. Detection of the poor metabolizer-associated CYP2D6 (D) gene deletion allele by long-PCR technology. Pharmacogenetics. 1995;5:215–223. doi: 10.1097/00008571-199508000-00005. [DOI] [PubMed] [Google Scholar]
- 19.Heim M, Meyer UA. Genotyping of poor metabolisers of debrisoquine by allele-specific PCR amplification. Lancet. 1990;336:529–532. doi: 10.1016/0140-6736(90)92086-w. [DOI] [PubMed] [Google Scholar]
- 20.Coller JK, Somogyi AA, Bochner F. Association between CYP2C19 genotype and proguanil oxidative polymorphism. Br J Clin Pharmacol. 1997;43:659–660. doi: 10.1046/j.1365-2125.1997.00596.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Newton DJ, Wang RW, Lu AY. Cytochrome P 450 inhibitors. Evaluation of specificities in the in vitro metabolism of therapeutic agents by human liver microsomes. Drug Metab Dispos. 1995;23:154–158. [PubMed] [Google Scholar]
- 22.Andersson T, Miners JO, Veronese ME, Birkett DJ. Diazepam metabolism by human liver microsomes is mediated by both S-mephenytoin hydroxylase and CYP3A isoforms. Br J Clin Pharmacol. 1994;38:131–137. doi: 10.1111/j.1365-2125.1994.tb04336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.von Moltke LL, Greenblatt DJ, Court MH, Duan SX, Harmatz JS, Shader RI. Inhibition of alprazolam and desipramine hydroxylation in vitro by paroxetine and fluvoxamine: comparison with other selective serotonin reuptake inhibitor antidepressants. J Clin Psychopharmacol. 1995;15:125–131. doi: 10.1097/00004714-199504000-00008. [DOI] [PubMed] [Google Scholar]
- 24.Ripamonti C, Zecca E, Bruera E. An update on the clinical use of methadone for cancer pain. Pain. 1997;70:109–115. doi: 10.1016/s0304-3959(96)03286-1. [DOI] [PubMed] [Google Scholar]
- 25.Shimada T, Yamazaki H, Mimura M, Inui Y, Guengerich FP. Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians. J Pharmacol Exp Ther. 1994;270:414–423. [PubMed] [Google Scholar]
- 26.Shimada T, Yamazaki H, Mimura M, et al. Characterization of microsomal cytochrome P450 enzymes involved in the oxidation of xenobiotic chemicals in human fetal liver and adult lungs. Drug Metab Dispos. 1996;24:515–522. [PubMed] [Google Scholar]
- 27.Kuhn KL, Halikas JA, Kemp KD. Carbamazepine treatment of cocaine dependence in methadone maintenance patients with dual opiate-cocaine addiction. NIDA Res Monogr. 1989;95:316–317. [PubMed] [Google Scholar]
- 28.Saxon AJ, Whittaker S, Hawker CS. Valproic acid, unlike other anticonvulsants, has no effect on methadone metabolism: two cases. J Clin Psychiatry. 1989;50:228–229. [PubMed] [Google Scholar]
- 29.Tong TG, Pond SM, Kreek MJ, Jaffery NF, Benowitz NL. Phenytoin-induced methadone withdrawal. Ann Intern Med. 1981;94:349–351. doi: 10.7326/0003-4819-94-3-349. [DOI] [PubMed] [Google Scholar]
- 30.Liu SJ, Wang RI. Case report of barbiturate-induced enhancement of methadone metabolism and withdrawal syndrome. Am J Psychiatry. 1984;141:1287–1288. doi: 10.1176/ajp.141.10.1287. [DOI] [PubMed] [Google Scholar]
- 31.Kreek MJ, Garfield JW, Gutjahr CL, Giusti LM. Rifampin-induced methadone withdrawal. N Engl J Med. 1976;294:1104–1106. doi: 10.1056/NEJM197605132942008. [DOI] [PubMed] [Google Scholar]
- 32.Kosten TR, Gawin FH, Morgan C, Nelson JC, Jatlow P. Evidence for altered desipramine disposition in methadone-maintained patients treated for cocaine abuse. Am J Drug Alcohol Abuse. 1990;16:329–336. doi: 10.3109/00952999009001594. [DOI] [PubMed] [Google Scholar]
- 33.Maany I, Dhopesh V, Arndt IO, Burke W, Woody G, O’Brien CP. Increase in desipramine serum levels associated with methadone treatment. Am J Psychiatry. 1989;146:1611–1613. doi: 10.1176/ajp.146.12.1611. [DOI] [PubMed] [Google Scholar]
- 34.Kerry NL, Somogyi AA, Bochner F, Mikus G. The role of CYP2D6 in primary and secondary oxidative metabolism of dextromethorphan: in vitro studies using human liver microsomes. Br J Clin Pharmacol. 1994;38:243–248. doi: 10.1111/j.1365-2125.1994.tb04348.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu D, Otton SV, Sproule BA, et al. Inhibition of human cytochrome P450 2D6 (CYP2D6) by methadone. Br J Clin Pharmacol. 1993;35:30–34. doi: 10.1111/j.1365-2125.1993.tb05666.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Otton SV, Brinn RU, Gram LF. In vitro evidence against the oxidation of quinidine by the sparteine/debrisoquine monooxygenase of human liver. Drug Metab Dispos. 1988;16:15–17. [PubMed] [Google Scholar]
- 37.Chang TK, Gonzalez FJ, Waxman DJ. Evaluation of triacetyloleandomycin, alpha-naphthoflavone and diethyldithiocarbamate as selective chemical probes for inhibition of human cytochromes P450. Arch Biochem Biophys. 1994;311:437–442. doi: 10.1006/abbi.1994.1259. [DOI] [PubMed] [Google Scholar]
- 38.Pond SM, Tong TG, Benowitz NL, Jacob PD, Rigod J. Lack of effect of diazepam on methadone metabolism in methadone-maintained addicts. Clin Pharmacol Ther. 1982;31:139–143. doi: 10.1038/clpt.1982.22. [DOI] [PubMed] [Google Scholar]
- 39.Preston KL, Griffiths RR, Cone EJ, Darwin WD, Gorodetzky CW. Diazepam and methadone blood levels following concurrent administration of diazepam and methadone. Drug Alcohol Depend. 1986;18:195–202. doi: 10.1016/0376-8716(86)90051-7. [DOI] [PubMed] [Google Scholar]
- 40.von Moltke LL, Greenblatt DJ, Cotreau Bibbo MM, Harmatz JS, Shader RI. Inhibitors of alprazolam metabolism in vitro: effect of serotonin-reuptake-inhibitor antidepressants, ketoconazole and quinidine. Br J Clin Pharmacol. 1994;38:23–31. doi: 10.1111/j.1365-2125.1994.tb04317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nemeroff CB, DeVane CL, Pollock BG. Newer antidepressants and the cytochrome P450 system. Am J Psychiatry. 1996;153:311–320. doi: 10.1176/ajp.153.3.311. [DOI] [PubMed] [Google Scholar]
- 42.Andersson T, Miners JO, Veronese ME, Birkett DJ. Identification of human liver cytochrome P450 isoforms mediating secondary omeprazole metabolism. Br J Clin Pharmacol. 1994;37:597–604. doi: 10.1111/j.1365-2125.1994.tb04310.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Andersson T, Miners JO, Veronese ME, et al. Identification of human liver cytochrome P450 isoforms mediating omeprazole metabolism. Br J Clin Pharmacol. 1993;36:521–530. doi: 10.1111/j.1365-2125.1993.tb00410.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ko J-W, Sukhova N, Thacker D, Chen P, Flockhart DA. Evaluation of omeprazole and lansoprazole as inhibitors of cytochrome P450 isoforms. Drug Metab Dispos. 1997;25:853–862. [PubMed] [Google Scholar]